

Abstract
PKR is an interferon-induced serine-threonine protein kinase that plays an important role in the mediation of the antiviral and antiproliferative actions of interferons. PKR is present at low basal levels in cells and its expression is induced at the transcriptional level by interferons. PKR’s kinase activity stays latent until it binds to its activator. In the case of virally infected cells, double-stranded (ds) RNA serves as PKR’s activator. The dsRNA binds to PKR via two copies of an evolutionarily conserved motif, thus inducing a conformational change, unmasking the ATP-binding site and leading to autophosphorylation of PKR. Activated PKR then phosphorylates the α-subunit of the protein synthesis initiation factor 2 (eIF2α) thereby inducing a general block in the initiation of protein synthesis. In addition to dsRNA, polyanionic agents such as heparin can also activate PKR. In contrast to dsRNA-induced activation of PKR, heparin-dependent PKR activation has so far remained uncharacterized. In order to understand the mechanism of heparin-induced PKR activation, we have mapped the heparin-binding domains of PKR. Our results indicate that PKR has two heparin-binding domains that are nonoverlapping with its dsRNA-binding domains. Although both these domains can function independently of each other, they function cooperatively when present together. Point mutations created within these domains rendered PKR defective in heparin-binding, thereby confirming their essential role. In addition, these mutants were defective in kinase activity as determined by both in vitro and in vivo assays.
Keywords: domain mapping, dsRNA, heparin, interferon, protein kinase
Interferons (IFNs) are cytokines with antiviral, antiproliferative and immunomodulatory properties, which they exert by inducing synthesis of several proteins [1,2]. One such protein, the IFN-induced, dsRNA-activated protein kinase, PKR, a serine/threonine kinase, is a major mediator of the antiproliferative and antiviral actions of IFN [3,4]. Although induced at transcriptional level by IFNs, PKR is present at a low, basal level in most cell types. PKR’s kinase activity stays latent until it binds to an activator, the well-characterized activator being dsRNA. However, other polyanionic agents such as heparin have also been shown to activate PKR in vitro [5]. In addition, we have identified PACT as a cellular, protein activator of PKR, which heterodimerizes with PKR and activates it in the absence of dsRNA [6,7], thereby playing an important role in PKR activation in response to stress signals [8]. The α-subunit of the eukaryotic protein synthesis initiation factor eIF-2 (eIF2α) is the most studied physiological substrate of PKR. Phosphorylation of eIF2α on Ser51 by PKR leads to inhibition of protein synthesis [9,10]. In addition to its central role in antiviral activity of IFNs, PKR is also involved in the regulation of apoptosis [11,12], cell-proliferation [13,14], signal transduction [12,15], and differentiation [16,17].
The dsRNA-mediated activation of PKR has been characterized in detail [18–25]. The dsRNA-binding domain (DRBD) of PKR is composed of two copies of the dsRNA binding domain, a sequence conserved in many RNA binding proteins [26,27]. Binding of dsRNA to PKR through these motifs causes a conformational change [28,29] that leads to unmasking of the ATP-binding site in the kinase domain and results in autophosphorylation of PKR on several sites [30–32]. The domains that are involved in dsRNA binding are also involved in mediating dimerization of PKR, which is essential for its kinase activity in the presence of dsRNA [33–36]. Although the same domain mediates PKR’s dsRNA binding and dimerization, distinct residues have been identified that contribute to one or both these properties [36].
Although dsRNA is the most widely studied activator of PKR, it has been known that PKR binds to heparin–Sepharose efficiently and its activation can also be achieved efficiently by heparin [5]. The minimum size of heparin required to efficiently activate PKR autophosphorylation has been shown to be heparin octasaccharide and the hexamer is a very poor activator [37]. Although heparin can activate PKR in vitro, its ability to act as a PKR activator in vivo was demonstrated only recently [38]. Heparin is a potent antiproliferative agent for vascular smooth muscle cells (VSMC) and has been shown to be effective in both tissue culture systems [39,40] and in patients [41,42]. As excessive VSMC proliferation is a major contributing factor in establishment and development of atherosclerotic lesions, antiproliferative agents that block VSMC proliferation are of therapeutic interest [43]. Heparin treatment of VSMC causes PKR activation by internalization and direct binding of heparin by PKR [38]. This heparin-induced PKR activation was essential for the cell cycle block induced by heparin. PKR null cells were found to be largely insensitive to heparin-induced block in G1 to S-phase transition. In order to understand the heparin-mediated PKR activation, we sought out to map the heparin-binding domain of PKR. By generating deletion mutants and assaying their heparin-binding activity, we have identified two heparin-binding domains in PKR, each one of which is sufficient for heparin-binding activity but the two domains act together to increase the affinity of binding. Comparison of these regions with other known heparin-binding proteins revealed a conserved motif. Specific point mutations within the identified domains resulted in both a loss of heparin binding and kinase activity of PKR. Further analysis revealed that the loss of kinase activity was due to a loss of ATP-binding activity when residue R297 was mutated, suggesting that it may contribute to both heparin and ATP binding.
Results
In order to map the heparin-binding domain of PKR, we generated several deletion mutants of PKR and tested their ability to bind heparin-agarose. Using a similar approach, the dsRNA-binding domain (DRBD) of PKR has been mapped to reside between the residues 1–170 [19]. Our previous results have indicated that heparin may interact with PKR through a domain that is nonoverlapping with its DRBD [23]. In order to confirm that DRBD does not contribute to PKR’s heparin-binding activity, we compared the heparin binding of three deletion mutants of PKR with that of the full length PKR protein using the heparin–agarose binding assay. The heparin-binding activity was assayed at two salt concentrations, 50 mM and 200 mM. wtPKR bound to heparin–agarose with high affinity at both salt concentrations (Fig. 1A, lanes 2 and 3). The two amino terminal deletion mutants Δ170 and Δ145, also bound with high efficiency under both conditions (lanes 5, 6, 8, and 9). A further deletion of 278 amino terminal residues showed no loss of heparin-binding activity (lanes 11 and 12). However, a deletion of 40 more amino acids (to the residue 318) resulted in a partial loss of heparin-binding activity. The deletion mutant Δ318 showed a strong binding at 50 mM salt (lane 14), but a dramatically reduced (6.5% of wild-type, Fig. 1B) binding at 200 mM salt (lane 15). These results strongly indicate that the residues between 278 and 318 participate in high affinity binding of PKR to heparin. On the other hand, the carboxy terminally deleted mutant DRBD, which retains residues 1–170, showed no binding at either salt concentration (lanes 17 and 18). These results demonstrate that the residues between 1 and 170 are dispensable for heparin-binding activity of PKR and that the heparin-binding domain of PKR lies between residues 171 and 551, with residues between 278 and 318 being essential for high affinity binding to heparin. A quantification of the binding assays is shown in Fig. 1B and a schematic drawing representing the different deletions is shown in Fig. 1C.
Fig. 1.

(A) Residues between 279 and 318 are important for heparin-binding activity of PKR. The wild-type PKR (wtPKR) and its deletion mutants were tested for heparin–agarose binding activity. In vitro translated proteins (5 μL) were bound to heparin–agarose in binding buffer and the proteins remaining bound to the beads after washing were analyzed by SDS/PAGE followed by phosphorimager analysis. Lanes 1, 4, 7, 10, 13 and 16 represent total proteins present in the translation mix. Lanes 2, 5, 8, 11, 14, and 17 represent proteins bound at 50 mM salt and lanes 3, 6, 9, 12, 15 and 18 represent proteins bound at 200 mM salt. The different proteins that were tested are as indicated at the bottom of the panels and positions of the proteins are indicated by arrows. Additional bands observed below the expected bands arise due to initiation of translation at internal AUG codons in rabbit reticulocyte system. (B) Quantification of heparin-binding activity of deletion mutants. The percentage binding of various deletion mutants was quantified by phosphorimager analysis. The binding activity of the wtPKR was taken as 100% and binding of mutants is represented relative to this value. The white bars represent binding at 50 mM salt concentration and the black bars represent the binding activity at 200 mM salt. Error bars represent SD calculated based on three experiments. (C) A schematic representation of the deletion mutants. The names of the mutants and the residues retained in each mutant are indicated on the right.
