

Summary
A key issue for sustainable culture of adult epithelial cells is enrichment for stem cell populations in tissue organoids. Gastrointestinal stem cells can be propagated using conditioned media from a supportive cell line (L-WRN). This protocol describes how to prepare conditioned media and culture stem cell-enriched epithelial organoids from the mouse gastrointestine. These organoids are also amenable to genetic modification with recombinant lentiviruses. This system enables many types of cell biological assays that have been performed with immortalized cell lines to be applied to organoids. Isolation of epithelial cell units from mice takes up to 2 hours and stem cell-enriched gastrointestinal organoids are obtained within 3 days. Genetically modified organoids with lentiviruses can be obtained in 2 weeks.
INTRODUCTION
The in vitro propagation of enriched populations of stem cells is essential valuable research tool. Long-term in vitro culture of gastrointestinal epithelial cells was first achieved using a growth factor-optimized media and basement membrane matrix (Matrigel)1,2. However, as the majority of the cells undergo differentiation, these methods remain challenging when specifically studying stem cells. Therefore, we devised a simple and robust method to culture and propagate enriched intestinal epithelial stem cells (ISCs) 3, aiming to remove significant roadblocks to understanding the basic properties of epithelial stem cells and to facilitate movement towards the long-term goal of utilizing these cells therapeutically.
Rationale for the development of the protocol
Recent studies identified three factors that permit culture of small intestinal and gastric antral epithelial cells1,2. Two of these factors, Wnts and R-spondins, can enhance canonical Wnt signaling, a pathway required for self-renewal of various tissue-specific stem cells including those of the gastrointestinal tract4,5. Canonical Wnts, such as Wnt3a, bind the frizzled receptor family and activate β-catenin-dependent transcription. Members of the R-spondin protein family are potent co-activators of canonical Wnt signaling in the intestine and are essential for isolation of small intestinal stem cells1,6. A third factor, noggin, a bone morphogenetic protein (BMP) signaling inhibitor, enables the maintenance and passage of small intestinal organoids in vitro1. Although these three factors are commercially available, it is costly to maintain the large scale cultures that are required for standard assays currently performed with immortalized cell lines.
Conditioned media has been used for many studies of the Wnt signaling pathway. For example, L-Wnt3a cells that originated from mouse L cells are an excellent source of the canonical Wnt ligand, Wnt3a7. We further modified this cell line to establish a new supportive cell line (L-WRN)3 (Supplementary Methods and Supplementary Fig. 1a) that secreted the three factors (Wnt, R-spondin and noggin) that are sufficient to propagate and maintain intestinal epithelial cells. We created crypt culture media that contained 20% fetal bovine serum (FBS) and recovered conditioned media from L-WRN cells. We optimized this system by selecting and screening single cell clones derived from the L-WRN parent line to identify lines that produced the highest functional titers of conditioned media3.
Using 50% conditioned media (1:1 dilution with fresh crypt culture media) and basement membrane matrix (Matrigel), we successfully maintained colonic epithelial organoids for >50 passages for >150 days. In 50% conditioned media, most cells were proliferative and organoids maintained high levels of expression of a stem cell marker, Lgr53,8. We then applied the same methodology to isolate epithelial units and grow epithelial organoids from the mouse stomach, small intestine, and cecum (Figs. 1 and 2). To maximize early growth of developing primary organoids from small intestinal and cecal crypts, the tissue media was supplemented with Y276321,9, an inhibitor for Rho-associated protein kinase (ROCK), and SB431542, an inhibitor for TGF-β type I receptor for 2–3 days (Fig. 1). After this initial treatment, these inhibitors were no longer required for subsequent culture (Fig. 2). Thus an advantage of this approach is that organoids can be propagated from different sites using the same media.
Figure 1.

Development of mouse gastrointestinal epithelial organoids. (a) Collagenase digestion of the mouse stomach, small intestine, cecum, and colon. Vigorous pipetting separated single epithelial units from minced tissue fragments in collagenase solution. Bars=1 mm. (b) Isolated gastrointestinal epithelial units. Bars=200 μm. (c) Primary epithelial organoids from gastrointestinal tissues. Isolated epithelial units were embedded in Matrigel and cultured with 50% L-WRN media for 3 days. 10 μM Y27632 and 10 μM SB431542 were added for the small intestine and cecum. Bars=1 mm.
Figure 2.
