

Abstract
Lysosomal storage disorders (LSDs) are a group of about fifty life-threatening conditions caused by genetic defects affecting lysosomal components. The underscoring molecular deficiency leads to widespread cellular dysfunction through most tissues in the body, including peripheral organs and the central nervous system (CNS). Efforts during the last few decades have rendered a remarkable advance regarding our knowledge, medical awareness, and early detection of these genetic defects, as well as development of several treatment modalities. Clinical and experimental strategies encompassing enzyme replacement, gene and cell therapies, substrate reduction, and chemical chaperones are showing considerable potential in attenuating the peripheral pathology. However, a major drawback has been encountered regarding the suboptimal impact of these approaches on the CNS pathology. Particular anatomical and biochemical constraints of this tissue pose a major obstacle to the delivery of therapeutics into the CNS. Approaches to overcome these obstacles include modalities of local administration, strategies to enhance the blood-CNS permeability, intranasal delivery, use of exosomes, and those exploiting targeting of transporters and transcytosis pathways in the endothelial lining. The later two approaches are being pursued at the time by coupling therapeutic agents to affinity moieties and drug delivery systems capable of targeting these natural transport routes. This approach is particularly promising, as using paths naturally active at this interface may render safe and effective delivery of LSD therapies into the CNS.
Keywords: Lysosomal storage disorders, blood-brain barrier, targeting, transcytosis, drug delivery systems
1. LYSOSOMAL STORAGE DISORDERS AND THE CENTRAL NERVOUS SYSTEM
Lysosomal storage disorders (LSDs) are a group of about fifty different life-threatening, genetically inherited diseases that affect both humans and several animal species [1]. They are characterized by a deficiency of lysosomal function within cells throughout the body [1, 2]. The following three sections will provide a brief description of these pathologies, focusing on their impact on the central nervous system (CNS) and current treatment modalities.
1.1. Lysosomal Storage Disorders: A Brief Overview
Lysosomes are intracellular membranous compartments involved in the degradation of biomolecules that are either self-elements of the cell or taken up from the extracellular milieu (necessary for molecular turnover, signaling, and metabolism), and also serve as a defense against foreign substances and pathogens entering the cell [3]. Degradation within the lysosome is catalyzed by hydrolytic enzymes, with the help of activator molecules and the acidic environment generated in this organelle by ion pumps, and transport proteins also being important [3] (Figure 1). Hence, LSDs can be caused by genetic defects affecting any of these elements and, as a consequence, are commonly characterized by aberrant accumulation of undegraded metabolites within lysosomal compartments. For each one of the LSDs, the substance(s) accumulated or “stored” within the lysosome depends on the particular enzyme or catabolic pathway affected, including several types of lipids, glycoproteins, or mucopolysaccharides [4]. Disease progression has also been associated to the appearance and accumulation of these substances in other cellular locations as well as in the circulation, possibly due to lysosomal disruption or aberrant transport to/from this compartment [1, 2].
Figure 1. Lysosomal storage disorder pathology.

LSDs are caused by genetic defects affecting the lysosomal machinery, including lysosomal enzymes, elements of the pathway regulating their glycosylation, lysosomal activators or cofactors, or membrane transporters located in this compartment. As a consequence, aberrant lysosomal accumulation of metabolites occurs, influencing the function of this organelle, as well as other processes involving transit of membranous vesicles (e.g., endocytosis, recycling, secretion, autophagy), as well as cell signaling and communication. Cell homeostasis is ultimately affected, leading to dysfunction affecting most tissues in the body, including peripheral organs and the CNS.
The initial biochemical dysfunction in each particular LSD causes imbalances not only in the associated metabolic route and possible downstream interaction with other pathways, but also causes secondary effects that more broadly impact cell homeostasis (Figure 1). For instance, because lysosomes are in many cases the last stage with regard to endocytic vesicular transport, endocytosis may be affected in these deficiencies [1, 2]. Also other functions depending on vesicular motility, and membrane fusion and fission events can be impacted, including autophagy, the secretory pathway, cell signaling and communication, etc. Altogether, this leads to cell dysfunction [1, 2].
Depending on the metabolic pathway directly affected by the genetic deficiency, particular cell types within the body can be more or less impacted in each LSD. However, because lysosomes are housekeeping organelles, these diseases typically affect multiple tissues, including both peripheral organs as well as the CNS [2, 4] (Figure 1). LSD clinical manifestations typically include neurological deterioration, as well as hepatic and splenic dysfunctions [5, 4]. Other functions and tissues influenced by LSDs include pulmonary, cardiovascular, and renal activities, muscle, bone, connective tissue, the immune system, and others [5, 4]. The biochemical and metabolic stresses generated also affect the overall functional status of these tissues, which often causes oxidative stress with leukocyte activation. This ultimately leads to an inflammatory component that exacerbates and “spreads” the initial pathology [5, 4].
Regarding the severity of LSDs, this depends on several parameters, including the particular pathway and main tissue(s) affected, the genetic deficiency leading to residual versus no enzyme activity, and epigenetic causes. In severe cases, the clinical phenotype manifests soon after birth and causes patient mortality within the newborn period, which is common for LSDs impacting the CNS [5, 4]. In less severe cases, the clinical manifestation may develop during infancy and progress to adulthood, which is often associated with LSDs characterized by non-neurological abnormalities affecting peripheral organs [5, 4]. Importantly, all LSDs are associated with high morbidity and premature mortality [5, 4].
1.2. General Strategies for Treatment of Lysosomal Storage Disorders
The development of effective therapies for LSDs has been historically hampered due to the low incidence of these illnesses, which ranges between 1:25,000 to 1:250,000 for each individual disease, with 1:5,000 to 1:7,000 affected newborns as a group [6]. This fact made it specially difficult to attract significant support for translational endeavors for LSDs. Also, inherent factors pertaining to the biological and medical complexity of many of these pathologies have rendered a limited number of treatments clinically available [7].
Classically, therapeutic strategies have consisted of symptomatic palliative approaches and in some cases liver, bone marrow, or mesenchymal stem cell transplantation, which has been explored in animal models and implemented in the clinics, for instance in the case of Gaucher disease, Pompe disease, Krabbe disease, mucopolysaccharidosis (MPS) I, Hurler syndrome, and others [5, 8]. However, lack of compatible donors and graft-versus-host disease hinders these approaches [9–11]. These strategies have been put to the practice in an attempt to overcome life-threatening damage directly affecting these tissues or their derived counterparts, and also because healthy cells expressing normal copies of a given lysosomal enzyme can theoretically correct the defect in the neighboring tissue via a phenomenon called “cross correction” [12]. The cross correction effect represents a major pillar in the design of several therapeutic strategies, hence, this will be described here in more detail.
When endogenous lysosomal enzymes are synthesized within a cell, their precursors are transported from the endoplasmic reticulum (ER) to the Golgi apparatus, and then the lysosome. In transit between the first two organelles, oligosaccharides containing mannose residues are added to lysosomal enzyme precursors [13, 14]. The glycosylated enzymes are additionally phosphorylated at their mannose residues, rendering forms that display mannose-6-phosphate (M6P) [14]. Through M6P residues, these enzymes bind to M6P receptors in the Golgi network, and the “bound” enzyme molecules are transported via clathrin-coated pits that ultimately fuse to lysosomes, releasing the enzyme from the receptor due to the acidic pH in this compartment [14]. Alternatively, due to saturation of M6P receptors in the Golgi system and/or incomplete phosphorylation of mannose residues, a fraction of the synthesized enzyme travels via the secretory pathway and is secreted to the extracellular environment [15]. These “secreted” enzyme molecules can act in a “paracrine” manner, as they can bind to mannose receptors (MR) expressed on monocytes and macrophages, or to M6P receptors that are also present on the plasma membrane of most cell types in the body [15, 16]. Binding to these cell surface receptors results in clathrin-mediated endocytosis with transport to lysosomes [15]. Hence, functional enzymes “secreted” by healthy transplanted cells can be internalized and transported to lysosomes in neighboring diseased cells, correcting their enzymatic deficiency [12].
Enzyme replacement therapy is another treatment strategy directly derived from the cross correction effect and is clinically available for a (still reduced) number of LSDs with no major CNS involvement [17, 18]. This includes six FDA-approved products in the United States, for treatment of diseases with no major CNS syndrome, such as MPS I (Hurler disease), MPS II (Hunter disease), MPS VI (Maroteaux-Lamy syndrome), Fabry disease, Gaucher Type I and III, and Pompe disease [5]. This strategy consist of infusion of tissue-extracted or, rather, recombinantly-produced lysosomal enzymes into the systemic circulation, with potential binding to mannose or M6P receptors on cells [17, 18]. Post-production modifications aiding in exposing these binding residues, pharmacological agents to increase receptor expression, and use of recombinant enzymes fused to peptides providing cell binding by glycosylation-independent mechanisms (discussed below), have been attempted in order to optimize the outcome of this therapy [19–26]. Importantly, low amounts of the “correcting” enzyme (≤ 10–20%) need to be delivered within lysosomes in order to observe a therapeutic effect, making this a relevant option[1].
Since LSDs are monogenic disorders, it is theoretically possible to correct the gene deficiency and, hence, the pathological outcome through gene therapy. This treatment option is currently under investigation in small and large animal models for a number of LSDs, including Pompe disease, Fabry disease, several MPS types, etc., and currently approved for clinical trials in the case of Sanfilippo and Pompe disease [5, 27]. This strategy includes somatic gene transfer using viral vectors in animal models using adenovirus, adeno-associated virus, herpes virus, retrovirus, and lentivirus vectors capable of integrating the exogenous gene sequences into the host genome for prolonged enzyme expression [28–31]. In principle, because of the multiplicity of tissues and organs affected in LSDs, it would be impractical to transform all cells affected in the body. Yet, due to cross correction this is not necessary, as transformation of key cell types may provide not only self rectification of the transformed cells, but also a long-term supply of active enzyme for neighboring cells. As in the case of enzyme replacement therapy, relatively low level of enzyme activity may attenuate these pathologies and delay their progression.
In the case of both enzyme replacement by infusion of recombinant enzymes or gene therapy modalities, several approaches are focusing on cross correction strategies where enzymes can enter deficient cells via glycosylation-independent mechanisms. This strategy represents an advantage in cases where recombinant enzymes are not properly glycosylated or to overcome low levels or function of M6P receptors observed in some LSDs [32, 33]. This can be done by designing fusion proteins containing an enzymatic moiety fused to a peptide that can be recognized by cell surface receptors in a glycosylation-independent manner. For instance, this is the case for chimeric β-glucuronidase (deficient MPS VII) fused to a peptide derived from the insulin-like growth factor II, which binds to a different domain of the M6P receptor, following the same clathrin-mediated pathway for lysosomal delivery [20]. This strategy was tested using skin fibroblasts from MPS VII patients and the corresponding mouse model, where this fusion protein showed enhanced binding despite removal M6P residues from the enzyme [20]. Similarly, some chimeric enzymes have been designed to display the receptor associated protein RAP [23]. This molecule is located in the endoplasmic reticulum, where it binds as a chaperone to newly synthesized LDLR family receptors, preventing premature association to their physiological ligands [24]. Cell surface LDLR family receptors targeted by RAP are also internalized by cells via clathrin-dependent endocytosis [24]. This strategy has been explored in cell cultures in the case of α-L-iduronidase or α-glucosidase (deficient in MPS I and Pompe disease, respectively) [24]. In addition to targeting to endocytic receptors, fusion enzymes containing protein transduction domains, such as peptides derived from the HIV transcription factor Tat, have been designed to improve delivery of recombinant lysosomal enzymes by simply providing charge-dependent binding to the plasma membrane [34–37]. Subsequently, internalization can occur via passive uptake within endocytic vesicles and/or via direct penetration through the plasmalemma. This strategy has been investigated in the case of gene therapy for β-glucuronidase, glucocerebrosidase, and galactocerebrosidase, deficient in MPS VII, Gaucher, and Krabbe diseases, respectively [34–37].