To map the carboxy terminal boundary of the heparin-binding domain, we then tested carboxy terminal deletions of Δ145. Deletion of carboxy terminal residues either between 480 and 551 or between 318 and 551 showed no loss of binding (Fig. 2A, lanes 2, 3, 5 and 6) but a further deletion to residue 277 showed extremely poor (7.6% of wild-type, Fig. 2B) heparin–agarose binding at 50 mM salt (lane 8) and no binding (0.5% of wild-type, Fig. 2B) at 200 mM salt (lane 9). A further deletion to residue 255 resulted in a complete loss of binding under both conditions (lanes 11 and 12). These results, in combination with the results shown in Fig. 1, suggest that the heparin binding occurs between residues 278 and 318.
Fig. 2.

(A) Heparin-binding activity of carboxy terminal deletion mutants of Δ145. The heparin–agarose binding activity of carboxy terminal deletion mutants of Δ145 was tested as described in Fig. 1 legend. Lane 1, 4, 7 and 10 represent total proteins in the translation mix. Lanes 2, 5, 8 and 11, binding performed at 50 mM salt; lanes 3, 6, 9 and 12, binding performed at 200 mM salt. Arrows indicate the positions of deletion mutants and the labels at the bottom of the panels show the residues retained in the deletion mutants. (B) A second region between residues 413 and 479 also contributes to heparin-binding activity of PKR. The heparin–agarose binding activity of carboxy terminal deletion mutants of Δ318 was tested. Lane 1 and 4, total proteins in the translation mix; lanes 2 and 5, binding performed at 50 mM salt; lanes 3 and 6, binding performed at 200 mM salt. Arrows indicate the positions of deletion mutants and the labels at the bottom of the panels show the residues retained in the deletion mutants. (C) The region between 318 and 412 is dispensable for heparin-binding activity of PKR. An internal deletion mutant of Δ278 was created that lacked amino acids between 318 and 412 (278-ID). The heparin–agarose binding activity of this mutant was tested. Lane 1, total protein in the translation mix; lane 2, protein bound to heparin–agarose at 50 mM salt; lane 3, protein bound to heparin–agarose at 200 mM salt. An arrow indicates the position of the deletion mutant. (D) Quantification of heparin-binding activity of deletion mutants. The percentage binding of various deletion mutants was quantified by phosphorimager analysis. The binding activity of the wt PKR was taken as 100% and binding of mutants is represented relative to this value. The white bars represent binding at 50 mM salt concentration and the black bars represent the binding activity at 200 mM salt. Error bars represent SD calculated based on three experiments. (E) A schematic representation of the deletion mutants. The names of the mutants (residues retained in each mutant) are indicated on the right.
The deletion mutant Δ318 showed weak binding to heparin–agarose at 50 mM salt, as opposed to deletion mutants containing residues 1–170 (DRBD) (Fig. 1) and 146–255 (Fig. 2A), which show no binding even at 50 mM salt. These observations suggested that additional domains downstream of residue 318 might contribute at least in part to heparin binding. To test this possibility, we tested carboxy terminal deletions of Δ318 mutant for heparin binding. A deletion to residue 479 did not show any loss of binding (97.6% of wild-type; Fig. 2B, lane 2) at 50 mM salt. This deletion showed very weak binding (56.4% of wild-type; Fig. 2B, lane 3) at 200 mM salt, similar to Δ318 mutant (Fig. 1A, lane 15). A further deletion to residue 412 from the carboxy-terminus, resulted in a total loss of binding (Fig. 2B, lanes 5 and 6), under both conditions. In order to confirm that the loss of binding was not due to the fact that this region encoded a protein that was too small and therefore did not bind efficiently, we created a fusion construct with residues 319–412 joined to luciferase coding region at the amino terminus. This chimeric protein showed no binding to heparin–agarose at both the salt concentrations (data not shown), thereby confirming that the residues between 319 and 412 did not show any heparin-binding activity. These results demonstrate that residues between 413 and 479 contribute to the low affinity binding of PKR to heparin. These results suggest that two noncontiguous regions, 278–318 and 413–479 contribute to PKR’s heparin-binding activity. A graph representing the binding efficiencies of different deletion constructs is shown in Fig. 2D and a schematic diagram representing the deletions is shown in Fig. 2E.
Our results have indicated that the two regions 278–318 and 413–479, in the carboxy terminal half of PKR can function independently of each other for binding to heparin. In order to determine if the residues between 318 and 412 are dispensable for heparin binding, we generated 278–318,412–551, an internal deletion mutant of Δ278. This internal deletion mutant bound efficiently to heparin agarose (Fig. 2C, lanes 2 and 3). This suggests that the residues between 318 and 412 are dispensable for heparin binding and that the spacing between the two heparin-binding domains of PKR can be varied without a loss of heparin-binding activity.
In order to determine further if the two regions that we have mapped as heparin-binding domains can function independently of each other, we tested whether these regions can confer the heparin-binding activity on a heterologous protein such as luciferase, which does not bind heparin. We designed fusion constructs such that they encoded proteins with either the amino terminal domain (ATD, residues 278–318), the carboxy terminal domain (CTD, residues 413–479), or both the ATD and CTD domains (HBD, residues 278–479) fused in frame to the amino-terminus of luciferase. The fusion proteins were tested for their heparin-binding activity by the heparin–agarose binding assay. All of the constructs encoded proteins of corresponding sizes (Fig. 3A). When tested for their heparin-binding activity, luciferase protein itself showed no heparin-binding activity (Fig. 3B, lanes 1–3). Both ATD (lanes 4–6) as well as CTD (lanes 7–9) fusion could confer heparin-binding activity to luciferase. However, fusion of HBD to luciferase showed the highest affinity binding to heparin–agarose (lanes 10–12). To determine the relative binding efficiencies, we calculated the percentage heparin binding at 200 mM salt concentration by phosphorimager analysis (Fig. 3C). Although in the context of PKR, ATD showed higher affinity binding as compared to CTD (Figs 1 and 2) when present as individual domains linked to luciferase, they both were capable of binding to heparin with nearly equal efficiencies. When present together, they act in a cooperative manner, increasing the binding efficiency significantly. In order to confirm the functional significance of the HBD, we performed kinase activity assays. We reasoned that HBD protein may inhibit heparin-mediated PKR activation by competing for heparin. We carried out PKR activity assays using in vitro translated, flag-tagged PKR protein. Effect of flag-tagged HBD protein was assayed on PKR activation by heparin. PKR activity was not affected by addition of flag-HBD when dsRNA was used to activate PKR (Fig. 3D, lanes 1–3). This is expected because the HBD protein does not compete for dsRNA binding. On the other hand, PKR activity was inhibited in a dose dependent manner by flag-HBD when heparin was used as an activator (lanes 4–6). As seen in lanes 7–8, addition of flag-tagged p56 protein did not inhibit PKR activity confirming the specificity of inhibition by flag-HBD.
Fig. 3.