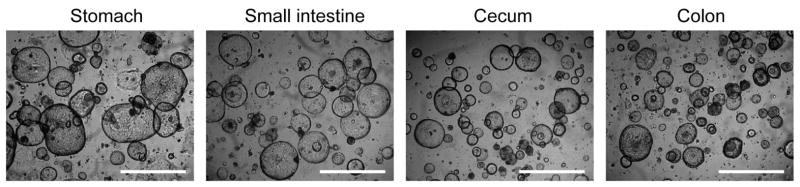
Typical morphology of gastroiontestinal organoids cultured in 50% L-WRN conditioned media. Bars=1 mm.
For passage, organoids were dissociated by incubation in 0.25% trypsin. The trypsinization process disrupted the spherical organoids into cell aggregates that were then embedded in fresh Matrigel. These aggregates developed into new spherical organoids (Fig. 2). Depending on cell density, gastrointestinal organoids should be passaged every 3 days using 1:4 to 1:8 dilutions. Organoids can be frozen in a standard cell freezing media containing 10% dimethyl sulfoxide (DMSO). For use in functional assays, organoids were trypsinized with vigorous pipetting which dissociated cells into homogeneous small aggregates. This method evenly distributes cells in the wells of 96-well plates for functional assays (Fig. 3).
Figure 3.
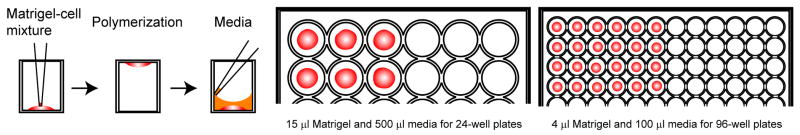
Schematic drawing of the stem cell culture system.
We also established a new cell line (L-WR) that expresses Wnt3a and R-spondin 3 (Supplementary Fig. 1b) as a substitute for L-WRN cells. We found that, in this system, noggin was not essential to maintain organoids from the mouse stomach, small intestine, cecum, and colon for over 3 months (>30 passages). One possible explanation for the fact that noggin is dispensable is that equivalent BMP inhibitors are present in the FBS or were secreted from the modified L cells.
The advantage of using conditioned media is that it provides relatively intact and high titer proteins compared with reconstituted media with purified proteins. Potential disadvantages included exhaustion of basal media and secretion of undesired factors from host cells. Therefore, we optimized the timing of harvest and dilution rate with fresh media. To determine the most efficient timing for harvest, we recovered conditioned media at different time points (1–3 days) and found that the activity of conditioned media did not change depending on the incubation time (Supplementary Fig. 2). Thus, in this protocol, conditioned media is recovered every 24 hours and four serial collections are combined into one batch to homogenize activity. We found that media could be serially collected for at least 12 days (3 batches) without a decrease in activity. If larger volumes of conditioned media are needed, one can adopt this protocol for large scale preparation using the Cell Factory system (Thermo Scientific). To evaluate the activity of each lot, we check the viability and growth of organoids for several passages. Lgr5 expression level in organoids is a quantitative indicator of media activity for maintaining stem cells (Supplementary Methods and Supplementary Fig. 2).
Although permanent gene transduction of small intestinal organoids with lentiviral infection has been reported10, the process can be simplified and enhanced to achieve similar efficiency to that observed when modifiying immortalized and cancer cell lines. To demonstrate the feasibility of genetic modification of colonic stem cells, we generated recombinant lentiviral particles expressing green fluorescent protein (GFP) and shRNA3. Because lentivirus particles can not penetrate the Matrigel layer, we incubated trypsinized organoids with viral particles in liquid media for 6 hours. Y276321,9 improved survival of dissociated epithelial cells during the infection step. After infection, cells were embedded in Matrigel and grown into organoids. Supplementation with Y27632 and SB431542 supported the formation of organoids that formed from infected small cell aggregates. Primary organoids (small intestinal and colonic) after infection show a high level of GFP signal (typically >50% of cells) (Fig. 4a, b and Supplementary Fig. 5). To generate spheres that were 100% GFP-positive, we trypsinized organoids and generated small cell aggregates, some of which consisted of only GFP-positive cells. We then picked 100% GFP-positive spheres using an epifluorescence microscope to guide selection (Fig. 4c). We confirmed significant GFP expression under the phosphoglycerate kinase (hPGK) promoter in gastric, small intestinal, cecal, and colonic organoids. We anticipate that this method will allow for loss and gain of function experiments for specific genes when genetically modified mice are not available. The RNAi Consortium shRNA Library is a great source of shRNA-expressing lentiviral plasmids (available from SIGMA or Thermo Scientific). We adapted these plasmids to our system by replacing a puromycin resistant gene in the pLKO.1 backbone plasmid with GFP as described7.