Apart from these strategies based on cross correction, a few other approaches capitalize on alternative elements [38]. For instance, substrate reduction therapy is based on the use of small molecules that inhibit biosynthesis of the metabolites accumulated in a particular LSD, hence helping in reducing their storage [39–41]. This approach holds great potential and is available clinically in some cases. This is the case for the use of Miglustat (N-butyldeoxinojirimycin), which inhibits the first step of the glycosphingolipid biosynthesis pathway, a common route for a number of deficiencies, such as Gaucher disease, Fabry disease, and type C Niemann-Pick [5]. Some inhibitor molecules also serve as chemical chaperones at sub-inhibitory concentrations, improving folding of mutated lysosomal enzymes. An example is that of the use of 1-deoxygalactonojirimycin in clinical trials for Fabry disease patients [5]. This has an application in the case of patients with misfolding defects, leading to improved lysosomal transport and residual activity [42–45].
A related experimental strategy capitalizes on cytosolic molecules that control intracellular trafficking of lysosomes, facilitating reduction of metabolites whose storage is caused by defective lysosomal transporters. This is the case for Rab9 that, when overexpressed in cell culture, attenuates the cellular phenotype associated to defects on lysosomal NPC1 or NPC2 transport proteins in type C Niemann-Pick disease, which is characterized by accumulation of cholesterol and sphingolipids [46]. Also to tackle said cholesterol accumulation, cyclodextrin derivatives are under investigation. Cyclodextrins are small sugar molecules with a ring-like structure capable of extracting cholesterol from the plasma membrane and intracellular compartments [47]. In the case of LSDs characterized by cholesterol accumulation, such as type C Niemann-Pick disease mentioned above, pharmacological reduction of the storage by cyclodextrin derivatives has been shown to help attenuate the disease phenotype in animal models [47]. This approach is currently under clinical trial [47].
Finally, modalities of combination therapy encompassing more than one of these approaches are recently being considered as an interesting alternative for the treatment of LSDs, which may provide synergistic benefits to patients. This includes, for example, the use of enzyme replacement therapy along with substrate reduction therapy, clinically implemented in type III Gaucher disease, or gene therapy along with stem cell transplantation, under investigation in animal models of MPS VII and types A and B Niemann-Pick disease [48].
1.3. Central Nervous System Pathology in LSDs and Obstacles to Treatment
Between 1/2 and 1/3 of all LSDs are characterized by neurological dysfunction, leading to developmental delay, movement disorders, ataxia, seizures, spasticity, psychiatric disease, visual loss, etc [49]. Neurodegenerative complications are characterized by both neuronal dysfunction as well as neuronal loss, the later often associated to Purkinje cells involved in motor coordination in the cerebellar cortex [8]. This has been observed in a number of syndromes, including gangliosidosis, types A and C Niemann-Pick disease, neuronal ceroid lipofuscinosis, etc [8]. Lysosomal storage has been observed to also occur in glial cells, e.g., microglia, whose storage could be contributed by clearance of apoptotic cells in the CNS, altering their function [8]. Storage in neurons can impact both anterograde and retrograde transport of vesicles along the axon, communication with the cell body, and synaptic function, with genesis and morphology of dendrites also being affected [8].
At the molecular level, LSDs involving accumulation of glycosphingolipids and related gangliosides, with strong a neurological component, have been related to disruption of lipid raft domains and their main function in terms of membrane trafficking and cell signaling. Altered calcium homeostasis, e.g., with depletion of calcium from intracellular (ER) stores, has also been observed in neurons, where this cationic second messenger is involved in cell signaling pathways and long-term plasticity and responses [50]. In turn, ER calcium depletion can cause ER stress and activate the unfolded protein response pathway, leading to apoptosis [51]. This has been reported in the case of animal models of Gaucher disease, Sandhoff disease, and GM1 gangliosidosis, and confirmed in several other neuropathies [8]. Concomitantly to these changes, there is an inflammatory phenotype mediated by the microglia as well as macrophages secondly recruited to the brain, and T lymphocytes being involved in advanced pathological stages. Oxidative stress associated to this inflammatory outcome exacerbates neurotoxicity, altogether contributing to disease progression. Unfortunately, most therapeutic modalities discussed in the previous section, although helpful regarding treatment of the condition in peripheral tissues, have shown rather suboptimal efficacy regarding treatment of the neurological pathology associated with LSDs [52].
Strategies such as those involving small drugs, namely substrate reduction therapy or chaperone-mediated approaches, seem more amenable to overcome the blood-brain barrier (BBB; discussed in detail in section 2.1) and are under investigation regarding their potential in the case of neuropathic disorders [43]. Some of these compounds, such as those that inhibit synthesis of glycosphingolipids, are being used in patients since they can be conveniently administered orally and have been previously shown to extend the pre-symptomatic period, also delaying disease progression and increasing life expectancy in animal models [53].
However, the BBB poses an impenetrable obstacle for efficient transport of other compounds, such as cyclodextrin derivatives, viral vectors, and enzymes from the circulation into the brain, which negatively impacts the efficacy of these strategies, primarily those based on the cross correction effect. For instance, in the case of bone marrow, stem cell or organ transplantation in peripheral regions outside the CNS, the enzymes “secreted” by the transplanted cells do not exert an efficient cross correction toward cells in the CNS since they cannot enter this compartment of the body [10, 54]. Similarly, recombinant enzymes injected into the circulation gain rapid access to cells in certain peripheral organs but do not access the brain. This is also the case for many strategies of gene therapy, which have shown to provide relevant cross correction in peripheral organs but are largely inefficient in the CNS [28]. In both cases, enzyme and gene therapies, correction of the defects seems to align somehow with the natural patterns of blood flow in the body, as expected. For instance, some targeting of the pulmonary capillary bed has been detected, as this constitutes a first-pass tissue after intravenous injection [27]. Also, this is the case for the RES such as in the spleen and liver, where the parenchyma becomes accessible to substances injected in the circulation, due to the presence of fenestrations in the endothelial lining [27]. Multiple i.v. injections with high doses of recombinant enzymes have been attempted in order to overcome this challenge and improve CNS delivery, yet clinical trials have not reflected a main advantage of this strategy [55]. In several cases, CNS expression of systemically administered viral vectors have been shown to be rather low and transient, if any, in part promoted by immune reactions [5].
To address these obstacles, a considerable effort is being devoted to develop strategies exploiting alternative administration routes or taking advantage of natural transport mechanisms into the CNS. These aspects will be developed in the following section.
2. MEANS OF DRUG DELIVERY INTO THE CENTRAL NERVOUS SYSTEM
As mentioned above, transport across the BBB is a formidable obstacle for treatment of medical conditions affecting the CNS, leading to a relatively low capability to effectively and safely deliver therapeutics into this tissue. The following sections will elaborate on the BBB and methods to overcome this obstacle.
2.1. Barriers of Transport into the Central Nervous System
Transport of fluid, macromolecules, and blood cells into tissues in the body typically occurs through capillaries of the vascular system where only a thin (200 – 300 nm) layer of endothelial cells separates the blood from the tissue parenchyma and its cell components [56, 57]. Transport through this endothelial layer is effective via passive passage through discontinuous vessels in the liver, spleen and organs of the RES, where large gaps or fenestrations exist in the endothelial lining [58, 59]. Contrarily, endothelial cells in other organs form a continuous layer with sealed junctions between the adjacent cells [58, 60]. These junctions are primarily formed by intercellular protein complexes located in areas of the plasma membrane between neighboring endothelial cells.
There are several types of these cell-cell contacts, most relevantly including tight junctions and adherens junctions. In both cases, these structures encompass transmembrane proteins (e.g., claudins, occludins and JAMs for tight junctions, and cadherins for adherens junctions), which are capable of homophilic interactions with their counterparts in the neighboring cells, forming a zipper-like attachment at the cell periphery in the cell-cell border area [58, 60]. The cytoplasmic domain of these “adhesive” transmembrane proteins typically interacts with “scaffold” protein partners. These in turn connect to cell signaling elements and the cytoskeleton, altogether allowing control over the formation and potential remodeling of the junction, as well as communication with the adjacent tissue environment [58, 60]. In vascular endothelial cells, contrarily to more proper epithelial linings, these two individual junctions are less distinguishable as separate entities, as their components are somehow intertwined [58, 60]. In any case, while adherens junctions primarily serve as an adhesive and regulatory contact interface, tight junctions constitute the real permeability barrier that hinders free transport of most substances between neighboring cells [58, 60]. Additionally, to control free transport of substances, these junctions help compartmentalize functional elements at the luminal versus abluminal endothelial membrane, contributing to interaction of the endothelium with both the blood and tissue parenchyma interfaces [58, 60].
Because of this and other reasons further discussed below, the endothelial layer is rather impermeable in the CNS. This anatomical (and biochemical) barrier is formed progressively during the developmental stages, with endothelial cells of the perineural vessels being initially fenestrated, despite displaying tight junctions [58, 60]. Then fenestrae disappear when endothelial cells establish contact with other cells of the CNS, stimulated also by local soluble factors [61]. In the adult CNS, this barrier is located at the interface of the endothelium with: (a) the brain parenchyma (BBB), (b) the choroid plexus epithelium (blood-cerebrospinal fluid (CSF) barrier), and (c) the arachnoid epithelium (meningeal barrier) [62, 63]. Additionally, this permerability barrier function is found not only in the brain, but also in the spinal cord and other neural tissues, including the retina, nerves, and the labyrinth [64]. The complexity of these interfaces becomes apparent when examining the BBB, one of the most studied blood-CNS barriers (Figure 2). The BBB is finely regulated by concerted interaction of the brain endothelium with astrocytes and pericytes, and chemical communication with other cells of the brain parenchyma, such as neurons and the microglia. Indeed, the expression “neurovascular unit” is currently preferred when describing this complex interface [61], very challenging to reproduce in experimental cell model systems.
Figure 2. Anatomy and transport in the blood-brain barrier.
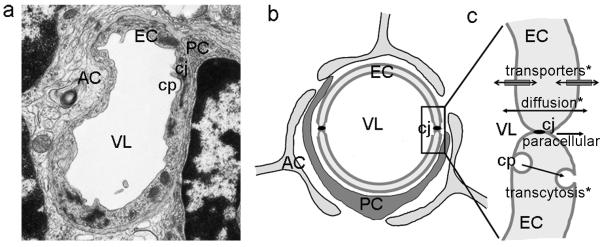
(a) Transmission electron microscopy of a capillary in the brain and its (b) schematic representation. Endothelial cells (EC) surrounding the vessel lumen (VL), with pericytes (PC) and feet of astrocytes (AC) adjacent to the endothelial layer. Endothelial cells display electron dense cell junctions (cj) sealing the cell-cell borders. A forming clathrin-coated pit (cp) can be observed in the endothelial luminal surface. (c) Transport of small and lipophilic molecules across the endothelial lining can occur between the cell junctions (the paracellular pathway, almost defunct in the BBB) or by diffusion across the cell. In addition, other pathways of transport across the cell layer include transporters located in the endothelia luminal and abluminal surfaces, as well as vesicular transcytosis (mainly mediated by clathrin-mediated mechanisms). * transcellular.