The two heparin-binding domains of PKR can confer heparin-binding activity to a heterologous protein. The region between residues 279 and 318 (ATD), region between residues 413 and 479 (CTD), and also the region between residues 279 and 479 (HBD) was fused in frame at the amino terminus of the luciferase coding region. The heparin–agarose binding activity of the resulting fusion proteins was tested as described in the legend to Fig. 1. (A) Expression of the corresponding fusion proteins in an in vitro translation system. Translation products (2 μL) were analyzed by SDS/PAGE. Lane 1, luciferase; lane 2, ATD-luciferase; lane 3, CTD-luciferase and lane 4, HBD-luciferase. The positions of the corresponding proteins are as indicated. (B) Heparin-binding activity of the fusion proteins. Lanes 1–3, Luciferase; lanes 4–6, ATD-Luciferase; lanes 7–9, CTD-luciferase and lanes 10–12, HBD-luciferase. Lanes 1, 4, 7 and 10 represent the total protein from the translation mix. Lanes 2, 5, 8 and 11 represent the proteins bound to heparin–agarose at 50 mM salt. Lanes 3, 6, 9 and 12 represent the proteins bound to heparin–agarose at 200 mM salt. Arrows indicate the corresponding bands. (C) Quantification of the heparin-binding activity. The percentage of protein bound to heparin–agarose at 200 mM salt concentration was calculated by phosphorimager analysis. (D) HBD inhibits PKR activation by heparin. The kinase activity of in vitro translated flag-tagged PKR protein was examined in the presence of dsRNA or heparin. Each lane contains translation of 200 ng of flag-PKR/BSIIKS+DNA. The flag-tagged HBD protein was cotranslated and coimmunoprecipitated as indicated in lanes 2, 3, 5 and 6. Lanes 2, 5 and 8 represent cotranslation with 100 ng of plasmid DNA and lanes 3, 6, and 9 represent cotranslation with 200 ng plasmid DNA. Lanes 7–9 represent a negative control with cotranslation of flag-p56 protein. In vitro translated PKR and HBD proteins (3 μL) were immunoprecipitated by anti-flag mAb conjugated to agarose. The kinase activity present in the immune complexes was assayed using either poly(I)·poly(C) (lanes 1–3) or heparin (lanes 4–9) as activator. Position of the autophosphorylated PKR band is indicated by arrows.
A schematic representation of the heparin-binding domains is shown in Fig. 4A. Cardin and Weintraub have aligned a broad collection of alleged heparin-binding sequences in order to identify common motifs [44]. In their study, the motifs (XBBXBX) and (XBBBXXBX) were identified, where B designates a basic amino acid and X indicates any other amino acid. Both the sequences that we have identified as heparin-binding domains within PKR contain the (XBBXBX) motif (Fig. 4B).
Fig. 4.
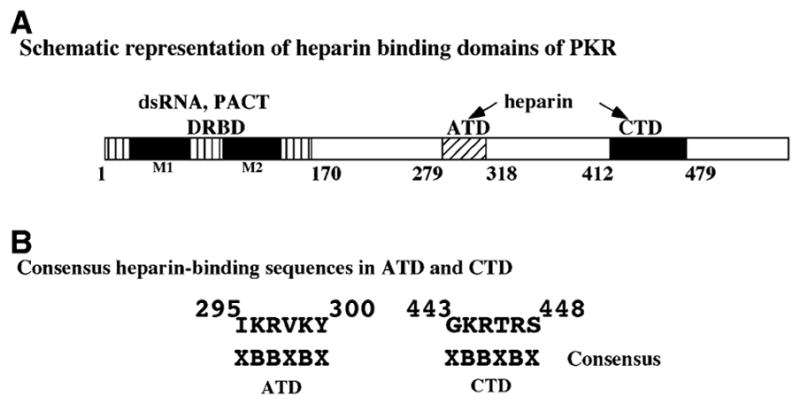
(A) Schematic representation of the heparin-binding domains of PKR. The relative positions of the domains involved in PKR binding to different activators are indicated. The dsRNA and PACT interacting domain is shown as a box with vertical lines and the two conserved motifs are shown as black boxes. The amino terminal heparin-binding domain (ATD heparin) is shown as a box with oblique hatch and the carboxy terminal heparin-binding domain (CTD heparin) is shown as a black box. (B) The two heparin-binding domains of PKR contain a consensus heparin-binding sequence XBBXBX. B indicates a basic residue and X indicates any residue. Residues 295–300 within ATD and residues 443–448 within CTD fit the consensus.
As a next step in understanding the importance of the identified heparin-binding domains in mediating PKR activation, we generated point mutations of the basic residues within the ATD and CTD. We generated two mutations in ATD, K299A and double mutant (DM) R297A,K299A and one in CTD (the hep2 triple mutant: K444E,R445E,R447E) (Fig. 5A). The heparin-binding activity of these mutants was tested (Fig. 5B) and all the mutations showed reduced binding to heparin compared to the wild-type PKR (Fig. 5E). K299A showed about 20% loss in heparin-binding, whereas the double mutant with both R297A and K299A mutations showed a 72% loss in heparin binding. The hep2 mutation by itself showed marginal loss of heparin binding with only 23% less activity than wild-type, which is consistent with its minor role in heparin binding (Fig. 2). However, when combined with the DM, it resulted in 86% loss of heparin binding. As the dsRNA–binding domain does not overlap with PKR’s heparin-binding domains, these mutations are expected to have no effect on dsRNA-binding. All mutants showed dsRNA-binding comparable to wild-type PKR (Fig. 5C). No binding of PKR to agarose beads alone was observed, thereby confirming that the binding was specific for dsRNA (data not shown). The kinase activity of PKR has been linked to its dimerization and we wanted to examine if any of these mutations affected its dimerization. All of the mutants showed dimerization activity and hep2 and DM mutations showed slightly enhanced dimerization activity (Fig. 5D). This dimerization assay has been characterized carefully and no binding of labeled PKR protein to the Ni-charged His-bind resin is observed in the absence of recombinant hexahistidine tagged PKR protein, thus demonstrating the specificity of the dimerization assay [33].
Fig. 5.

(A) Point mutations in ATD and CTD. The amino acids targeted by mutations within ATD and CTD are underlined. The individual mutations are as described in the text. (B) Heparin-binding activity of the mutants. The heparin–agarose binding activity of the point mutants was tested. The T lanes represent total proteins in the translation mix. The B lanes represent binding performed at 200 mM salt. Arrows indicate the positions of PKR mutants and the mutant name is shown at the bottom of the panels. (C) dsRNA-binding activity of the mutants. The dsRNA-binding activity of the point mutants was tested by their binding to poly(I)·poly(C)-agarose. The T lanes represent total proteins in the translation mix. The B lanes represent binding performed at 300 mM salt. Arrows indicate the positions of PKR mutants and the mutant name is shown at the bottom of the panels. (C) Dimerization activity of the point mutants. The ability of point mutants to dimerize was tested using the in vitro dimerization assay. The T lanes represent total proteins in the translation mix. The B lanes represent binding to PKR immobilized on Ni-charged His-bind resin performed at 200 mM salt. Arrows indicate the positions of PKR mutants and the mutant name is shown at the bottom of the panels. (D) Quantification of the heparin-binding activity of point mutants. The data shown in panel A was quantified using a phosphorimager analysis. The binding activities of mutants are represented as a percentage of wild-type PKR. The error bars represent SD from three separate experiments.
To test the effect of the mutations on the kinase activity of PKR, we tested its activity in vitro by activity assays and in vivo both in mammalian and yeast systems. The K299A mutant retained its ability to be activated both by dsRNA and heparin (Fig. 6A). The DM and hep2 showed a loss of kinase activity both in the presence of dsRNA or heparin, thereby indicating that these mutations resulted in a loss of kinase function due to a perturbation of PKR’s catalytic activity. The heparin-binding defective mutants are expected to be activated normally by dsRNA, unless the mutation results in a loss of an essential catalytic function. Similar results were obtained in the in vivo activity assay as judged by inhibition of plasmid-driven translation. This in vivo translation inhibition assay has been widely used by us and others in the field to determine if a particular mutation renders the PKR molecule catalytically inactive [45–49]. In this assay, the expression of a reporter gene expressed from a constitutive promoter such as cytomegalovirus (CMV), is down-regulated when it is cotransfected with a PKR expression construct. This down-regulation occurs at the translational level due to activation of the PKR encoded by the expression construct due to the transfection process. In this system, cotransfection of an expression construct of a trans-dominant negative PKR mutant such as K296R results in an up-regulation of the reporter gene activity due to inhibition of endogenous PKR activity in the transfected cells. Cotransfection with wild-type (wt) PKR resulted in an expected down-regulation of the luciferase reporter activity (Fig. 6B). K299A mutant also showed reduction of luciferase activity indicating that it was an active kinase. All three other mutants DM, hep2, and the double mutant DM,hep2 showed an up-regulation of luciferase activity, thereby indicating that these mutations were not only inactive as kinases, but also resulted in rendering them transdominant negative. Expression of the mutant proteins was quantified by western blot analysis of the extracts (Fig. 6C) and all of the mutants were expressed at the same level in HT1080 cells. The kinase activity of the mutants was further tested by assaying the effect of their expression on yeast growth (Fig. 6D). It has been established that expression of wtPKR causes a slow-growth phenotype in yeast [24,36,50]. We have expressed the various PKR mutants from a galactose-inducible promoter in pYES2 plasmid. Growth on glucose-containing medium is not affected due to a lack of expression from the galactose-inducible promoter in the presence of glucose (Fig. 6D). However, when galactose is used as the only carbon source in the growth medium, expression of wtPKR causes a significant reduction in growth compared to the inactive K296R mutant, confirming the previously reported results [36]. K299A expression also resulted in significant inhibition of yeast growth, thereby indicating that this mutation does not result in a loss of PKR kinase activity. The DM and hep2 mutants both showed no inhibition of yeast growth, thereby indicating that these were inactive kinase mutants. Double mutant DM,hep2 also showed no growth inhibition as expected from the phenotype of the individual mutations. Thus, based on our results from the in vitro activity assays and the in vivo assays in both mammalian and yeast systems, it can be concluded that the mutations in the two identified heparin-binding domains lead to a complete loss of kinase activity, even in response to activation by dsRNA.