Figure 4.
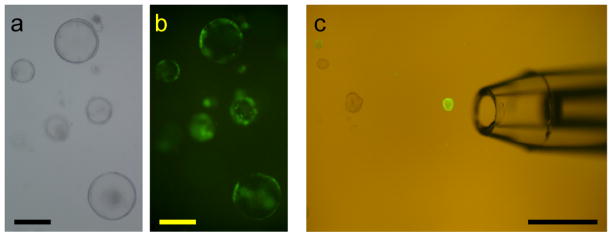
Isolation of lentivirus-infected organoids. (a, b) Primary organoids after infection showed mosaic GFP expression. Representative fluorescence (a) and bright field images (b) are shown. Bars=200 μm. (c) Hand-selection of secondary organoids that consist of GFP-positive cells. Bar=1 mm.
Taken together, this protocol greatly facilitates study of emerging tissue-specific stem cell populations in the gastrointestine and facilitates the use of these cells to tackle a variety of problems including transplantation. Any individuals who have experience in cell culture study can easily access this system without any complicated devices or techniques.
Experimental Design
Basal media for the conditioned media
In this protocol, we use Advanced DMEM/F12 as basal media according a previous report1. We add 20% FBS in the media to support secretion of recombinant factors from L cells and growth of epithelial organoids. We use this basal media to isolate and expand intestinal epithelial stem cells. For some downstream functional studies, it is possible to perform experiments in reduced levels of serum. Serum-free media did not affect the viability of L-WRN and L-WR cells. However, the activity of serum-free conditioned media will be lower than that of serum-containing media because serum is required for the secretion of Wnt ligands7.
Applicability of the protocol to non-gastrointestinal tissues and tumors
Although this method was developed to culture gastrointestinal stem cells, it also allows for growth of other endoderm-derived stem cells. Thus far, we have established epithelial organoids from pancreas, trachea, lung, and thymus using 50% L-WRN conditioned media containing 10 μM Y27632 and 10 μM SB431542 (Supplementary Fig. 3a). We have also cultured tumor cells from intestinal polyps developed in ApcMin and azoxymethane (AOM)/dextran sodium sulphate (DSS)-treated wild type mice using basal media (0% conditioned media) containing 10 μM Y27632 and 10 μM SB431542 (Supplementary Fig. 3b, c). Some tumors and non-gastrointestinal tissues contain greater amounts of mesenchymal cells that are difficult to separate from epithelial units. Here, we also provide a protocol to expand epithelial organoids that become free from mesenchymal contaminating cells (Box 1).
Box 1. Purifying organoids from stromal cell contamination ● TIMING 40–60 min.
Scratch and suspend Matrigel in culture media (with a 1,000 μl pipette). Transfer organoid mixture to a 6 cm dish with 5 ml washing media.
Pick up epithelial organoids under a dissection microscope using forged glass capillaries connected to a mouth pipette.
Collect organoids in a 1.5 ml test tube with ~100 μl washing media.
Spin down organoids at 200 g for 5 min.
Aspirate supernatant carefully using a 200 μl pipette.
Add ~200 μl PBS-EDTA.
Spin down organoids at 200 g for 5 min.
Aspirate supernatant carefully using a 200 μl pipette.
Add 20 μl trypsin-EDTA.
Incubate tubes in the 37 °C water bath for 2 min.
Add 200 μl washing media and dissociate organoids by vigorous pipetting.
Add 500 μl washing media.
Centrifuge at 200 g for 5 min.
Aspirate supernatant completely. Suspend cells in 15 μl Matrigel
Place Matrigel-cell mixture in the 24-well plate. Incubate the plate in the cell culture incubator until Matrigel polymerizes (Turn the plates upside down).
Add 500 μl 50% conditioned media to the well. Continue the routine passage procedure (Steps 38–53).