The presence of these blood-CNS barriers compartmentalizes and protects the CNS tissue from potential interference of substances and cells circulating in the bloodstream with respect to the neural function. This homeostasis is disrupted under certain pathological stimuli, such as the presence of glioblastomas, stroke, degenerative diseases and inflammatory processes, which may lead to enhanced permeability [63, 64]. Parasites, bacteria, and viruses can also invade the CNS by bypassing this barrier, either as free entities or carried within immune cells [61]. These insults lead to an altered architecture of the endothelial tight junctions and/or expression of transporters, increasing movement of immune cells across the blood-CNS barrier, along with secretion and transport of neuroinflammatory substances [63, 64].
However, except for such pathological scenarios, transport from the circulation into the CNS parenchyma is highly controlled and restricted to particular elements (Figure 2). For these substances, transport occurs via several mechanisms, including the paracellular route between the junctions described above and the transcellular route, which encompasses transmembrane diffusion, saturable transporters, and vesicular transcytosis (the latter pathway is in many cases the only one referred to as “transcellular”) [65]. Drug delivery into the CNS can either exploit these options or bypass the blood-CNS barrier by local administration of therapeutic agents into the CNS [65–67]. Yet, altogether, the presence of the blood-CNS barrier hinders penetration of most (~98+%) drugs and biotechnological agents currently available.
2.2. Local Administration into the Central Nervous System
Therapeutic agents can be directly delivered within the CNS using surgical procedures, mostly involving drilling and injecting through the cranium into the brain or between the vertebrae in the spinal cord [68, 69]. This encompasses modalities such as administration through a catheter into the epidural space over the dura mater, or intrathecal administration in the leptomeningeal space under the arachnoid (hence, in the CSF), which are most typically dispensed along the spinal cord. Injections in the brain are also performed, which can deliver therapeutics into the CSF in the brain ventricles (intra-ventricular or -cerebroventricular administration) or directly into the brain parenchyma (intra-cerebral or -parenchymal administration) [68, 69].
A major downside of these local administration techniques resides in the sub-optimal diffusion of drug from the point of administration through the CNS tissue. For instance, with respect to intraventricular injections, the rate of CSF clearance from the brain has been reported to be significantly faster than the rate of solute diffusion from the ependymal surface into the brain parenchyma [68]. In some cases, drug release into the blood via this mechanism appears to achieve greater levels than those attained within the brain [70]. Similarly, parenchymal drug diffusion upon intracerebral injection decreases exponentially with increasing distance, which has been speculated to translate into a 90% reduction in drug concentration after traveling just 500 μm distance from the injection point [71]. To compensate for these deficiencies, higher drug doses need to be administered, hence most of these delivery modalities render relatively high drug concentrations in the delivery site, often resulting in toxicity [72].
These drawbacks can be partially overcome by brain implantation of convention-enhanced diffusion devices [73]. In this case, the drug is continuously or sequentially infused at a given rate into the brain via a catheter affixed to a pump and a reservoir containing the drug. The goal of this approach is to use convection to supplement diffusion, so that the drug can penetrate further from the administration site. Although this technique certainly improves this aspect, the resistance to fluid flow once the drug enters the brain tissue hinders deep penetration [74]. Also, this generates an increase in the intraparenchymal fluid, since the CNS lacks effective systems of drainage of exogenously added fluid, rendering fluid movement into the white mater. This restricts the application of this technique and also impacts the appearance of potential side effects, such as astroglia activation and demyelination [69].
Nevertheless, approaches of local administration within the CNS have shown therapeutic utility and, as for other applications, are being implemented for delivery of treatments for neuropathic LSDs. For instance, CSF injections have been largely explored in animal models, for experimental gene therapies using viral vectors as well as enzyme replacement therapies with recombinant lysosomal enzymes [27]. This is also the case for clinical trials of intrathecal therapies for Hurler syndrome, MPS I and MPS III [27]. However, intraventricular or intrathecal administration of enzymes leads to a relatively small fraction of the injected dose effectively entering the brain parenchyma; yet in some cases this appears to be sufficient to detect enzyme entering neurons and oligodendrocytes in the brain, attaining measurable therapeutic benefit [75, 76]. Similarly, intra-CSF injection of viral vectors has been observed to render transduction of cells in the ventricular system, including those of the ependymal layer, the choroid plexus, and the central canal in the spinal cord [27]. This has been speculated to generate a relatively stable source of therapeutic enzyme being released into the CSF, with the capability of cross correcting the lysosomal defect in cells of the brain parenchyma by the mechanisms described above [27, 29, 31, 77].
Intraparenchymal administration has also been explored and offers benefits for both enzyme and gene therapies, including examples of implantation of cells transduced ex vivo [31, 54, 78]. This has been explored in the case of rodent models of several MPS syndromes, type A Niemann-Pick disease, gangliosidosis, and others [27]. As the case of intra-CSF delivery, the cross correction effect provides a means to benefit the tissue adjacent to the application site, and recombinant enzymes have been observed to enter neurons using axonal transport mechanisms [79]. However, given the chronic nature of LSDs, recurrent intraparenchymal administration of exogenous enzymes (versus viral vectors or transducted cells) using such invasive procedures is impractical. With regard to gene therapy, viral tropism seems to have an important impact on the therapeutic outcome of this strategy, where serotypes can be selected or engineered to transduce particular cellular types which represent main targets for intervention in certain LSDs [5, 78, 80].
Noticeably, these procedures involving intra-CNS administration are relatively invasive and, therefore, not amenable for repeated implementation (needed in the case of chronic diseases), due to the safety risks and high cost involved. As a consequence, peripheral administration of therapeutics for transport into the CNS is preferable.
2.3 Peripheral Administration
Transport of substance from peripheral tissues into the CNS can occur through the paracellular route between junctions or the transcellular route that encompasses transmembrane diffusion, saturable transporters, and vesicular transcytosis. In addition, although less characterized, the intranasal route seems amenable for noninvasive transport of certain substances into the CNS, which takes place by diffusion across the nasal mucosa, transit into the perivascular channels in the lamina propia or along the olfactory or trigeminal nerves, finally reaching the CNS and olfactory bulbs [81]. A number of therapeutic agents have been shown to reach the CNS in this manner, including small lipophilic molecules and peptide hormones [65, 82–84].
With respect to the paracellular pathway, this seems particularly restricted in the case of the blood-CNS barrier, as compared to other vascular endothelial beds or other epithelial linings where the junctions can be regulated to allow transport of some substances. Several approaches have aimed to transiently disrupt this barrier, e.g., by using hyperosmotic solutions, vasoactive agents, vaccine adjuvants, ultrasound irradiation, and optical techniques [68, 85, 86]. A number of works employing administration of therapeutics in the presence of common solvents and stabilizers, such as SDS, DMSO, ethanol, polysorbate-80, glycerol, etc., have also shown improved CNS delivery by causing transient disruption of the permability barrier [87–89]. Although this can certainly increase transport into the brain, lack of specificity causes leakage of blood substances into the brain parenchyma, with side effects. For instance, albumin leakage into the brain has been shown to cause astrocyte toxicity and opening of the barrier also causes chronic neuropathological changes [90, 91].
Regarding diffusion through the endothelial cell membrane at the blood-CNS interface, this is a non-saturable pathway that depends largely on physicochemical nature of the substances to be transported [65, 68]. In order to cross the phospholipid bilayer in the plasmalemma, molecules can be designed to have a low molecular weight ≤ 400–600 Da, yet this is a parameter not fully understood since some peptides cross the BBB via diffusion despite their larger size [64]. Molecules should also have relatively high lipid solubility, with total capacity for hydrogen bonding with water ≤8. Yet, the lipid solubility cannot exceed a threshold or otherwise the molecule to be transported would remain at the endothelial layer, unable to cross and be released into the brain parenchyma [65]. An additional obstacle to using this transport route is the existence of efflux transporters (e.g., P-glycoprotein), which limits the amount of drug capable of entering the brain via this mechanism [68].
Efflux transporters are just one of the transporter systems located in the BBB, while other examples exert an influx activity. These are saturable pathways where transporter proteins located at the plasmalemma facilitate direct uptake into the cell cytoplasma and/or out into the tissue [65]. Also, in most cases they offer selectivity toward the molecule being transported, e.g., those for glucose, L-DOPA, vitamin B12, cationic or large neutral amino acids, etc. Transport rates via this route are much higher than that of simple diffusion mechanisms, hence beneficial from the perspective of drug delivery. Nevertheless, larger molecules cannot use this route, which can additionally be regulated up or down depending on physiological and pathological status of the system [65, 68, 92, 93].
Perhaps one of the most promising means for non-invasive, safe, and relatively efficient transport into the CNS is provided by vesicular transcytosis. This is a natural route permitting transit of relatively large molecules, which largely controls transport between the extra and intra CNS compartment [68, 94]. This occurs via membranous vesicles that are typically induced by binding of specific ligands to their receptors located in the endothelial plasmalemma. Binding leads to engulfment of the ligand by the plasma membrane, with formation of vesicles that pinch off into the cytosol, through a process globally call endocytosis [94, 95]. These vesicles are then trafficked across the cell body, sometimes through intermediate compartments, and finally fuse to the plasma membrane in the opposite side of the cell (abluminal or luminal, depending whether transport is into or from the CNS), releasing the cargo [68, 94, 95]. There are two main mechanisms serving the transcytosis route in most cell layers in the body, the so called clathrinand caveolar-mediated pathways [68, 94, 95]. Caveolar-mediated transport occurs through caveoli, small (≤100 nm diameter) flask-shaped invaginations of the plasma membrane, characterized by the presence of the protein caveolin in specialized cholesterol- and sphingolipid-enriched membrane domains, referred to as lipid rafts. Although the caveolar-mediated mechanism appears to be the primary transcytosis pathway in other vascular endothelial beds, such as the case of the lung, it is almost inexistent in the case of the BBB [68, 96]. Instead, clathrin-mediated transcytosis operates in this endothelium. In this case, ligands that bind to their plasmalemma receptors are internalized within slightly larger (100 – 200 nm diameter) vesicles called clathrin-coated pits, in virtue of a characteristic cytosolic lattice of the clathrin proteins [68, 94, 95]. Several receptors have been indentified to serve this pathway in the BBB, including receptors for transferrin, lipoproteins, insulin, insulin-like growth factors, and leptin, among others [68, 97, 98].
A number of works have capitalized in this modality of receptor-mediated transport into the CNS to develop treatment approaches for neurological conditions, including LSDs. For instance, some recombinant lysosomal enzymes have been shown to be transported in this manner via the M6PR, such as the case of β-glucoronidase (deficient in MPS VII) and phosphorylated sulfaminase (deficient in MPS IIIA), studied in mouse models [99]. However, results in this study indicated that such a strategy may only be efficient in neonates, e.g., in mice the M6PR is downregulated in the BBB soon after birth [99]. Instead, the insulin receptor and transferrin receptor are active in adult brain endothelium, and this approach has been used with recombinant fusion proteins where the lysosomal enzymes are tagged by antibody-derived peptides targeting these receptors [23, 100, 101]. Similar targeting strategies for transport of gene therapy viral vectors across endothelial barriers via receptor-mediated transcytosis have also been reported [102, 103].
This route has been speculated to be used also by exosome-mediated strategies, which represents a particularly interesting new modality of therapeutic delivery into the CNS. Exosomes are natural membranous vesicles ~50–100 nm in diameter, which are secreted by fusion of multivesicular bodies to the plasma membrane in certain cells (e.g., mast cells, dendritic cells, etc) [104]. They naturally contain specific arrays of nucleic acids and proteins, can be loaded with therapeutics, and can be recombinantly modified to display targeting moieties in their membrane [104]. A recent study showing 60% mRNA and protein knowdown of a therapeutic target in the brain after systemic injection of siRNA-loaded exosomes, highlights the great potential of this strategy. Although this remains to be characterized, receptor-mediated transport by transcytosis across the BBB seems to mediate entrance of exosomes from the bloodstream into the CNS [104].