Fig. 6.

A. In vitro kinase activity of the mutants. The kinase activity of the in vitro translated proteins was examined in the presence of dsRNA or heparin. In vitro translated proteins (3 μL) were immunoprecipitated by anti-PKR mAb and protein A-Sepharose. The kinase activity present in the immune complexes was assayed. The positions of PKR and eIF2α bands are indicated by arrows. The different mutants are as indicated under the panels. (B) In vivo PKR activity assayed by translation inhibition assay. The transfections were performed using HT-1080 cells grown in six-well plates. The reporter used was pGL2C. An 800 ng aliquot of pGL2C was cotransfected using Lipofectamine reagent with 200 ng of the expression constructs for the proteins indicated. At 24 h after transfection, luciferase activity was measured in the cell extracts and normalized to the amount of total protein in the extract. All expression constructs were in pCDNA3; Control indicates the empty-vector (pCDNA3) control. Each sample was assayed in triplicate and the data represent means of six samples from two separate experiments. Error bars indicate SD. The expression of all proteins was ascertained to be at the same level by western blot analysis. (C) Western blot analysis was performed on the HT1080 cell extracts. All the expression constructs were in pCDNA3 and encoded flag-tagged PKR proteins. The western blot analysis was performed with anti-flag mAb and the same blot was stripped and reprobed with anti-β-actin mAb to ensure equal loading in all lanes. (D) Yeast growth phenotype of the PKR mutants. Growth of transformed INVSc1 yeast strain containing wtPKR/pYES2 (wt), K296R/pYES2 (K296R), K299A/pYES2 (K299A), DM/pYES2 (DM), hep2/pYES2 (hep2), and DM,hep2/pYES2 (DM,hep2). Cells were grown for 2 days at 30 °C on synthetic medium lacking uracil with 2% glucose (bottom panel) or 10% galactose (top panel) as sole carbon source.
It is possible that the observed loss of kinase activity results from a loss of one of the domains important for the catalytic activity of PKR, as unlike the dsRNA-binding domains, heparin-binding domains are located within the catalytic half of the PKR molecule. It has been reported previously by George et al. that the K296R mutant cannot be trans-phosphorylated by wtPKR when activated by heparin [37]. In addition, these authors also reported that in order for PKR to bind ATP and get activated, ATP had to be present at the time of incubation with heparin. Pre-incubation of PKR with heparin in the absence of ATP rendered PKR unresponsive to activation even when ATP was provided at a later step. In the view of our results presented here, we reasoned that the lysine at position 296 could also be involved in heparin-binding as it lies within the consensus motif BBXB within the ATD involved in heparin-binding. The mutation K296R is not expected to result in a loss of heparin binding as it retains a basic residue in position 296. We therefore tested the heparin-binding property of K296P mutant. Both K296R and K296P showed good binding to heparin-agarose (Fig. 7A), thereby indicating that the lysine at position 296 is dispensable for the interaction of PKR with heparin. The ATD maps within the conserved catalytic domain II described for several kinases [51]. This domain is known to be involved in ATP-binding and the actual phosphotransfer reaction, thereby explaining why the K296R mutation is a trans-dominant negative mutation in PKR. In order to examine if the loss of kinase activity in DM and hep2 mutants was due to a loss in ATP-binding, we performed ATP-binding assays using the in vitro translated mutant proteins. wtPKR showed binding to ATP-agarose in the presence of dsRNA and heparin (Fig. 7B). The hep2 mutant also showed significant binding to ATP-agarose in the presence of both the activators. However, the DM mutant showed no binding above the background levels in presence of either activators, thereby indicating that possible reason for the lack of kinase activity of DM mutant could be the loss of ATP-binding activity in addition to a loss of heparin-binding ability. The loss in kinase activity of the hep2 mutant may be due to a change in conformation of the catalytic domain or loss of crucial residues needed for PKR’s catalytic activity. Thus, due to the position of heparin-binding domain within PKR’s catalytic domain, it does not appear to be possible to generate a mutation that would allow for dsRNA-dependent activation of PKR, but prevent heparin-dependent activation. Such a mutant would be valuable in understanding the contribution of dsRNA-vs. heparin-dependent activation of PKR in cells. However, as the ATD and CTD are located in regions involved in ATP-binding, phosphotransfer reaction, and catalytic functions a mutation of these regions results in an inactive kinase.
Fig. 7.

(A) K296 does not contribute to PKR’s heparin-binding activity. The heparin–agarose binding activity of K296R and K296P mutants was tested at 200 mM salt. T lanes represent 2 μL of the total proteins in the translation mix. The B lanes represent bound proteins at 200 mM salt. The top band indicates the positions of point mutants and the additional bands below the full-length protein band arise from initiations of translation at the internal methionines. (B) ATP-binding activity of hep2 and DM mutants. ATP-agarose binding was assayed for the mutants. 4 μL of the in vitro translated proteins were bound to ATP-agarose in binding buffer either in the absence of any activator or in the presence of 0.1 mg·mL−1 poly(I)·poly(C) or 50 mg·mL−1 of heparin. T lanes, total proteins present in the translation mixture; –lanes, proteins bound to the beads in the absence of activator; ds lanes, proteins bound to the beads in the presence of dsRNA and hep lanes, proteins bound to the beads in the presence of heparin. The names of the proteins are indicated below the panels and the additional bands observed below the expected bands in T lanes arise due to initiation of translation at internal AUG codons in rabbit reticulocyte system.
Discussion
Among all the known activators of PKR, its activation by dsRNA has been studied the most. DsRNA binds to PKR through the amino terminal DRBD (1–170 residues), which contains two copies of the evolutionarily conserved dsRNA binding motif [18–21,26,27]. The second, carboxy terminal copy within DRBD has been shown to interact with the catalytic domains of PKR, thereby masking its ATP-binding site [29]. The binding of dsRNA to these motifs has been shown to lead to a conformational change in PKR protein, which unmasks the ATP-binding site by relieving the interaction between the catalytic domain and the second copy of the conserved motif [29,52]. PKR has also been shown to function as a dimer and two different regions have been shown to be involved in dimerization [33,53]. The DRBD domain was shown to be essential for PKR’s dimerization [33–35,54] and an additional dimerization domain was also mapped between residues 244 and 296 [53]. Dimerization of PKR through its DRBD has been shown to be essential for its catalytic activity [36]. Although heparinmediated PKR activation has not been studied much, there are several differences between the dsRNA mediated and heparin-mediated activation of PKR in vitro. The general conclusions that activators dsRNA and heparin involve quite distinct mechanisms is supported by several observations in the literature. Our previous studies have indicated that heparin can activate PKR deletion mutants that are devoid of the DRBD [23,36]. In these studies we showed that several point mutants of PKR that were defective in dimerization through the two conserved dsRNA binding/dimerization motifs could bind heparin effectively and get activated. Furthermore, their binding to heparin did not lead to dimerization thereby indicating that heparin dependent activation of PKR may be primarily brought about by intramolecular autophosphorylation [36]. Studies of George et al. [37] on heparin-activated PKR have demonstrated that unlike dsRNA activated PKR, heparin activated PKR cannot phosphorylate the K296R mutant, raising a possibility that heparin activates intramolecular autophosphorylation and dsRNA promotes intermolecular phosphorylation.