Functional assays
The simplest method to analyze organoids is to determine the mRNA expression levels for genes of interest. One can test the effects of chemicals, growth factors, and cytokines on the downstream gene expression associated with specific signaling pathways. Enzymatic assays that utilize chemicals such as luciferase and MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) would be more suitable using these cells for high throughput screening. Real-time imaging of fluorescent proteins is a useful tool to analyze functions of specific targets in live cells. Fluorescent protein- and luciferase-expressing organoids can be obtained from transgenic mice or by infecting organoids with lentiviruses. Fluorescence-activated cell sorting (FACS) analyses would also be useful to analyze cell surface markers and cell cycle.
Histological analyses
We have reported whole mount immunostaining of organoids3 for which we applied a modified staining method for use with mouse early embryos11. Maintaining organoids in Matrigel during the staining process causes uneven staining because antibodies were not able to penetrate Matrigel after fixation. In such cases, Matrigel should be removed with incubating in Cell Recovery Solution (BD: 354253). Histological sections can be cut from frozen samples in optimal cutting temperature (OCT) compound (Sakura Finetek: 4583) as well as paraffin-embedded samples.
Comparison with other methods
Most of the recent studies using gastrointestinal organoids use the method developed by Sato et al1. They reconstitute the essential conditions for long-term maintenance of gastrointestinal organoids using defined chemicals and proteins. In developing our protocol, we followed Sato et als. fundamental principle that maintenance of normal gastrointestinal stem cells needs canonical Wnt agonists and extracellular matrix (e.g. Matrigel) in addition to the basic requirements for cell culture. A significant advantage of our protocol is that the conditioned media contains non-purified Wnt3a and R-spondin 3 which may provide higher levels of canonical Wnt activity that leads to enhanced stem cell propagation. We reported that 50% L-WRN conditioned media stimulates robust Lgr5 expression compared with reasonable concentrations of purified Wnt3a (100 ng/ml) and R-spondin 1 (500 ng/ml), indicating that commercial sources can not maximize canonical Wnt activity in organoids3. Another difference is that our protocol utilizes serum-containing media. Generally non-serum (or serum-reduced) media is preferred for stem cell culture because animal serum contains undefined differentiation factors. However, non-serum media requires additional supplementary factors including recombinant growth factors (e.g., EGF, FGF, and insulin), minerals and vitamins. For example, the colonic organoid culture media developed by Sato et al. contains R-spondin 1, Wnt3a, noggin, EGF, N-2 supplement, and B-27 supplement in addition to basal media12. The complexity and availability of specialized media is a substantial hurdle to starting stem cell culture, even for researchers who have experience in cell biology. Meanwhile, any laboratories that utilize basic cell culture systems can easily produce conditioned media themselves in liter quantities at a reasonable cost. However, there are limitations to using conditioned media that must be borne in mind. Optimal culture media for each specific assay should be determined individually. Undefined factors derived from host cells and serum could affect some signaling pathways. If assays that require stringent conditions need to be performed, the reconstituted media should be used according to previous reports1,2,12. It is also noted that organoids in 50% conditioned media contain only a small number of fully differentiated cells. The conditions for inducing differentiation may differ for individual lineages. Nevertheless, this protocol provides a simple and accessible method to obtain a sufficient number of stem cells for experiments such as high-throughput screening and transplantation13.
Generally puromycin selection or cell sorting is used for enrichment of the stably transduced population after lentiviral infection. With the puromycin selection method, it is difficult to determine the best concentration of puromycin that will achieve the high level of shRNA expression required for efficient gene silencing. Cell sorting is a more reliable method to enrich the high-expressing population of transduced cells, but organoids need to be dissociated into single cells. This process dramatically decreases the viability of stem cells and efficiency of organoid formation and growth. Therefore, we adopted a method in which we hand-pick 10–20 transduced organoids under visible GFP fluorescence. After infection, a transduced cell expands as a cluster within an organoid. Following trypsinization clonal cell aggregates are generated that develop into clonal organoids. These independent organoids are easily picked up with a pipette tip (Fig. 4c).
In conclusion, our protocol is specialized for the establishment, expansion, and genetic modification of normal tissue stem cells. Our aim is to provide the basic tools for further analyses using normal tissue stem cells.
Controls
It must be noted that diversity of a stem cell population decreases during long-term culture, though there is no sign of transformation. Clonal lineages with higher proliferation rates dominate organoids by neutral competition14. When organoids are established from genetically modified mice, organoids from littermate wild type mice must be cultured in parallel and analyzed as a control.