Finally, as described below, this route also constitutes one of the most promising for transport of nanomedicines and drug delivery systems across the blood-CNS barrier.
3. DRUG TARGETING AND DELIVERY SYSTEMS FOR TRANSPORT OF THERAPEUTICS INTO THE BRAIN
Targeting strategies and drug delivery vehicles consist of either molecular conjugates (the “Trojan horse” approach) or macromolecular assemblies (referred to as carriers) that can incorporate imaging and therapeutic compounds. These systems can improve several aspects of therapeutics, including their solubility, stability, circulation, biodistribution, subcellular transport, and/or release rate, altogether enhancing their bioavailability and efficacy [105–108]. The following section will touch upon these aspects, with a particular focus on transport into the CNS.
3.1. Types and Properties of Drug Targeting and Delivery Systems
Targeting of drugs to areas of the body where their action is required can be facilitated by improving their local retention and bioadhesion, a relevant aspect in order to attain enhanced therapeutic outcomes. This can be achieved by tagging drugs with moieties that display affinity toward general biological features, e.g., positively charged peptides (HIV Tat, RGD sequences, and others) can provide affinity to the negative-charged plasma membrane of cells [37, 69]. Alternatively, drugs can be tagged with affinity moieties that recognize specific markers on cells, including antibodies and their fragments, proteins and peptides, sugars, vitamins, aptamers, etc [69]. Coupling of the targeting and therapeutic units can be done by direct synthesis/production, for instance in the case of recombinant proteins (e.g., recombinant lysosomal enzymes described in section 2.2.), as well as chemical conjugation, which can be based on covalent linkage, electrostatic or hydrophobic interaction, or through interactive biological molecules, such as the biotin-avidin pair [69, 109–114]. In addition, these conjugates can be targeted to cellular receptors involved in endocytic transport or coupled to cell penetrating peptides, providing delivery to a variety of intracellular compartments [94, 95].
Apart from these targeting strategies, drug delivery systems offer several other advantages to formulation of therapeutics (Figure 3). Drug delivery carriers can be fabricated using biological, synthetic, and semi-synthetic materials. They can be assembled in linear, branched or dendrimer structures, spherical or filamentous micelles, hollow capsules, and porous or solid particles. Furthermore, their functionality and performance can be tuned by modulating their physicochemical properties [115–119]. The most typical size of drug delivery systems rages under a micrometer in size, for which they are often referred to as “nanocarriers”. These two terms will be used hereafter.
Figure 3. Functionalization and transport of drug delivery systems.
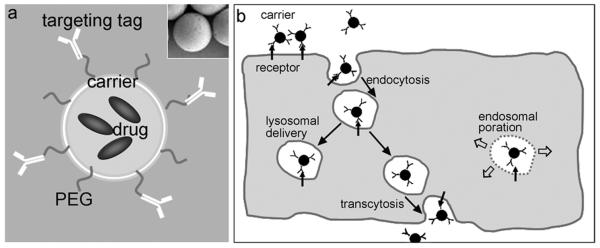
(a) A schematic representation of a model drug delivery system is shown. Drugs can be encapsulated, while the carrier shell can be functionalized to display poly(ethylene-glycol) (PEG) for stealth properties and prolonged circulation, as well as affinity moieties to target cells and tissues of therapeutic interest. (b) Drug carriers that are targeted to cell receptors involved in endocytosis can be internalized by cells. This often leads to transport into endo-lysosomal vesicles for: (i) pH dependent release of drugs aimed to act within this compartment or the cytosol (if permeable); (ii) endosomal poration by reactive materials, allowing leakage of drugs or biologicals into the cytoso; or (iii) transcytosis with release in the abluminal side of the cell.
Liposomes are likely the oldest and most popularly recognized drug delivery system. They consist of phospholipid-based capsules, that can be loaded with either hydrophilic compounds in their inner aqueous lumen or onto their surface, while allowing loading of hydrophobic counterparts in the lipid bilayer [119–121]. Polymerosomes are the polymer analog of liposomes, formed from block co-polymers bearing polymer chains composed of both hydrophobic and hydrophilic moieties, with higher functionalization capacity [115, 122]. Polymers can also be formulated as linear or branched structures conjugated (covalently or not) to drugs, to improve their bioavailabilty. Dendrimers are tree-like hyperbranched polymer carriers with a very high surface-to-volume ratio along with good drug complexation capacity and easy diffusion through tissues, due to their small size and architecture [123, 124]. Nanoparticles of different sizes and shapes can also be fabricated with either natural polymers or natural molecular blocks reacted to form polymers, and can display solid, hollow, or multi-porous structures [107, 125]. Some examples of the materials employed in the fabrication of these carriers include chemically inert and non-toxic counterparts (e.g., poly(methyl methacrylate), poly(acrylic acid), poly(vinyl alcohol), poly(ethylene glycol)), natural polysaccharide and polypeptide polymers (e.g., chitosan, alginate, gelatin, albumin), or polymerized organic molecules (polycaprolactone, polylactides, polyglycolides) [119].
Drug delivery carriers can be functionalized to prolong circulation and enhance biodistribution in the body at the tissue, cellular, and sub-cellular level (Figure 3). For instance, often the body recognizes foreign materials injected in the circulation, leading to relatively rapid clearance mediated by the RES in the liver and spleen, other elements of the immune system, and kidney filtration, among others [120]. This can be attenuated by coupling to poly(ethylene glycol) or PEG either directly or after encapsulation in nanocarriers, which minimizes interactions with plasma opsonins, complement, phagocytic cells, and lymphocytes related to specific immunity [120, 126] (Figure 3). Coupling of drug carriers to CD47, a transmembrane protein that acts like a marker of the “self” to avoid clearance by elements of the innate immune system, is another alternative for prolonging circulation and inhibiting inflammatory reactions [127]. Apart from systemically administered formulations, nanocarriers can also improve the drug efficacy upon release in the case of local administration, e.g., by loading drugs within materials capable of releasing the therapeutic agent in response to properties of the microenvironment, or by displaying a sustained release pattern over prolonged periods of time [128].
These drug delivery system can also take advantage of the targeting strategies described above. This can be achieved by formulating them to display affinity toward general biological features, such as the case for positively-charged polymers or carriers coupled to positively charged peptides already mentioned [107, 116]. Importantly (discussed in the following section), as compared to coupling of therapeutic agents directly to affinity moieties, carriers bearing multiple copies of a given targeting molecule display greater avidity due to this multivalency [129] (Figure 3).
3.2. Drug Targeting and Delivery Systems for LSDs
The advantages described above can clearly enhance current formulation of small chemical drugs employed in treatment of LDSs, as well as enzyme or gene therapy modalities, evolving into new or complementary interventions. For instance, carrier coupling or encapsulation can help prolong circulation and sustain the release of therapies, lowering the frequency of administration and potential side effects. Indeed, one of the drawbacks of enzyme replacement therapies injected systemically is the appearance of immunological effects that ultimately hinder the transport into the cell and/or enzymatic activity [130, 131]. It is likely that this can be overcome by encapsulating or crosslinking recombinant enzymes using biodegradable carriers, where they can be masked from the immune system and released upon carrier degradation within the lysosome [132–135].
Also, when glycosylation-independent targeting is to be exploited (section 2.2.), this can be achieved relatively simply by coupling existing recombinant enzymes to targeted drug delivery carriers. Such carriers can be used as generic vehicles avoiding the need to design and produce individual fusion proteins or conjugates, where fusion or conjugation signifies an extra effort and may ultimately hinder the enzymatic activity. Also, multivalency of targeted carriers is an important parameter that can be regulated. For instance the avidity of multivalent targeted formulations needs to overcome the threshold for effective targeting in a physiological environment, e.g., under dragging forces of the shear stress caused by the blood flow in the circulation or in the presence of natural ligands for the selected receptor. Conversely, if the avidity is too high, then binding to off-target areas may occur (e.g., if the receptor targeted is expressed is low amounts in other organs or tissues) or binding can be impaired due to steric hindrances that depend of the expression level and location of the receptor on the cell surface [129, 137, 138].
In addition to the properties discussed above, conjugates and drug delivery systems can be targeted to cellular receptors involved in endocytic transport or coupled to cell penetrating peptides, providing delivery to a variety of intracellular compartments [94, 95]. Endocytic transport of carriers and conjugates often provides endo-lysosomal delivery, which can be exploited for delivery of enzyme replacement therapies [7]. Furthermore, targeting cell receptors associated to the transcytosis pathway described in section 2.1. helps transport the carried drugs across cellular layers, such as the epithelial lining in the gastrointestinal track for oral delivery or the blood-CNS barriers in the case of systemic administration of drugs for neurological conditions [68]. This is the case for brain delivery of recombinant lysosomal enzymes targeted to the LDL receptor family, insulin-like growth factor receptor, or the transferrin receptor described in section 2.3. [23, 100, 101], and that of lysosomal enzymes delivered via ICAM-1-targeting nanocarriers (section 3.3.). For instance, it was recently shown that α-L-iduronidase fused to a peptide derived from an antibody against the human insulin receptor accumulated in the brain of adult Rhesus monkey following intravenous administration [20]. This chimeric enzyme was also capable of attenuating about 70% of the glycosaminoglycan accumulation typical in MPS I. A similar strategy tested in the MPS I mouse model demonstrated that delivery of a plasmid codifying for a fusion protein comprising transferrin and α-L-iduronidase was also effective in attenuating glycosaminoglycan in the mouse cerebellum [23].
Additionally, transport into lysosomes by conjugates and nanocarriers can be used to render cytosolic accumulation of small cell permeable compounds upon entering this compartment. This can be achieved by designing formulations such that the drug moiety can be released from the targeting moiety, e.g., by proteolytic cleavage of the conjugate within this organelle [119]. Also, a variety of other strategies can be used in this direction, including elements derived from biological molecules, such as pathogen toxins, or materials that experience changes in their physicochemical properties upon contact with this low pH compartment, releasing their cargo within the lysosome [117]. Then membrane permeable compounds can penetrate into the cell cytosol and exert a therapeutic action. Even though a priori it may appear that small membrane permeable compounds (e.g., those employed in substrate reduction therapy or chaperone-mediated strategies) would not need intracellular delivery by these means, such an approach has been shown to help avoiding rapid drug removal from the intracellular milieu via efflux pumps, which can be beneficial in several tissues and the BBB [65, 69]. More obviously, gene therapies requiring cytosolic delivery can also be enhanced by such designs, as well as materials that can help permeate the plasmalemma.
In this direction, endo-lysosomal escape can be achieved by a diversity of materials, e.g., it has been observed for biocompatible PLGA carriers [139] and it is a common property of cationic polymers, such as poly-L-lysine and poly(ethylenimine), whose amine groups become protonated at endosomal pH, resulting in osmotic swelling that bursts the endosome [140]. Since these cationic polymers display toxicity, other experimental formulations more amenable for in vivo applications. This is the case for pH-sensitive poly(acrylic acid) derivative carriers that act as proton sponges within lysosomes, and temperature-responsive polyelectrolyte hydrogels whose hydration rate varies with temperature, leading to changes in the carrier volume within the endo-lysosomal compartment [116, 141, 142]. Positively-charged cell penetrating peptides (such as RGD and Tat peptides) also bind to the cell surface due to electrostatic interactions and can facilitate cytosolic delivery of cargoes [143, 144]. Carriers can also be coupled to fusogenic peptides derived from bacterial toxins (e.g., hemaglutinnin and GALA peptides), which can induce poration of endosomal membrane in response to gradual pH lowering in these compartments [145].