In addition to these in vitro studies, recent results from our lab have shown that treatment of VSMC with heparin results in PKR activation that is brought about by direct binding to PKR after its internalization [38]. Proliferation of VSMC is a key step in the pathogenesis of atherosclerosis or restenosis after vascular interventions such as angioplasty [43]. Much attention has been focused on the search for an antiproliferative agent to regulate VSMC proliferation. Heparin is also known to inhibit VSMC proliferation in vivo [40] after invasive vascular surgeries in animal models [39,55]. In addition, heparin is currently used as one of the local-delivery drugs after invasive procedures in some cases [56]. Although heparin is present mainly in the extracellular matrix, VSMCs are known to synthesize heparin as they cease proliferation [57]. It is therefore possible that heparin serves as a natural activator of PKR under certain situations. Heparin treatment of VSMC results in inhibition of proliferation due to a block at the G1 to S phase transition and PKR activation is essential at least in part for this cell-cycle block [38]. In addition, heparin-induced cell-cycle block is mediated by an increase in p27kip1 protein levels that occurs by stabilization of p27kip1 protein in a PKR-dependent and independent manner (our unpublished results).
As a first step in understanding the mechanism of heparin-induced PKR activation, here we present evidence that PKR has two separate heparin-binding domains (ATD and CTD), both of which are nonoverlapping with its DRBD (Fig. 4A). Although each one of these domains is sufficient for heparin binding, they work in cooperation to enhance the affinity of heparin binding of full length PKR. Both domains function with equal efficiency and independently of each other when removed from their natural context. However, when present together in PKR, the ATD seems to confer higher affinity for binding to heparin. This may be due to contribution from the neighboring basic residues (outside of 279–318) upstream of the ATD to heparin binding, although these residues remain to be identified. It has been reported before that for certain proteins, the heparin-binding residues come from concontiguous regions of the protein [58–61]. Although in case of PKR, defined heparin-binding domains can be identified; additional contribution from residues upstream of ATD to enhance its binding cannot be ruled out. As the deletion mutant containing residues 146–277 shows extremely weak binding to heparin, we know that this region by itself is not sufficient for efficient heparin binding. However, it may participate in strengthening the binding of ATD (278–318), because of the fact that the deletion mutant 146–318 shows better binding to heparin–agarose than the deletion mutant 279–318 or 279–412 (data not shown).
Heparin is a negatively charged polymer of a regular disaccharide repeat sequence that has a high degree of sulfation [62]. Thus, many proteins are expected to bind heparin via electrostatic interactions. Several studies with a diverse set of proteins have indicated the importance of positively charged amino acids for heparin binding [44,63]. Cardin and Weintraub aligned a broad collection of alleged heparin-binding sequences in order to identify common motifs [44]. In their study, the motifs (XBBXBX) and (XBBBXXBX) were identified, where B designates a basic amino acid and X indicates any other amino acid. Both of the sequences that we have identified as heparin-binding domains within PKR contain the (XBBXBX) motif (Fig. 4B). In another study, a more stringent approach was taken to analyze heparin-binding sequences and only the segments which were directly shown to be involved in heparin binding were analyzed structurally [63]. Using a 3D graphics technique, these authors also identified a distinct spatial pattern in the distribution of basic residues within these segments. As no structural information is available at present for the regions important for heparin binding within PKR, it cannot be predicted at present if these domains conform to the spatial patterns noted by Margalit et al. [63].
Point mutations of basic amino acids in the identified ATD and CTD regions led to a loss of heparin binding. The hep2 mutations are in a region between the conserved kinase subdomains VII and VIII within PKR’s catalytic domain. Although the subdomains VI and VII are known to be involved in ATP-binding [4], hep2 mutation located downstream did not alter PKR’s ATP-binding significantly. The mutation R297A showed a significant reduction in ATP binding, suggesting that this residue may contribute to some extent to ATP binding. At present we do not know the significance of this result, although arginine residues in ATPase enzymes are involved in ATP-binding [64,65]. In the case of PKR, it may be possible that mutation of R297 causes a local structural perturbation leading to a loss of ATP-binding although K296 has been shown to be the conserved lysine involved in ATP interaction. Mutations in the two heparin-binding domains resulted in a loss of kinase activity in response to both heparin and dsRNA. Although a mutant that can be activated normally by dsRNA but is unresponsive to heparin would be extremely valuable for understanding the role of PKR activation in heparin-induced cell-cycle block, the possibility of generating such a mutation seems unlikely due to the fact that heparin-binding domains also overlap with kinase domains crucial for the catalytic function. In this regard, it is worth noting that George et al. reported that heparin-activated PKR could not catalyze transmolecular phosphorylation of the inactive K296R mutant [37]. These authors also reported that preincubation of PKR with heparin in the absence of ATP blocked subsequent autophosphorylation of PKR mediated either by dsRNA or heparin in the presence of ATP. In view of our results presented here, one likely explanation for this could be that once heparin is bound to PKR in the absence of ATP, it precludes ATP-binding when it is added later due to an overlap in the heparin-binding and ATP-binding sites. Our results indicate that the ATP-binding residues and heparin-binding residues are present within the aminoterminal heparin-binding domain (ATD) of PKR.
Thus, in contrast to the dsRNA-binding domain of PKR, its heparin-binding domains are located in the carboxy-terminal half of the molecule. The aminoterminal heparin-binding domain (ATD) overlaps with the ATP-binding catalytic subdomain II and the carboxy-terminal heparin-binding domain (CTD) is located between the conserved kinase subdomains VII and VIII. Point mutations within these domains resulted in a loss of heparin-binding and also caused perturbations in the catalytic functions of PKR leading to a loss of kinase activity in response to both dsRNA and heparin.
Experimental procedures
Generation of amino terminal deletion mutants
The different amino terminal deletions were created by sub-cloning the fragments from PKR/BSIIKS+ DNA [19] into the appropriate expression vectors to maintain the reading frame. Δ145 has been described before [19]. Δ278, and Δ318 (the numbers represent the number of amino terminal residues deleted) were constructed by subcloning the MscI-BamHI and SspI-BamHI fragments generated from the coding region of PKR into SmaI-BamHI sites of pGEM3–5T and pGEM3–9T [19], respectively. Δ170 was generated by PCR amplification of the region encoding residues 171–551 of the protein and subcloning it into BSIIKS+. The Kozak consensus and translation start codon were added as a part of the upstream primer. All mutant constructs were confirmed by sequencing.
Generation of carboxy terminally deleted proteins
The PKR/BSIIKS+ DNA [19] was cut with appropriate restriction enzymes and the resulting templates were used for in vitro transcription/translation using the TNT T7 system (Promega, Madison, WI, USA) to generate truncated proteins as described before [6,36].
Generation of 278-ID mutant
The Δ278 construct was digested with SspI and BglII. This releases a fragment encoding residues 318–412. The BglII 5′ overhangs were filled in with Klenow DNA polymerase. The larger piece was gel-purified and self ligated to generate a plasmid with an insert that lacks the piece encoding residues 318–412.
Generation of luciferase fusion constructs
The ATD (residues 278–318) and CTD (residues 410–479) regions of PKR were amplified by PCR using the following primers. For subcloning purposes, restriction enzyme sites were added to the primers. To generate the HBD fragment, which contains the region between residues 278–479, we used the primers ATD sense and CTD antisense for PCR amplification.