MATERIALS
REAGENTS
-
Mice >8 weeks of age (Jackson Laboratory: C57BL6/J)
! Caution All experiments using animals must follow national and institutional regulations.
Advanced DMEM/F12 (Invitrogen: 12634-010)
200 mM L-glutamine (SIGMA: G7513)
10,000 units/ml penicillin and 10 mg/ml streptomycin (SIGMA: P4333)
Fetal Bovine Serum (FBS) (SIGMA: F6178)
Dulbecco’s Phosphate Buffered Saline (PBS) (SIGMA: D8537)
0.5 M EDTA solution (Invitrogen: 15575020)
2.5% Trypsin (Invitrogen: 15090046)
Preparation of conditioned media
L-WRN (ATCC: XXX) or L-WR (ATCC: XXX) cells (low passage cells are utilized)
DMEM high glucose (SIGMA D6429)
50 mg/ml G418 (SIGMA: G8168)
100 mg/ml hygromycin (InvivoGen: ant-hg-1)
Organoid culture
DMEM/F12 with HEPES (SIGMA D6421)
Collagenase type I (Invitrogen: 17100-017)
50 mg/ml gentamicin (Invitrogen: 15750060)
Basement Membrane Matrix (Matrigel) (BD: 356234, 354234, or 356235)
-
Dimethyl sulfoxide (DMSO) (SIGMA: D2650)
! Caution Harmful.
Y27632 (R&D: 1254)
SB431542 (R&D: 1614)
Stable gene transfection of organoids with lentiviruses
293FT cell line (Invitrogen: R70007)
Lentiviral vector plasmid (GFP-expressing)
psPAX2 packaging plasmid (Addgene: 12260)
pMD2.G envelope plasmid (Addgene: 12259)
OPTI-MEM serum-free media (Invitrogen: 31985062)
Lipofectamine 2000 transfection reagent (Invitrogen: 11668027)
Hexadimethrine bromide (SIGMA: H9268)
PEG-it Virus Precipitation Solution (System Biosciences: LV810A-1)
Optional reagents
Recombinant Mouse R-Spondin 1 (R&D: 3474-RS-050)
Recombinant Human Wnt-3a (R&D: 5036-WN-010)
EQUIPMENT
Common equipment
0.5–10 μl (10 μl), 2–20 μl (20 μl), 20–200 μl (200 μl), and 100–1,000 μl (1,000 μl)
pipettes (Nichiryo: Nichipet EX)
Pipet-Aid XP (Drummond)
5, 10 and 25 ml pipettes
15 ml and 50 ml centrifuge tubes
1.5 ml test tubes
Biological hood
Dissection microscope
Inverted phase microscope Centrifuge (Thermo Scientific: Sorvall Legend)
37°C water bath
37°C cell culture incubator with 5% CO2
Preparation of conditioned media
150 cm2 cell culture flasks
1 L storage bottles (CORNING: 430518) or autoclaved 1 L glass media bottles
Large scale preparation of conditioned media
EasyFill Cell Factory Nunclon (10 trays) (Thermo Scientific: 140400)
Autoclaved 2 L glass media bottles
Bottle-top dispenser (Nichiryo: Dispet EX, DPX-500)
Organoid culture
9 cm petri dishes
3.5 cm petri dishes
24-well cell culture plates
96-well cell culture plates
2 ml cryotubes
40 μm cell strainers (BD: 352340)
70 μm cell strainers (BD: 352350)
10 ml syringes
0.22 μm syringe filters
Blunt needles 19g X 1 ½″ (BRICO Medical Supplies: BN1915)
Fine scissors
Fine forceps
200 μl calibrated pipetes (Drummond: 2-000-200)
Lentiviral infection
0.45 μm syringe filters
Fluorescence microscope (inverted or stereoscopic)
REAGENT SETUP
L-cell media
Add penicillin (100 units/ml), streptomycin (0.1 mg/ml), and FBS (10%) to DMEM. Store at 4 °C and use within 2 weeks.
Washing media
Add penicillin (100 units/ml), streptomycin (0.1 mg/ml), L-glutamine (2 mM), and FBS (10%) to DMEM/F12 with HEPES. Store at 4 °C and use within 2 weeks.
Primary culture media
Add penicillin (100 units/ml), streptomycin (0.1 mg/ml), L-glutamine (2 mM), and FBS (20%) to Advanced DMEM/F-12. Keep at 4 °C during CM preparation.