Some of these concepts are being explored, e.g., coupling of enzymes to PEG, PEG-modified liposomes, and polymer nanoparticles targeted to endocytic receptors for systemic administration, or entrapment into biocompatible polymers for local implantation [146–154]. Needless to say, a thorough investigation on the potential short and/or chronic effects of these carrier-mediated applications will be required in order to optimize their properties. This is compelling in order to advance their translation, since it has been observed that deposition of insoluble or colloidal nanomaterials in the CNS can prompt production of reactive oxygen species, causing inflammation and contributing to neurodegeneration [155, 156]. Yet, this is also the case regarding side effects of viral gene therapies applied to the CNS, and should be manageable by controlling the degradation of the carriers. Hence, these strategies hold considerable promise in modulating the biodistribution pattern within the body, and rendering enhanced and prolonged enzyme activity in tissues. The following section will elaborate deeper on a relevant example of these applications.
3.3. ICAM-1-Targeting for Delivery and Treatment of LSDs
As described above, the LSDs are characterized by multi-organ dysfunction, with most tissues of the body being affected at some extent. Hence, an interesting generic target for developing LSD treatment modalities [2, 4] would be a molecule broadly expressed and capable of accessing both peripheral organs and the CNS after systemic administration. Enhanced expression in areas particularly affected by pathology versus those less severely impacted, would also provide benefit. Preferentially, this molecular target should be accessible to conjugates and larger drug delivery systems, to increase the level of versatility of the intended applications, and should also be able to induce transport across cellular barriers and into cells, for safe and efficient therapeutic delivery. Intercellular adhesion molecule 1 (ICAM-1), an immunoglobulin-like transmembrane glycoprotein that mediates adhesion of fibrinogen and leukocytes to inflammation sites [157, 158], seems to be a particularly interesting target for lysosomal interventions as it meets these requirements.
ICAM-1 is expressed by most LSD target cells, including vascular endothelial cells of vessels irrigating all organs, leukocytes, epithelial cells, glial cells, neurons, muscle cells, etc [157], and is accessible from the intravascular, intratracheal, or intracerebral routes [159]. Its expression is enhanced by many pathological stimuli concurring with LSDs, including metabolic imbalance, oxidative stress, inflammation, and others [157, 160, 161]. Targeting to this molecule is being explored for treatment of chronic inflammatory conditions, infection by certain pathogens, and cancer [157, 160, 161]. Numerous works by our group and others have shown successful ICAM-1 targeting in the case of antibodies and peptides, isotopes and contrast probes, biosensors, biologicals (including enzymes) and other therapeutics, liposomes and polymer nanocarriers [129, 153, 162, 163], in cell cultures, animals models, and clinical trials [152, 153, 162–154]. This is also the case for lysosomal enzymes, such as acid sphingomyelinase deficient in types A and B Niemann-Pick disease, α-galactosidase deficient in Fabry disease, and α-glucosidase deficient in Pompe disease [151–154, 170]. Targeting to ICAM-1 has been shown to provide a remarkable enhancement in the amount of enzymes accumulated in organs after intravenous administration in mice as compared to free counterparts, e.g., ~200-fold and ~15-fold in the lung and liver, major targets for type B Niemann-Pick disease, ~5-fold and ~1.5-fold in the heart and kidneys, major targets for Fabry disease, and ~10-fold and ~6-fold in the heart and skeletal muscle, major targets in Pompe disease [151–153].
Although ICAM-1 is not an endocytic receptor per se, binding of conjugates and drug delivery systems to this molecule induces vesicular transport into cells, resulting in efficient intracellular delivery of therapeutic and diagnostic agents [151–153, 167, 171–173]. Interestingly, uptake via ICAM-1 is mediated via a unique pathway called cell adhesion molecule or CAM-mediated endocytosis, which is distinct from clathrin- and caveolar-mediated pathways described in section 2.1, and also from macropinocytosis and phagocytosis typical of immune cells [173, 174] (Figure 4). Binding of carriers to ICAM-1 leads to interaction with the sodium-proton exchanger molecule NHE1 and formation of engulfment areas on the plasmalemma [173, 174]. These regions are enriched in cholesterol, sphingomyelin and gangliosides, with specific recruitment of intracellular sphingomyelinases and concomitant enrichment of ceramide [175]. Ceramide appearance at sites of engulfment of ICAM-1 targeting likely regulates physicochemical properties of the plasma membrane and signals for reorganization of actin cytoskeleton. Tyrosine phosphorylation, activation of protein kinase C, Src kinases, and Rho dependent kinase are also involved in the signaling cascade leading to uptake [174–176]. As a consequence, the actin cytoskeleton reorganizes into stress fibers, which attach to the cytosolic domain of the ICAM-1/NHE1 complex [173 174]. Ceramide-mediated progression of invaginations at binding sites, dynamic changes of actin filaments, and activity of dynamin result in endocytosis [173, 175]. Interestingly, targeting of lysosomal enzymes to ICAM-1 has shown to follow the CAM-mediated pathway, bypassing clathrin-coated pits used by free lysosomal enzymes [151, 152, 154].
Figure 4. ICAM-1 mediated pathway for transport of therapeutic carriers.

(a) The initial events elicited upon binding of ICAM-1-targeted carriers to endothelial cells include interaction between ICAM-1 and the sodium/proton exchanger protein NHE1 in lipid raft domains enriched in sphingomyelin. This leads to recruitment of acid sphingomyelinase to carrier binding sites, acidification of the immediate extracellular region by NHE1, and ceramide generation. Ceramide facilitates formation of engulfment structures and re-organization of the actin cytoskeleton into stress fibers, altogether leading to endocytosis of the drug carrier. (b) The signaling cascade governing this pathway is shown, including activation of protein kinase C (PKC), Src kinase, Rho and Rho-dependent kinase (ROCK). This cascade leads to formation of endocytic structures that pinch off the plasmalemma helped by dynamin. Subsequently, carriers are transported to early endosomes, where ICAM-1 separates from ICAM-1-targeted carriers. ICAM-1 recycles to the plasma membrane and carriers are transported to both lysosomes and the abluminal membrane (by transcytosis).
This aspect is important because, in contrast to clathrin and caveolar endocytosis, characterized by vesicles ~100–150 nm and ~50–80 nm diameter, CAM-endocytosis renders uptake of carriers with various sizes and shapes, e.g., 100 nm to 5 μm size, spherical, elongated, or irregular [170, 174, 175]. This feature is relevant since carriers with high surface to volume ratios, such as filamentous micelles with ≤ 20 nm in diameter and several micrometers in length, appear to offer enhanced circulation and minimal unspecific interactions in animal models. Additionally, after uptake of drug carriers, ICAM-1 recycles to the plasma membrane and hence provides sustained targeting by circulating vehicles [172] (Figure 4). In addition, conjugates and carriers targeted to ICAM-1 traffic to lysosomes (Figure 4), which has proven beneficial from the perspective of delivery of lysosomal enzymes for replacement therapies. Indeed, delivery of acid sphingomyelinase, α-galactosidase, and α-glucosidase by this route has been shown to enhance lysosomal degradation of sphingomyelin, globotriaosylceramide, and glycogen as compared to free enzymes [151, 152, 154, 170]. Moreover, the rate of endocytosis and lysosomal traffic via the CAM-mediated pathway can be modulated by varying several parameters, such as the molecular epitope to which carriers binding [177], the carrier size and shape [170], and pharmacological agents impacting the cell machinery that regulates endocytic transport [171, 173].
Importantly, in a cell culture model of a permeability barrier composed of epithelial cells, ICAM-1-targeted carriers transport significant amounts of lysosomal enzymes across the cell monolayer, which occurs via a transcytosis pathway related to CAM endocytosis, without opening of the tight junctions or unspecific leakage (Figure 4). This result pairs well with enhanced accumulation of lysosomal enzymes in mouse brain after a single intravenous administration, e.g., ~7-fold for acid sphingomyelinase, ~4-fold for α-galactosidase, and ~6-fold for α-glucosidase, suggesting that ICAM-1-targeting may provide a new avenue for delivery of therapeutic lysosomal enzymes to the CNS [151, 152].
4. Final Remarks
The CNS represents a major target tissue for therapeutic intervention of numerous LSDs. Yet, effective treatment of the pathology at this site is largely hampered by particular limitations regarding the architecture of this tissue and the highly controlled transport of substances between the blood and the CNS parenchyma. Overcoming these obstacles will require a concerted and multidisciplinary effort aimed to provide a deep understanding of the anatomical and physiological regulation of this tissue and the natural transport mechanisms at this interface, along with technological development of drug delivery approaches.
ACKNOWLEDGEMENTS
This work was funded by the National Institutes of Health grant R01-HL98416.