ATD sense: 5′-GCTCTAGAGCCATGGGCCAAGTT TTCAAAGCAAAAC-3′,
ATD antisense: 5′-GCGGATCCCATTTACATGATCA AGTTTTGC-3′;
CTD sense: 5′-GCTCTAGAGCCATGGCGATTCATA GAGATCTTAAGCC-3′,
CTD antisense: 5′-GCGGATCCGAAGTTCAGCAAGA ATTAGCCC-3′.
The corresponding regions were amplified using PCR reaction on template PKR/ BS and the resulting fragments were subcloned into BSIIKS+ at XbaI and BamHI sites. The luciferase coding region was excised from T7 control luciferase plasmid (Promega) with BamHI and SacI and joined to the ATD, CTD or HBD regions in frame by subcloning into BamHI and SacI sites. This results in an in-frame fusion to the amino terminus of luciferase. The constructs were confirmed by sequencing.
Heparin-binding assay
Binding to heparin–agarose was assayed at two different salt concentrations. The binding and washing was carried out in the same buffer, e.g. for binding assayed at 50 mM salt concentration, both binding and washing was performed with 50 mM NaCl. Heparin agarose beads (25 μL; Sigma, St. Louis, MO, USA) were washed three times with 500 μL of binding buffer [20 mM Hepes, pH 7.5, 50 mM/ 200 mM NaCl, 5 mM magnessium acetate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5% (v/v) NP-40, 10% (v/v) glycerol] and suspended in 25 μL binding buffer. 35S-Methionine labeled mutant proteins were synthesized by in vitro translation using the TNT T7 quick system (Promega). Translation products (5 μL) diluted to 300 μL with the binding buffer were used for each binding assay. The heparin–agarose beads were mixed with the translation products and incubated at 30 °C for 30 min on a rotating wheel. The beads were then washed in 500 μL binding buffer three times. After the last wash, the beads were suspended in 30 μL of 1 × SDS sample loading buffer, boiled for 5 min, centrifuged at room temperature in a microfuge, and the eluted proteins were analyzed on a 12% SDS/polyacrylamide gel by phosphorimager analysis.
Generation of point mutants
The point mutant of PKR were produced using the PKR/ BSIIKS+ construct using the GeneEditor in vitro site-directed mutagenesis system (Promega) and the following mutation oligonucleotides according to the instructions provided with the kit:
K299A: 5′-GTTATTAAACGTGTTGCATATAATAAC GAG-3′; DM: 5′-ACTTACGTTATTGCACGTGTTGCAT ATAATAACGAG-3′; hep2: 5′-CTGAAAAATGATGGAG AGCTCACAGAGAGTAAGGGAACTTTG-3′.
The mutations generated were confirmed by sequencing and were used for further analysis.
Poly(I)·poly(C)-agarose binding assay
The in vitro translated, 35S-labeled PKR deletion proteins were synthesized using the TNT T7 coupled reticulocyte lysate system from Promega. The dsRNA-binding activity was measured using a poly(I)·poly(C)-agarose binding assay. A 4 mL aliquot of the translation products was diluted with 25 mL of binding buffer [20 mM Tris/HCl, pH 7.5, 0.3 M NaCl, 5 mM MgCl2, 1mM dithiothreitol (dithiothreitol), 0.1 mM phenylmethylsulfonyl fluoride, 0.5% (v/v) IGEPAL, 10% (v/v) glycerol], mixed with 25 mL of poly(I)·poly(C)-agarose (Pharmacia, Amersham Biosciences Corp., Piscataway, NJ, USA) beads and incubated at 30 °C for 30 min with intermittent shaking. The beads were then washed four times with 500 mL of binding buffer. The proteins bound to the beads after washing were analyzed by SDS /PAGE followed by fluorography.
In vitro dimerization assay
The in vitro PKR–PKR interaction assay was performed as described previously [36]. The proteins were in vitro-translated and 35S-methionine labeled. Four mililitres of the translation mix were incubated with 1 mg of pure recombinant hexahistidine tagged PKR protein and 20 mL of Ni-charged His-bind resin (Novagen, Madison, WI, USA) at 30 °C for 2 h in binding buffer [5 mM imidazole, 200 mM NaCl, 20 mM Tris/HCl, pH 7.9, and 0.5% (v/v) Nonidet P-40]. After binding, the beads were washed with 500 mL of wash buffer [60 mM imidazole, 200 mM NaCl, 20 mM Tris/HCl, pH 7.9, and 0.5% (v/v) Nonidet P-40] six times. The washed beads were then boiled in Laemmli buffer [150 mM Tris/HCl, pH 6.8, 5% (w/v) SDS, 5% (v/v) 2-mercaptoethanol, and 20% (v/v) glycerol] for 2 min and analyzed by SDS /PAGE on a 12% gel. Fluorography was performed at −80 °C with intensifying screens.
Kinase activity assay
PKR activity assays were performed as described previously [23] using an anti-PKR monoclonal Ig (Ribogene, Hayward, CA, USA; 71/10). The proteins were produced by in vitro translation using the TNT T7 quick translation system (Promega) without any labeled amino acid. Non-radioactive methionine was added to the translation mix. A 3 mL aliquot of total protein was immunoprecipitated using anti-PKR monoclonal Ig (71 : 10) in high-salt buffer (20 mM Tris/HCl, pH 7.5, 50 mM KCl, 400 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, 100 units·mL−1 aprotinin, 0.2 mM phenylmethylsulfonyl fluoride, 20% glycerol, 1% Triton X-100) at 4 °C for 30 min on a rotating wheel followed by addition of 10 mL of Protein A-Sepharose slurry and incubation for another hour. The Protein A–Sepharose beads were washed four times in 500 mL of high-salt buffer and twice in activity buffer (20 mM Tris/HCl, pH 7.5, 50 mM KCl, 2 mM MgCl2, 2 mM MnCl2, 100 units·mL−1 aprotinin, 0.1 mM phenylmethylsulfonyl fluoride, 5% glycerol). The PKR assay was performed with PKR still bound to the beads in activity buffer containing 250 ng of purified eIF2 (kindly provided by Dr William Merrick, Case Western Reserve University, Cleveland, OH, USA), 0.1 mM ATP and 3700 Bq of [32P]ATP[γ] at 30 °C for 10 min. The standard activator of the enzyme was 0.1 mg·mL−1 poly(I)·poly(C) or 50 mg·mL−1 of heparin. For the inhibition assays with HBD protein, the flag-tagged PKR and HBD proteins were cotranslated using the TNT T7 quick translation system (Promega) without any labeled amino acid. The PCR amplified HBD domain was cloned into BSIIKS+ (Stratagene) and a flag epitope was introduced at the amino-terminus by inserting the corresponding oligonucleotide at the 5′-end. Flag-p56/BSIIKS+ construct encodes another interferon-induced protein p56 and it was used as a negative control. Non-radioactive methionine was added to the translation mix. A 3 mL aliquot of total protein was immunoprecipitated using antiflag monoclonal antibody agarose (Sigma) and the rest of the kinase assay protocol was same as above except that no eIF2 was used as substrate.
In vivo translation inhibition assay
The translation-inhibiting activity of PKR and its point mutants was tested using a translation inhibition assay as described previously [45]. In this assay, the effect of cotransfection with an effector plasmid on translation of a reporter such as luciferase is tested. HT-1080 fibrosarcoma cells were transfected in six-well plates in triplicate with 800 ng of pGL2-luciferase reporter plasmid and 200 ng of effector plasmid (expression constructs of various point mutants of PKR) DNAs using the Lipofectamine (Invitrogen, Carlsbad, CA, USA) reagent. Cells were harvested 24 h after transfection and assayed for luciferase activity.
Yeast growth phenotype assay
Wild-type PKR and the point mutants were subcloned into the pYES2 yeast expression plasmid (Invitrogen), giving us galactose inducible expression of these proteins. The constructs were than introduced into the yeast strain INVsc1 (Invitrogen) using the lithium acetate method. Transformed yeast strains were grown to an D600 of about 2 in yeast extract, peptone and dextrose medium. From this culture 1 mL was then pelleted and resuspended in the appropriate amount of distilled water to yield an D600 of 10. For example, a 1 mL culture with an D600 of 2 would be pelleted and resuspended in 200 μL of distilled water, thus yielding an D600 of 10. Serial dilutions were then made to yield D600 values of 1, 0.1, and 0.01. 10 μL of each dilution was then spotted onto synthetic medium lacking uracil and containing either glucose or galactose/raffinose as a carbon source (Clontech, Palo Alto, CA, USA).
ATP-binding assay
Binding to ATP-agarose was assayed at a 50 mM salt concentration (20 mM Hepes, pH 7.5, 50 mM NaCl, 5 mM Mg acetate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5% NP-40, 10% glycerol). The same buffer was used both for the binding of the mutants to ATP-agarose and the subsequent washes. 35S-methionine labeled wild-type PKR and mutant PKR proteins were synthesized by in vitro translation using the TNT T7 Quick system (Promega). ATP-agarose beads (20 μL; Sigma) were used for each binding reaction. From the translation product, 4 μL was diluted to 50 μL with 50 mM binding buffer and was used in each binding reaction. In appropriate reactions, either heparin or dsRNA was added to the binding reaction as an activator of PKR. The ATP-agarose beads were mixed with the translation products and incubated for 30 min at 30 °C on a rotating wheel. The beads were then washed with 50 mM binding buffer once. After washing, the beads were suspended in 20 μL 1 × SDS sample loading buffer, boiled for 5 min, centrifuged at 16 000 g at room temperature in a microfuge, and the eluted proteins were analyzed on a 12% SDS/polyacrylamide gel by phosphorimager analysis.
Acknowledgments
The authors would like to thank Anna McNeal for excellent technical assistance. This work was supported by a US Public Health Service grant R01 HL63359 (National Heart, Lung, and Blood Institute) to RCP.
Abbreviations
- ATD
amino terminal domain
- CTD
carboxy terminal domain
- DRBD
double stranded RNA binding domain
- ds
double stranded
- eIF2α
α-subunit of the eukaryotic initiation factor 2
- HBD
heparin-binding domain
- IFN
interferon
References
- 1.de Veer MJ, Holko M, Frevel M, Der Walker ES, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 2.Sen GC, Ransohoff RM. Interferon-induced antiviral actions and their regulation. Adv Virus Res. 1993;42:57–102. doi: 10.1016/s0065-3527(08)60083-4. [DOI] [PubMed] [Google Scholar]
- 3.Clemens MJ. PKR – a protein kinase regulated by double-stranded RNA. Int J Biochem Cell Biol. 1997;29:945–949. doi: 10.1016/s1357-2725(96)00169-0. [DOI] [PubMed] [Google Scholar]
- 4.Meurs E, Chong K, Galabru J, Thomas NS, Kerr IM, Williams BR, Hovanessian AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 5.Hovanessian AG, Galabru J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur J Biochem. 1987;167:467–473. doi: 10.1111/j.1432-1033.1987.tb13360.x. [DOI] [PubMed] [Google Scholar]
- 6.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. Embo J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999;274:15427–15432. doi: 10.1074/jbc.274.22.15427. [DOI] [PubMed] [Google Scholar]
- 8.Patel CV, Handy I, Goldsmith T, Patel RC. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- 9.Colthurst DR, Campbell DG, Proud CG. Structure and regulation of eukaryotic initiation factor eIF-2. Sequence of the site in the alpha subunit phosphorylated by the haem-controlled repressor and by the double-stranded RNA-activated inhibitor. Eur J Biochem. 1987;166:357–363. doi: 10.1111/j.1432-1033.1987.tb13523.x. [DOI] [PubMed] [Google Scholar]
- 10.Samuel CE. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J Biol Chem. 1993;268:7603–7606. [PubMed] [Google Scholar]
- 11.Jagus R, Joshi B, Barber GN. PKR, apoptosis and cancer. Int J Biochem Cell Biol. 1999;31:123–138. doi: 10.1016/s1357-2725(98)00136-8. [DOI] [PubMed] [Google Scholar]
- 12.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 13.Koromilas AE, Roy S, Barber GN, Katze MG, Sonenberg N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257:1685–1689. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- 14.Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams BR. Signal integration via PKR. Sci STKE 2001. 2001;RE2 doi: 10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 16.Judware R, Petryshyn R. Partial characterization of a cellular factor that regulates the double-stranded RNA-dependent eIF-2 alpha kinase in 3T3-F442A fibroblasts. Mol Cell Biol. 1991;11:3259–3267. doi: 10.1128/mcb.11.6.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salzberg S, Mandelbaum M, Zalcberg M, Shainberg A. Interruption of myogenesis by transforming growth factor beta 1 or EGTA inhibits expression and activity of the myogenic-associated (2′-5′) oligoadenylate synthetase and PKR. Exp Cell Res. 1995;219:223–232. doi: 10.1006/excr.1995.1222. [DOI] [PubMed] [Google Scholar]
- 18.Katze MG, Wambach M, Wong ML, Garfinkel M, Meurs E, Chong K, Williams BR, Hovanessian AG, Barber GN. Functional expression and RNA binding analysis of the interferon-induced, doublestranded RNA-activated, 68,000-Mr protein kinase in a cell-free system. Mol Cell Biol. 1991;11:5497–5505. doi: 10.1128/mcb.11.11.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel RC, Sen GC. Identification of the double-stranded RNA-binding domain of the human interferon-inducible protein kinase. J Biol Chem. 1992;267:7671–7676. [PubMed] [Google Scholar]
- 20.Green SR, Mathews MB. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev. 1992;6:2478–2490. doi: 10.1101/gad.6.12b.2478. [DOI] [PubMed] [Google Scholar]
- 21.McCormack SJ, Thomis DC, Samuel CE. Mechanism of interferon action: identification of a RNA binding domain within the N-terminal region of the human RNA-dependent P1/eIF-2 alpha protein kinase. Virology. 1992;188:47–56. doi: 10.1016/0042-6822(92)90733-6. [DOI] [PubMed] [Google Scholar]
- 22.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. Embo J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patel RC, Stanton P, Sen GC. Role of the amino-terminal residues of the interferon-induced protein kinase in its activation by double-stranded RNA and heparin. J Biol Chem. 1994;269:18593–18598. [PubMed] [Google Scholar]
- 24.Romano PR, Green SR, Barber GN, Mathews MB, Hennebusch AG. Structural requirements for double-stranded RNA binding, dimerization, and activation of the human eIF2a kinase DAI in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:365–378. doi: 10.1128/mcb.15.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmedt C, Green SR, Manche L, Taylor DR, Ma Y, Mathews MB. Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR) J Mol Biol. 1995;249:29–44. doi: 10.1006/jmbi.1995.0278. [DOI] [PubMed] [Google Scholar]
- 26.St Johnston D, Brown NH, Gall JG, Jantsch M. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci USA. 1992;89:10979–10983. doi: 10.1073/pnas.89.22.10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 28.Carpick BW, Graziano V, Schneider D, Maitra RK, Lee X, Williams BR. Characterization of the solution complex between the interferon-induced, double-stranded RNA-activated protein kinase and HIV-I trans-activating region RNA. J Biol Chem. 1997;272:9510–9516. doi: 10.1074/jbc.272.14.9510. [DOI] [PubMed] [Google Scholar]
- 29.Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. Embo J. 2000;19:5567–5574. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol. 1998;18:7304–7316. doi: 10.1128/mcb.18.12.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor DR, Lee SB, Romano PR, Marshak DR, Hinnebusch AG, Esteban M, Mathews MB. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol Cell Biol. 1996;16:6295–6302. doi: 10.1128/mcb.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem. 2001;276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 33.Patel RC, Stanton P, McMillan NM, Williams BR, Sen GC. The interferon-inducible double-stranded-RNA-activated protein kinase self-associates in vitro and in vivo. Proc Natl Acad Sci USA. 1995;92:8283–8287. doi: 10.1073/pnas.92.18.8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cosentino GP, Venkatesan S, Serluca FC, Green SR, Mathews MB, Sonenberg N. Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc Natl Acad Sci USA. 1995;92:9445–9449. doi: 10.1073/pnas.92.21.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Kaufman RJ. Double-stranded (ds) RNA binding and not dimerization correlates with the activation of the dsRNA-dependent protein kinase (PKR) J Biol Chem. 1996;271:1756–1763. doi: 10.1074/jbc.271.3.1756. [DOI] [PubMed] [Google Scholar]