REFERENCES
- 1.Ballabio A, Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta. 2009;1793(4):684–96. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5(7):554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 3.Sabatini DD, Adesnick MB. The metabolic and molecular bases of inherited disease. 2001. The biogenesis of membranes and organelles; pp. 433–520. [Google Scholar]
- 4.Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–80. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- 5.Seregin SS, Amalfitano A. Gene therapy for lysosomal storage diseases: progress, challenges and future prospects. Curr Pharm Des. 2011;17(24):2558–74. doi: 10.2174/138161211797247578. [DOI] [PubMed] [Google Scholar]
- 6.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. Jama. 1999;281(3):249–54. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 7.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2(2):189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6(9):713–25. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- 9.Jin HK, Carter JE, Huntley GW, Schuchman EH. Intracerebral transplantation of mesenchymal stem cells into acid sphingomyelinase-deficient mice delays the onset of neurological abnormalities and extends their life span. J Clin Invest. 2002;109(9):1183–91. doi: 10.1172/JCI14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malatack JJ, Consolini DM, Bayever E. The status of hematopoietic stem cell transplantation in lysosomal storage disease. Pediatr Neurol. 2003;29(5):391–403. doi: 10.1016/j.pediatrneurol.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Vellodi A, Young EP, Cooper A, Wraith JE, Winchester B, Meaney C, et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child. 1997;76(2):92–9. doi: 10.1136/adc.76.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barton RW, Neufeld EF. The Hurler corrective factor. Purification and some properties. J Biol Chem. 1971;246(24):7773–9. [PubMed] [Google Scholar]
- 13.Kornfeld S, Reitman ML, Varki A, Goldberg D, Gabel CA. Steps in the phosphorylation of the high mannose oligosaccharides of lysosomal enzymes. Ciba Found Symp. 1982;(92):138–56. doi: 10.1002/9780470720745.ch8. [DOI] [PubMed] [Google Scholar]
- 14.Rosenfeld MG, Kreibich G, Popov D, Kato K, Sabatini DD. Biosynthesis of lysosomal hydrolases: their synthesis in bound polysomes and the role of co- and post-translational processing in determining their subcellular distribution. J Cell Biol. 1982;93(1):135–43. doi: 10.1083/jcb.93.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16(1):77–84. [PubMed] [Google Scholar]
- 16.Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gaucher's disease. Lancet. 1996;348(9041):1555–9. doi: 10.1016/S0140-6736(96)04451-0. [DOI] [PubMed] [Google Scholar]
- 17.Brady RO. Enzyme replacement therapy: conception, chaos and culmination. Philos Trans R Soc Lond B Biol Sci. 2003;358(1433):915–9. doi: 10.1098/rstb.2003.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3(12):954–66. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 19.Achord DT, Brot FE, Bell CE, Sly WS. Human beta-glucuronidase: in vivo clearance and in vitro uptake by a glycoprotein recognition system on reticuloendothelial cells. Cell. 1978;15(1):269–78. doi: 10.1016/0092-8674(78)90102-2. [DOI] [PubMed] [Google Scholar]
- 20.LeBowitz JH, Grubb JH, Maga JA, Schmiel DH, Vogler C, Sly WS. Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci U S A. 2004;101(9):3083–8. doi: 10.1073/pnas.0308728100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, et al. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987;329(6137):301–7. doi: 10.1038/329301a0. [DOI] [PubMed] [Google Scholar]
- 22.Murray GJ. Lectin-specific targeting of lysosomal enzymes to reticuloendothelial cells. Methods Enzymol. 1987;149:25–42. doi: 10.1016/0076-6879(87)49041-1. [DOI] [PubMed] [Google Scholar]
- 23.Osborn MJ, McElmurry RT, Peacock B, Tolar J, Blazar BR. Targeting of the CNS in MPS-IH using a nonviral transferrin-alpha-L-iduronidase fusion gene product. Mol Ther. 2008;16(8):1459–66. doi: 10.1038/mt.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prince WS, McCormick LM, Wendt DJ, Fitzpatrick PA, Schwartz KL, Aguilera AI, et al. Lipoprotein receptor binding, cellular uptake, and lysosomal delivery of fusions between the receptor-associated protein (RAP) and alpha-L-iduronidase or acid alpha-glucosidase. J Biol Chem. 2004;279(33):35037–46. doi: 10.1074/jbc.M402630200. [DOI] [PubMed] [Google Scholar]
- 25.Sands MS, Vogler CA, Ohlemiller KK, Roberts MS, Grubb JH, Levy B, et al. Biodistribution, kinetics, and efficacy of highly phosphorylated and non-phosphorylated beta-glucuronidase in the murine model of mucopolysaccharidosis VII. J Biol Chem. 2001;276(46):43160–5. doi: 10.1074/jbc.M107778200. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Y, Li X, Schuchman EH, Desnick RJ, Cheng SH. Dexamethasone-mediated up-regulation of the mannose receptor improves the delivery of recombinant glucocerebrosidase to Gaucher macrophages. J Pharmacol Exp Ther. 2004;308(2):705–11. doi: 10.1124/jpet.103.060236. [DOI] [PubMed] [Google Scholar]
- 27.Gritti A. Gene therapy for lysosomal storage disorders. Expert Opin Biol Ther. 2011;11(9):1153–67. doi: 10.1517/14712598.2011.582036. [DOI] [PubMed] [Google Scholar]
- 28.Ellinwood NM, Vite CH, Haskins ME. Gene therapy for lysosomal storage diseases: the lessons and promise of animal models. J Gene Med. 2004;6(5):481–506. doi: 10.1002/jgm.581. [DOI] [PubMed] [Google Scholar]
- 29.Haskins M. Gene therapy for lysosomal storage diseases (LSDs) in large animal models. ILAR J. 2009;50(2):112–21. doi: 10.1093/ilar.50.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karolewski BA, Wolfe JH. Genetic correction of the fetal brain increases the lifespan of mice with the severe multisystemic disease mucopolysaccharidosis type VII. Mol Ther. 2006;14(1):14–24. doi: 10.1016/j.ymthe.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13(5):839–49. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Cardone M, Porto C, Tarallo A, Vicinanza M, Rossi B, Polishchuk E, et al. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics. 2008;1(1):6. doi: 10.1186/1755-8417-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dhami R, Schuchman EH. Mannose 6-phosphate receptor-mediated uptake is defective in acid sphingomyelinase-deficient macrophages: implications for Niemann-Pick disease enzyme replacement therapy. J Biol Chem. 2004;279(2):1526–32. doi: 10.1074/jbc.M309465200. [DOI] [PubMed] [Google Scholar]
- 34.Lee KO, Luu N, Kaneski CR, Schiffmann R, Brady RO, Murray GJ. Improved intracellular delivery of glucocerebrosidase mediated by the HIV-1 TAT protein transduction domain. Biochem Biophys Res Commun. 2005;337(2):701–7. doi: 10.1016/j.bbrc.2005.05.207. [DOI] [PubMed] [Google Scholar]
- 35.Xia H, Mao Q, Davidson BL. The HIV Tat protein transduction domain improves the biodistribution of beta-glucuronidase expressed from recombinant viral vectors. Nat Biotechnol. 2001;19(7):640–4. doi: 10.1038/90242. [DOI] [PubMed] [Google Scholar]
- 36.Zhang XY, Dinh A, Cronin J, Li SC, Reiser J. Cellular uptake and lysosomal delivery of galactocerebrosidase tagged with the HIV Tat protein transduction domain. J Neurochem. 2008;104(4):1055–64. doi: 10.1111/j.1471-4159.2007.05030.x. [DOI] [PubMed] [Google Scholar]
- 37.Orii KO, Grubb JH, Vogler C, Levy B, Tan Y, Markova K, et al. Defining the pathway for Tat-mediated delivery of beta-glucuronidase in cultured cells and MPS VII mice. Mol Ther. 2005;12(2):345–52. doi: 10.1016/j.ymthe.2005.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smid BE, Aerts JM, Boot RG, Linthorst GE, Hollak CE. Pharmacological small molecules for the treatment of lysosomal storage disorders. Expert Opin Investig Drugs. 19(11):1367–79. doi: 10.1517/13543784.2010.524205. [DOI] [PubMed] [Google Scholar]
- 39.Butters TD, Dwek RA, Platt FM. New therapeutics for the treatment of glycosphingolipid lysosomal storage diseases. Adv Exp Med Biol. 2003;535:219–26. doi: 10.1007/978-1-4615-0065-0_14. [DOI] [PubMed] [Google Scholar]
- 40.Platt FM, Jeyakumar M. Substrate reduction therapy. Acta Paediatr Suppl. 2008;97(457):88–93. doi: 10.1111/j.1651-2227.2008.00656.x. [DOI] [PubMed] [Google Scholar]
- 41.Radin NS. Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj J. 1996;13(2):153–7. doi: 10.1007/BF00731489. [DOI] [PubMed] [Google Scholar]
- 42.Alfonso P, Pampin S, Estrada J, Rodriguez-Rey JC, Giraldo P, Sancho J, et al. Miglustat (NB-DNJ) works as a chaperone for mutated acid beta-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells Mol Dis. 2005 doi: 10.1016/j.bcmd.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki Y, Ogawa S, Sakakibara Y. Chaperone therapy for neuronopathic lysosomal diseases: competitive inhibitors as chemical chaperones for enhancement of mutant enzyme activities. Perspect Medicin Chem. 2009;3:7–19. doi: 10.4137/pmc.s2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. Faseb J. 2005;19(1):12–8. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- 45.Porto C, Cardone M, Fontana F, Rossi B, Tuzzi MR, Tarallo A, et al. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther. 2009;17(6):964–71. doi: 10.1038/mt.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narita K, Choudhury A, Dobrenis K, Sharma DK, Holicky EL, Marks DL, et al. Protein transduction of Rab9 in Niemann-Pick C cells reduces cholesterol storage. Faseb J. 2005 doi: 10.1096/fj.04-2714fje. [DOI] [PubMed] [Google Scholar]
- 47.Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, et al. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001;70(2):131–42. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- 48.Hawkins-Salsbury JA, Reddy AS, Sands MS. Combination therapies for lysosomal storage disease: is the whole greater than the sum of its parts? Hum Mol Genet. 20(R1):R54–60. doi: 10.1093/hmg/ddr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bellettato CM, Scarpa M. Pathophysiology of neuropathic lysosomal storage disorders. J Inherit Metab Dis. 33(4):347–62. doi: 10.1007/s10545-010-9075-9. [DOI] [PubMed] [Google Scholar]
- 50.Korkotian E, Schwarz A, Pelled D, Schwarzmann G, Segal M, Futerman AH. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J Biol Chem. 1999;274(31):21673–8. doi: 10.1074/jbc.274.31.21673. [DOI] [PubMed] [Google Scholar]
- 51.Tessitore A, del PMM, Sano R, Ma Y, Mann L, Ingrassia A, et al. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15(5):753–66. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 52.Begley DJ, Pontikis CC, Scarpa M. Lysosomal storage diseases and the blood-brain barrier. Curr Pharm Des. 2008;14(16):1566–80. doi: 10.2174/138161208784705504. [DOI] [PubMed] [Google Scholar]
- 53.Schiffmann R. Therapeutic approaches for neuronopathic lysosomal storage disorders. J Inherit Metab Dis. 33(4):373–9. doi: 10.1007/s10545-010-9047-0. [DOI] [PubMed] [Google Scholar]
- 54.Hodges BL, Cheng SH. Cell and gene-based therapies for the lysosomal storage diseases. Curr Gene Ther. 2006;6(2):227–41. doi: 10.2174/156652306776359522. [DOI] [PubMed] [Google Scholar]
- 55.Shire Human Genetics Therapies I Open-label extension study of recombinant human arylsulfatase A (HGT-1111) in late infantile metachromatic leukodystrophy. ClinicalTrialsgov. NCT00681811. Available at: http://clinicaltrials.gov/ct2/show/NCT00681811?term=NCT00681811&rank=1.
- 56.Minshall RD, Tiruppathi C, Vogel SM, Malik AB. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol. 2002;117(2):105–12. doi: 10.1007/s00418-001-0367-x. [DOI] [PubMed] [Google Scholar]
- 57.Stevens T, Garcia JG, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L419–22. doi: 10.1152/ajplung.2000.279.3.L419. [DOI] [PubMed] [Google Scholar]
- 58.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5(4):261–70. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 59.Mehta D, Bhattacharya J, Matthay MA, Malik AB. Integrated control of lung fluid balance. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1081–90. doi: 10.1152/ajplung.00268.2004. [DOI] [PubMed] [Google Scholar]
- 60.Blasig IE, Haseloff RF. Tight junctions and tissue barriers. Antioxid Redox Signal. 15(5):1163–6. doi: 10.1089/ars.2011.4003. [DOI] [PubMed] [Google Scholar]
- 61.Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. 2005;25(1):25–39. doi: 10.1007/s10571-004-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rapoport SI. Opening of the blood-brain barrier by acute hypertension. Exp Neurol. 1976;52(3):467–79. doi: 10.1016/0014-4886(76)90218-1. [DOI] [PubMed] [Google Scholar]
- 63.Sharma HS. Blood-CNS barrier, neurodegeneration and neuroprotection: recent therapeutic advancements and nano-drug delivery. J Neural Transm. 2011;118(1):3–6. doi: 10.1007/s00702-010-0542-0. [DOI] [PubMed] [Google Scholar]
- 64.Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7(1):84–96. doi: 10.1016/S1474-4422(07)70326-5. [DOI] [PubMed] [Google Scholar]
- 65.Banks WA. Blood-brain barrier as a regulatory interface. Forum Nutr. 2009;63:102–10. doi: 10.1159/000264398. [DOI] [PubMed] [Google Scholar]
- 66.Kabanov AV, Batrakova EV. New technologies for drug delivery across the blood brain barrier. Curr Pharm Des. 2004;10(12):1355–63. doi: 10.2174/1381612043384826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cornford EM, Cornford ME. New systems for delivery of drugs to the brain in neurological disease. Lancet Neurol. 2002;1(5):306–15. doi: 10.1016/s1474-4422(02)00136-9. [DOI] [PubMed] [Google Scholar]
- 68.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 69.Pardridge WM. Biopharmaceutical drug targeting to the brain. J Drug Target. 2010;18(3):157–67. doi: 10.3109/10611860903548354. [DOI] [PubMed] [Google Scholar]
- 70.Aird RB. A study of intrathecal, cerebrospinal fluid-to-brain exchange. Exp Neurol. 1984;86(2):342–58. doi: 10.1016/0014-4886(84)90192-4. [DOI] [PubMed] [Google Scholar]
- 71.Fung LK, Shin M, Tyler B, Brem H, Saltzman WM. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res. 1996;13(5):671–82. doi: 10.1023/a:1016083113123. [DOI] [PubMed] [Google Scholar]
- 72.Day-Lollini PA, Stewart GR, Taylor MJ, Johnson RM, Chellman GJ. Hyperplastic changes within the leptomeninges of the rat and monkey in response to chronic intracerebroventricular infusion of nerve growth factor. Exp Neurol. 1997;145(1):24–37. doi: 10.1006/exnr.1997.6448. [DOI] [PubMed] [Google Scholar]
- 73.Patel MM, Goyal BR, Bhadada SV, Bhatt JS, Amin AF. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs. 2009;23(1):35–58. doi: 10.2165/0023210-200923010-00003. [DOI] [PubMed] [Google Scholar]
- 74.Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459–66. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 75.Dickson PI. Novel treatments and future perspectives: outcomes of intrathecal drug delivery. Int J Clin Pharmacol Ther. 2009;47(Suppl 1):S124–7. [PubMed] [Google Scholar]
- 76.Calias P, Papisov M, Pan J, Savioli N, Belov V, Huang Y, et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS One. 2012;7(1):e30341. doi: 10.1371/journal.pone.0030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Watson G, Bastacky J, Belichenko P, Buddhikot M, Jungles S, Vellard M, et al. Intrathecal administration of AAV vectors for the treatment of lysosomal storage in the brains of MPS I mice. Gene Ther. 2006;13(11):917–25. doi: 10.1038/sj.gt.3302735. [DOI] [PubMed] [Google Scholar]
- 78.Sands MS, Haskins ME. CNS-directed gene therapy for lysosomal storage diseases. Acta Paediatr Suppl. 2008;97(457):22–7. doi: 10.1111/j.1651-2227.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Passini MA, Macauley SL, Huff MR, Taksir TV, Bu J, Wu IH, et al. AAV vector-mediated correction of brain pathology in a mouse model of Niemann-Pick A disease. Mol Ther. 2005;11(5):754–62. doi: 10.1016/j.ymthe.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 80.Vandenberghe LH, Wilson JM, Gao G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009;16(3):311–9. doi: 10.1038/gt.2008.170. [DOI] [PubMed] [Google Scholar]
- 81.Dhuria SV, Hanson LR, Frey WH., 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–73. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 82.Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 2004;29(10):1326–34. doi: 10.1016/j.psyneuen.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 83.Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL. Sniffing neuropeptides: a transnasal approach to the human brain. Nat Neurosci. 2002;5(6):514–6. doi: 10.1038/nn849. [DOI] [PubMed] [Google Scholar]
- 84.Westin U, Piras E, Jansson B, Bergstrom U, Dahlin M, Brittebo E, et al. Transfer of morphine along the olfactory pathway to the central nervous system after nasal administration to rodents. Eur J Pharm Sci. 2005;24(5):565–73. doi: 10.1016/j.ejps.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 85.Choi M, Ku T, Chong K, Yoon J, Choi C. Minimally invasive molecular delivery into the brain using optical modulation of vascular permeability. Proc Natl Acad Sci USA. 2011;108(22):9256–61. doi: 10.1073/pnas.1018790108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund's adjuvant in mice provoke leakage of serum proteins through the blood-brain barrier without inducing reactive gliosis. Brain Res. 1999;832(1–2):84–96. doi: 10.1016/s0006-8993(99)01479-1. [DOI] [PubMed] [Google Scholar]
- 87.Azmin MN, Florence AT, Handjani-Vila RM, Stuart JF, Vanlerberghe G, Whittaker JS. The effect of non-ionic surfactant vesicle (niosome) entrapment on the absorption and distribution of methotrexate in mice. J Pharm Pharmacol. 1985;37(4):237–42. doi: 10.1111/j.2042-7158.1985.tb05051.x. [DOI] [PubMed] [Google Scholar]
- 88.Hanig JP, Morrison JM, Jr., Krop S. Ethanol enhancement of blood-brain barrier permeability to catecholamines in chicks. Eur J Pharmacol. 1972;18(1):79–82. doi: 10.1016/0014-2999(72)90134-3. [DOI] [PubMed] [Google Scholar]
- 89.Kobiler D, Lustig S, Gozes Y, Ben-Nathan D, Akov Y. Sodium dodecylsulphate induces a breach in the blood-brain barrier and enables a West Nile virus variant to penetrate into mouse brain. Brain Res. 1989;496(1–2):314–6. doi: 10.1016/0006-8993(89)91079-2. [DOI] [PubMed] [Google Scholar]
- 90.Nadal A, Fuentes E, Pastor J, McNaughton PA. Plasma albumin is a potent trigger of calcium signals and DNA synthesis in astrocytes. Proc Natl Acad Sci USA. 1995;92(5):1426–30. doi: 10.1073/pnas.92.5.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Salahuddin TS, Johansson BB, Kalimo H, Olsson Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. An electron microscopic study. Acta Neuropathol. 1988;77(1):5–13. doi: 10.1007/BF00688236. [DOI] [PubMed] [Google Scholar]
- 92.Boado RJ, Wu D, Windisch M. In vivo upregulation of the blood-brain barrier GLUT1 glucose transporter by brain-derived peptides. Neurosci Res. 1999;34(4):217–24. doi: 10.1016/s0168-0102(99)00056-5. [DOI] [PubMed] [Google Scholar]
- 93.Cornford EM, Hyman S, Landaw EM. Developmental modulation of blood-brain-barrier glucose transport in the rabbit. Brain Res. 1994;663(1):7–18. doi: 10.1016/0006-8993(94)90457-x. [DOI] [PubMed] [Google Scholar]
- 94.Muro S, Koval M, Muzykantov V. Endothelial endocytic pathways: gates for vascular drug delivery. Curr Vasc Pharmacol. 2004;2(3):281–99. doi: 10.2174/1570161043385736. [DOI] [PubMed] [Google Scholar]
- 95.Jones AT. Gateways and tools for drug delivery: endocytic pathways and the cellular dynamics of cell penetrating peptides. Int J Pharm. 2008;354(1–2):34–8. doi: 10.1016/j.ijpharm.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 96.Schnitzer JE. Caveolae: from basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo. Adv Drug Deliv Rev. 2001;49(3):265–80. doi: 10.1016/s0169-409x(01)00141-7. [DOI] [PubMed] [Google Scholar]
- 97.Boado RJ. Brain-derived peptides increase blood-brain barrier GLUT1 glucose transporter gene expression via mRNA stabilization. Neurosci Lett. 1998;255(3):147–50. doi: 10.1016/s0304-3940(98)00731-9. [DOI] [PubMed] [Google Scholar]
- 98.Duffy KR, Pardridge WM, Rosenfeld RG. Human blood-brain barrier insulin-like growth factor receptor. Metabolism. 1988;37(2):136–40. doi: 10.1016/s0026-0495(98)90007-5. [DOI] [PubMed] [Google Scholar]
- 99.Urayama A, Grubb JH, Sly WS, Banks WA. Mannose 6-phosphate receptor-mediated transport of sulfamidase across the blood-brain barrier in the newborn mouse. Mol Ther. 2008;16(7):1261–6. doi: 10.1038/mt.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boado RJ, Zhang Y, Xia CF, Wang Y, Pardridge WM. Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol Bioeng. 2008;99(2):475–84. doi: 10.1002/bit.21602. [DOI] [PubMed] [Google Scholar]
- 101.Boado RJ, Zhang Y, Wang Y, Pardridge WM. Engineering and expression of a chimeric transferrin receptor monoclonal antibody for blood-brain barrier delivery in the mouse. Biotechnol Bioeng. 2009;102(4):1251–8. doi: 10.1002/bit.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Di Pasquale G, Chiorini JA. AAV transcytosis through barrier epithelia and endothelium. Mol Ther. 2006;13(3):506–16. doi: 10.1016/j.ymthe.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 103.Xia H, Anderson B, Mao Q, Davidson BL. Recombinant human adenovirus: targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J Virol. 2000;74(23):11359–66. doi: 10.1128/jvi.74.23.11359-11366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lakhal S, Wood MJ. Exosome nanotechnology: an emerging paradigm shift in drug delivery: exploitation of exosome nanovesicles for systemic in vivo delivery of RNAi heralds new horizons for drug delivery across biological barriers. Bioessays. 2011;33(10):737–41. doi: 10.1002/bies.201100076. [DOI] [PubMed] [Google Scholar]
- 105.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2(5):347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 106.Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10. [PubMed] [Google Scholar]
- 107.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–47. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 108.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005;19(3):311–30. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 109.Lee HJ, Zhang Y, Zhu C, Duff K, Pardridge WM. Imaging brain amyloid of Alzheimer disease in vivo in transgenic mice with an Abeta peptide radiopharmaceutical. J Cereb Blood Flow Metab. 2002;22(2):223–31. doi: 10.1097/00004647-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 110.Wu D, Pardridge WM. Neuroprotection with noninvasive neurotrophin delivery to the brain. Proc Natl Acad Sci USA. 1999;96(1):254–9. doi: 10.1073/pnas.96.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Murciano JC, Muro S, Koniaris L, Christofidou-Solomidou M, Harshaw DW, Albelda SM, et al. ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood. 2003;101(10):3977–84. doi: 10.1182/blood-2002-09-2853. [DOI] [PubMed] [Google Scholar]
- 112.Shuvaev V, Dziubla T, Wiewrodt R, Muzykantov VM. Streptavidin-Biotin crosslinking of therapeutic enzymes with carrier antibodies: nanoconjugates for protection against endothelial oxidative stress. Methods in molecular biology. Bioconjugation protocols. 2004 doi: 10.1385/1-59259-813-7:003. [DOI] [PubMed] [Google Scholar]
- 113.Debbage P. Targeted drugs and nanomedicine: present and future. Curr Pharm Des. 2009;15(2):153–72. doi: 10.2174/138161209787002870. [DOI] [PubMed] [Google Scholar]
- 114.Veronese FM, Morpurgo M. Bioconjugation in pharmaceutical chemistry. Farmaco. 1999;54(8):497–516. doi: 10.1016/s0014-827x(99)00066-x. [DOI] [PubMed] [Google Scholar]
- 115.Discher DE, Eisenberg A. Polymer vesicles. Science. 2002;297(5583):967–73. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 116.El-Sayed ME, Hoffman AS, Stayton PS. Smart polymeric carriers for enhanced intracellular delivery of therapeutic macromolecules. Expert Opin Biol Ther. 2005;5(1):23–32. doi: 10.1517/14712598.5.1.23. [DOI] [PubMed] [Google Scholar]
- 117.Langer R. Drug delivery. Drugs on target. Science. 2001;293(5527):58–9. doi: 10.1126/science.1063273. [DOI] [PubMed] [Google Scholar]