- 36.Patel RC, Sen GC. Requirement of PKR dimerization mediated by specific hydrophobic residues for its activation by double-stranded RNA and its antigrowth effects in yeast. Mol Cell Biol. 1998;18:7009–7019. doi: 10.1128/mcb.18.12.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.George CX, Thomis DC, McCormack SJ, Svahn CM, Samuel CE. Characterization of the heparinmediated activation of PKR, the interferon-inducible RNA-dependent protein kinase. Virology. 1996;221:180–188. doi: 10.1006/viro.1996.0364. [DOI] [PubMed] [Google Scholar]
- 38.Patel RC, Handy I, Patel CV. Contribution of double-stranded RNA-activated protein kinase toward antiproliferative actions of heparin on vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2002;22:1439–1444. doi: 10.1161/01.atv.0000028817.20351.fe. [DOI] [PubMed] [Google Scholar]
- 39.Castellot JJ, Jr, Addonizio ML, Rosenberg R, Karnovsky MJ. Cultured endothelial cells produce a heparinlike inhibitor of smooth muscle cell growth. J Cell Biol. 1981;90:372–379. doi: 10.1083/jcb.90.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clowes AW, Karnowsky MJ. Suppression by heparin of smooth muscle cell proliferation in injured arteries. Nature. 1977;265:625–626. doi: 10.1038/265625a0. [DOI] [PubMed] [Google Scholar]
- 41.Kleiman NS, Weitz JI, Campbell GR, Campbell JH, Woods TC, Blystone CR, Yoo J, Edelman ER, Cassady KA, Gross M. Putting heparin into perspective: its history and the evolution of its use during percutaneous coronary interventions. J Invasive Cardiol. 2000;12:20F–26. [PubMed] [Google Scholar]
- 42.Young JJ, Kereiakes DJ, Grines CL. Low-molecular-weight heparin therapy in percutaneous coronary intervention: the NICE 1 and NICE 4 trials. National investigators collaborating on enoxaparin investigators. J Invasive Cardiol. 2000;12:E14–E18. discussion E25–18. [PubMed] [Google Scholar]
- 43.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 44.Cardin AD, Weintraub HJ. Molecular modeling of protein–glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- 45.Patel RC, Stanton P, Sen GC. Specific mutations near the amino terminus of double-stranded RNA-dependent protein kinase (PKR) differentially affect its double-stranded RNA binding and dimerization properties. J Biol Chem. 1996;271:25657–25663. doi: 10.1074/jbc.271.41.25657. [DOI] [PubMed] [Google Scholar]
- 46.Kaufman RJ, Murtha P. Translational control mediated by eucaryotic initiation factor-2 is restricted to specific mRNAs in transfected cells. Mol Cell Biol. 1987;7:1568–1571. doi: 10.1128/mcb.7.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies MV, Furtado M, Hershey JW, Thimmappaya B, Kaufman RJ. Complementation of adenovirus virus-associated RNA I gene deletion by expression of a mutant eukaryotic translation initiation factor. Proc Natl Acad Sci USA. 1989;86:9163–9167. doi: 10.1073/pnas.86.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park H, Davies MV, Langland JO, Chang HW, Nam YS, Tartaglia J, Paoletti E, Jacobs BL, Kaufman RJ, Venkatesan S. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci USA. 1994;91:4713–4717. doi: 10.1073/pnas.91.11.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21:1908. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong KL, Feng L, Schappert K, Meurs E, Donahue TF, Friesen JD, Hovanessian AG, Williams BR. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. Embo J. 1992;11:1553–1562. doi: 10.1002/j.1460-2075.1992.tb05200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 52.Galabru J, Hovanessian A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J Biol Chem. 1987;262:15538–15544. [PubMed] [Google Scholar]
- 53.Tan SL, Gale MJ, Jr, Katze MG. Double-stranded RNA-independent dimerization of interferon-induced protein kinase PKR and inhibition of dimerization by the cellular P58IPK inhibitor. Mol Cell Biol. 1998;18:2431–2443. doi: 10.1128/mcb.18.5.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ortega LG, McCotter MD, Henry GL, McCormack SJ, Thomis DC, Samuel CE. Mechanism of interferon action. Biochemical and genetic evidence for the intermolecular association of the RNA-dependent protein kinase PKR from human cells. Virology. 1996;215:31–39. doi: 10.1006/viro.1996.0004. [DOI] [PubMed] [Google Scholar]
- 55.Hoover RL, Rosenberg R, Haering W, Karnovsky MJ. Inhibition of rat arterial smooth muscle cell proliferation by heparin. II. In vitro studies. Circ Res. 1980;47:578–583. doi: 10.1161/01.res.47.4.578. [DOI] [PubMed] [Google Scholar]
- 56.Brieger D, Topol E. Local drug delivery systems and prevention of restenosis. Cardiovasc Res. 1997;35:405–413. doi: 10.1016/s0008-6363(97)00155-7. [DOI] [PubMed] [Google Scholar]
- 57.Fritze LM, Reilly CF, Rosenberg RD. An antiproliferative heparan sulfate species produced by post-confluent smooth muscle cells. J Cell Biol. 1985;100:1041–1049. doi: 10.1083/jcb.100.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y, Henderson HE, Liu MS, Zhang H, Forsythe IJ, Clarke-Lewis I, Hayden MR, Brunzell JD. Mutagenesis in four candidate heparin binding regions (residues 279–282, 291–304, 390–393, and 439–448) and identification of residues affecting heparin binding of human lipoprotein lipase. J Lipid Res. 1994;35:2049–2059. [PubMed] [Google Scholar]
- 59.Sendak RA, Bensadoun A. Identification of a heparin-binding domain in the distal carboxyl-terminal region of lipoprotein lipase by site-directed mutagenesis. J Lipid Res. 1998;39:1310–1315. [PubMed] [Google Scholar]
- 60.Sendak RA, Berryman DE, Gellman G, Melford K, Bensadoun A. Binding of hepatic lipase to heparin. Identification of specific heparin-binding residues in two distinct positive charge clusters. J Lipid Res. 2000;41:260–268. [PubMed] [Google Scholar]
- 61.Thompson LD, Pantoliano MW, Springer BA. Energetic characterization of the basic fibroblast growth factor–heparin interaction: identification of the heparin binding domain. Biochemistry. 1994;33:3831–3840. doi: 10.1021/bi00179a006. [DOI] [PubMed] [Google Scholar]
- 62.Jackson RL, Busch SJ, Cardin AD. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- 63.Margalit H, Fischer N, Ben-Sasson SA. Comparative analysis of structurally defined heparin binding sequences reveals a distinct spatial distribution of basic residues. J Biol Chem. 1993;268:19228–19231. [PubMed] [Google Scholar]
- 64.Lansky Z, Kubala M, Ettrich R, Kuty M, Plasek J, Teisinger J, Schoner W, Amler E. The hydrogen bonds between Arg423 and Glu472 and other key residues, Asp443, Ser477, and Pro489, are responsible for the formation and a different positioning of TNP-ATP and ATP within the nucleotide-binding site of Na(+)/K(+)-ATPase. Biochemistry. 2004;43:8303–8311. doi: 10.1021/bi0496485. [DOI] [PubMed] [Google Scholar]
- 65.Morgan CT, Tsivkovskii R, Kosinsky YA, Efremov RG, Lutsenko S. The distinct functional properties of the nucleotide-binding domain of ATP7B, the human copper-transporting ATPase: analysis of the Wilson disease mutations E1064A, H1069Q, R1151H, and C1104F. J Biol Chem. 2004;279:36363–36371. doi: 10.1074/jbc.M404553200. [DOI] [PubMed] [Google Scholar]