- 118.Stayton PS, El-Sayed ME, Murthy N, Bulmus V, Lackey C, Cheung C, et al. `Smart' delivery systems for biomolecular therapeutics. Orthod Craniofac Res. 2005;8(3):219–25. doi: 10.1111/j.1601-6343.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 119.Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006;58(14):1532–55. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 120.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–78. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 121.Minko T, Pakunlu RI, Wang Y, Khandare JJ, Saad M. New generation of liposomal drugs for cancer. Anticancer Agents Med Chem. 2006;6(6):537–52. doi: 10.2174/187152006778699095. [DOI] [PubMed] [Google Scholar]
- 122.Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Current Opinion in Solid State and Materials Science. 2002;6:319–27. [Google Scholar]
- 123.Ghandehari H. Materials for advanced drug delivery in the 21st century: a focus area for Advanced Drug Delivery Reviews. Adv Drug Deliv Rev. 2008;60(9):956. doi: 10.1016/j.addr.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kitchens KM, El-Sayed ME, Ghandehari H. Transepithelial and endothelial transport of poly (amidoamine) dendrimers. Adv Drug Deliv Rev. 2005;57(15):2163–76. doi: 10.1016/j.addr.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 125.Dziubla TD, Karim A, Muzykantov VR. Polymer nanocarriers protecting active enzyme cargo against proteolysis. J Control Release. 2005;102(2):427–39. doi: 10.1016/j.jconrel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 126.Musacchio T, Torchilin VP. Recent developments in lipid-based pharmaceutical nanocarriers. Front Biosci. 2011;16:1388–412. doi: 10.2741/3795. [DOI] [PubMed] [Google Scholar]
- 127.Stachelek SJ, Finley MJ, Alferiev IS, Wang F, Tsai RK, Eckells EC, et al. The effect of CD47 modified polymer surfaces on inflammatory cell attachment and activation. Biomaterials. 2011;32(19):4317–26. doi: 10.1016/j.biomaterials.2011.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tokarev I, Minko S. Stimuli-responsive porous hydrogels at interfaces for molecular filtration, separation, controlled release, and gating in capsules and membranes. Adv Mater. 2010;22(31):3446–62. doi: 10.1002/adma.201000165. [DOI] [PubMed] [Google Scholar]
- 129.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, et al. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther. 2006;317(3):1161–9. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 130.Matzner U, Matthes F, Weigelt C, Andersson C, Eistrup C, Fogh J, et al. Non-inhibitory antibodies impede lysosomal storage reduction during enzyme replacement therapy of a lysosomal storage disease. J Mol Med. 2008;86(4):433–42. doi: 10.1007/s00109-008-0309-3. [DOI] [PubMed] [Google Scholar]
- 131.Ohashi T, Iizuka S, Ida H, Eto Y. Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. Mol Genet Metab. 2008;94(3):313–8. doi: 10.1016/j.ymgme.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 132.Torchilin V. Intracellular delivery of protein and peptide therapeutics. Drug Discovery Today. 2008;5(2–3):e95–e103. doi: 10.1016/j.ddtec.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 133.Solaro R, Chiellini F, Battisti A. Targeted Delivery of Protein Drugs by Nanocarriers. Materials. 2010;3:1928–80. [Google Scholar]
- 134.Klyachko NL, Manickam DS, Brynskikh AM, Uglanova SV, Li S, Higginbotham SM, et al. Cross-linked antioxidant nanozymes for improved delivery to CNS. Nanomedicine. 2012;8(1):119–29. doi: 10.1016/j.nano.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tong J, Luxenhofer R, Yi X, Jordan R, Kabanov AV. Protein modification with amphiphilic block copoly(2-oxazoline)s as a new platform for enhanced cellular delivery. Mol Pharm. 2010;7(4):984–92. doi: 10.1021/mp100102p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Calderon AJ, Bhowmick T, Leferovich J, Burman B, Pichette B, Muzykantov V, et al. Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J Control Release. 2011;150(1):37–44. doi: 10.1016/j.jconrel.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vyas SP, Singh A, Sihorkar V. Ligand-receptor-mediated drug delivery: an emerging paradigm in cellular drug targeting. Crit Rev Ther Drug Carrier Syst. 2001;18(1):1–76. [PubMed] [Google Scholar]
- 138.Shokeen M, Pressly ED, Hagooly A, Zheleznyak A, Ramos N, Fiamengo AL, et al. Evaluation of multivalent, functional polymeric nanoparticles for imaging applications. ACS Nano. 2011;5(2):738–47. doi: 10.1021/nn102278w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. Faseb J. 2002;16(10):1217–26. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 140.Sonawane ND, Szoka FC, Jr., Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J Biol Chem. 2003;278(45):44826–31. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 141.Yessine MA, Leroux JC. Membrane-destabilizing polyanions: interaction with lipid bilayers and endosomal escape of biomacromolecules. Adv Drug Deliv Rev. 2004;56(7):999–1021. doi: 10.1016/j.addr.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 142.Choi SH, Lee SH, Park TG. Temperature-Sensitive Pluronic/Poly(ethylenimine) Nanocapsules for Thermally Triggered Disruption of Intracellular Endosomal Compartment. Biomacromolecules. 2006;7(6):1864–70. doi: 10.1021/bm060182a. [DOI] [PubMed] [Google Scholar]
- 143.Magzoub M, Pramanik A, Graslund A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry. 2005;44(45):14890–7. doi: 10.1021/bi051356w. [DOI] [PubMed] [Google Scholar]
- 144.Suk JS, Suh J, Choy K, Lai SK, Fu J, Hanes J. Gene delivery to differentiated neurotypic cells with RGD and HIV Tat peptide functionalized polymeric nanoparticles. Biomaterials. 2006 doi: 10.1016/j.biomaterials.2006.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kakudo T, Chaki S, Futaki S, Nakase I, Akaji K, Kawakami T, et al. Transferrin-modified liposomes equipped with a pH-sensitive fusogenic peptide: an artificial viral-like delivery system. Biochemistry. 2004;43(19):5618–28. doi: 10.1021/bi035802w. [DOI] [PubMed] [Google Scholar]
- 146.Ansari NH, He Q, Cook JD, Wen J, Srivastava SK. Delivery of liposome-sequestered hydrophobic proteins to lysosomes of normal and Batten disease cells. J Neurosci Res. 1997;47(3):341–7. doi: 10.1002/(sici)1097-4547(19970201)47:3<341::aid-jnr12>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 147.Barrias CC, Lamghari M, Granja PL, Sa Miranda MC, Barbosa MA. Biological evaluation of calcium alginate microspheres as a vehicle for the localized delivery of a therapeutic enzyme. J Biomed Mater Res A. 2005;74(4):545–52. doi: 10.1002/jbm.a.30348. [DOI] [PubMed] [Google Scholar]
- 148.Mumtaz S, Bachhawat BK. Enhanced intracellular stability and efficacy of PEG modified dextranase in the treatment of a model storage disorder. Biochim Biophys Acta. 1994;1199(2):175–82. doi: 10.1016/0304-4165(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 149.Steger LD, Desnick RJ. Enzyme therapy. VI: Comparative in vivo fates and effects on lysosomal integrity of enzyme entrapped in negatively and positively charged liposomes. Biochim Biophys Acta. 1977;464(3):530–46. doi: 10.1016/0005-2736(77)90028-1. [DOI] [PubMed] [Google Scholar]
- 150.Weissmann G, Bloomgarden D, Kaplan R, Cohen C, Hoffstein S, Collins T, et al. A general method for the introduction of enzymes, by means of immunoglobulin-coated liposomes, into lysosomes of deficient cells. Proc Natl Acad Sci U S A. 1975;72(1):88–92. doi: 10.1073/pnas.72.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hsu J, Northrup L, Bhowmick T, Muro S. Enhanced delivery of alpha-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine. 2012 doi: 10.1016/j.nano.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, et al. Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149(3):323–31. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, et al. Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther. 2008;325(2):400–8. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 154.Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther. 2006;13(1):135–41. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 155.Rollerova E, Scsukova S, Jurcovicova J, Mlynarcikova A, Szabova E, Kovriznych J, et al. Polymeric nanoparticles - targeted drug delivery systems for treatment of CNS disorders and their possible endocrine disrupting activities. Endocr Regul. 2011;45(1):49–60. [PubMed] [Google Scholar]
- 156.Bondy SC. Nanoparticles and colloids as contributing factors in neurodegenerative disease. Int J Environ Res Public Health. 2011;8(6):2200–11. doi: 10.3390/ijerph8062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Muro S. Endothelial Biomedicine. 2007. VCAM-1 and ICAM-1; pp. 1058–70. [Google Scholar]
- 158.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. 1986;137(4):1270–4. [PubMed] [Google Scholar]
- 159.Muzykantov V. Targeting drugs to pulmonary endothelium. Expert Opinion Drug Delivery. 2005;2:909–26. doi: 10.1517/17425247.2.5.909. [DOI] [PubMed] [Google Scholar]
- 160.DeGraba T, Azhar S, Dignat-George F, Brown E, Boutiere B, Altarescu G, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. 2000;47(2):229–33. [PubMed] [Google Scholar]
- 161.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76(2):301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 162.Bloemen PG, Henricks PA, van Bloois L, van den Tweel MC, Bloem AC, Nijkamp FP, et al. Adhesion molecules: a new target for immunoliposome-mediated drug delivery. FEBS Lett. 1995;357(2):140–4. doi: 10.1016/0014-5793(94)01350-a. [DOI] [PubMed] [Google Scholar]
- 163.Sakhalkar HS, Dalal MK, Salem AK, Ansari R, Fu J, Kiani MF, et al. Leukocyte-inspired biodegradable particles that selectively and avidly adhere to inflamed endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2003;100(26):15895–900. doi: 10.1073/pnas.2631433100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Villanueva FS, Jankowski RJ, Klibanov S, Pina ML, Alber SM, Watkins SC, et al. Microbubbles targeted to intercellular adhesion molecule-1 bind to activated coronary artery endothelial cells. Circulation. 1998;98(1):1–5. doi: 10.1161/01.cir.98.1.1. [DOI] [PubMed] [Google Scholar]
- 165.Weiner RE, Sasso DE, Gionfriddo MA, Thrall RS, Syrbu S, Smilowitz HM, et al. Early detection of oleic acid-induced lung injury in rats using (111)In-labeled anti-rat intercellular adhesion molecule-1. J Nucl Med. 2001;42(7):1109–15. [PubMed] [Google Scholar]
- 166.Weller GE, Villanueva FS, Tom EM, Wagner WR. Targeted ultrasound contrast agents: In vitro assessment of endothelial dysfunction and multi-targeting to ICAM-1 and sialyl Lewis(x) Biotechnol Bioeng. 2005 doi: 10.1002/bit.20625. [DOI] [PubMed] [Google Scholar]
- 167.Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. Eur J Pharm Sci. 2009;37(2):141–50. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Finikova OS, Lebedev AY, Aprelev A, Troxler T, Gao F, Garnacho C, et al. Oxygen microscopy by two-photon-excited phosphorescence. Chemphyschem. 2008;9(12):1673–9. doi: 10.1002/cphc.200800296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rossin R, Muro S, Welch MJ, Muzykantov VR, Schuster DP. In vivo imaging of 64Cu-labeled polymer nanoparticles targeted to the lung endothelium. J Nucl Med. 2008;49(1):103–11. doi: 10.2967/jnumed.107.045302. [DOI] [PubMed] [Google Scholar]
- 170.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, et al. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16(8):1450–8. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Muro S, Cui X, Gajewski C, Murciano JC, Muzykantov VR, Koval M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am J Physiol Cell Physiol. 2003;285(5):C1339–47. doi: 10.1152/ajpcell.00099.2003. [DOI] [PubMed] [Google Scholar]
- 172.Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105(2):650–8. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 173.Muro S, Mateescu M, Gajewski C, Robinson M, Muzykantov VR, Koval M. Control of intracellular trafficking of ICAM-1-targeted nanocarriers by endothelial Na+/H+ exchanger proteins. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L809–17. doi: 10.1152/ajplung.00311.2005. [DOI] [PubMed] [Google Scholar]
- 174.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, et al. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116(Pt 8):1599–609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 175.Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler Thromb Vasc Biol. 2012;32(5):1178–85. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Garnacho C, Shuvaev V, Thomas A, McKenna L, Sun J, Koval M, et al. RhoA activation and actin reorganization involved in endothelial CAM-mediated endocytosis of anti-PECAM carriers: critical role for tyrosine 686 in the cytoplasmic tail of PECAM-1. Blood. 2008;111(6):3024–33. doi: 10.1182/blood-2007-06-098657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Garnacho C, Albelda SM, Muzykantov VR, Muro S. Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J Control Release. 2008;130(3):226–33. doi: 10.1016/j.jconrel.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]