CM-free organoid culture media
Add recombinant Wnt3a (100 ng/ml) and R-spondin 1 (500 ng/ml) to primary culture media.
▲ CRITICAL It must be made fresh.
Collagenase solution
Add collagenase type I powder (2 mg/ml) and gentamicin (50 μg/ml) to washing media. Filtrate with a 0.22 μm syringe filter. Aliquots can be stored at −20 °C for one month.
Freezing media
Mix 8 volume of washing media, 1 volume of FBS, and 1 volume of DMSO
▲ CRITICAL It must be made fresh.
PBS-EDTA
Add EDTA (0.5 mM) to PBS. Store at room temperature.
Trypsin-EDTA
Add trypsin (0.25%) to PBS-EDTA. Aliquots can be stored at −20 °C for one month.
Matrigel
Thaw the original bottle over night at 4 °C on ice. Mix the solution well by pipetting and make 1 ml aliquots in 1.5 ml test tubes. Aliquots can be stored at −20 °C until the expiration date.
▲ CRITICAL After thawing, keep the tube on ice and use within one week.
Y27632 stock solution
Make 10 mM stock solution in water. Aliquots can be stored at −20 °C for one month.
SB431542 stock solution
Make 10 mM stock solution in DMSO. Aliquots can be stored at −20 °C for one month.
Hexadimethrine bromide
Make 2 mg/ml solution in water. Aliquots can be stored at −20 °C for one month.
Glass capillary pipettes
Forge capillaries over the flame using 200 μl calibrated pipettes
PROCEDURES
Preparation of conditioned media ● TIMING 10–23 d
-
1|
Pre-warm 25 ml L-cell media in a 50 ml centrifuge tube in the 37 °C water bath.
-
2|
Thaw a cryotube of L-WRN or L-WR cells in the 37 °C water bath.
-
3|
Transfer cell solution to pre-warmed media immediately after ice disappears.
▲ CRITICAL STEP Because cells are very fragile after thawing, centrifugation and excess pipetting must be avoided.
-
4|
Transfer media in a 150 cm2 cell culture flask.
-
5|
Incubate the flask in the cell culture incubator for 1 d.
-
6|
Change media and add G418 (500 μg/ml) and hygromycin (500 μg/ml).
-
7|
Grow cells until they become confluent (2 or 3 d).
-
8|
Wash cells with 20 ml PBS-EDTA and aspirate.
-
9|
Add 1 ml trypsin-EDTA. Tap the flask several times to coat the plate surface with the trypsin-containing solution.
-
10|
Incubate the flask for 3–5 min at 37 °C.
-
11|
Suspend cells in 12 ml L-cell media.
-
12|
Choose option depending on the scale of conditioned media production. 1–3 L and 8–24 L of 50% conditioned media can be produced using options A and B, respectively.
(A) Small scale preparation ● TIMING 7–16 d
Add 120 ml L-cell media into the flask. Aliquot cell suspension in five 150 cm2 cell culture flasks (25 ml/flask).
-
Incubate flasks in the cell culture incubator until cells become over-confluent and a number of cell aggregates come off (3 or 4 d).
▲ CRITICAL STEP Cells should be cultured without G418 and hygromycin to prevent carryover of drugs in the conditioned media.
Wash cells with 10 ml (per flask) primary culture media and aspirate.
Add 25 ml (per flask) primary culture media.
Incubate flasks in the cell culture incubator for 24 h.
Recover conditioned media into 50 ml centrifuge tubes and add new media to flasks. Return flasks to the cell culture incubator.
Centrifuge tubes at 2,000 g for 5 min and decant supernatant carefully in a 1 L bottle (total ~125 ml). Store the bottle at 4 °C (1st conditioned media).
Every 24 h, collect 2nd, 3rd and 4th conditioned media in the same bottle.
After 4th collection, add equal volume (~500 ml) of primary culture media to the bottle (final concentration: 50%), mix well, and aliquot media into 15 ml or 50 ml centrifuge tubes. Store media at −20 °C.
Collect 5th to 8th and 9th to 12th media if desired
(B) Large scale preparation ● TIMING 9–19 d
Add 140 ml L-cell media, hygromycin (500 μg/ml), and G418 (500 μg/ml) to the flask. Aliquot cell suspension in six 150 cm2 cell culture flasks (25 ml pre flask).
Incubate flasks in the cell culture incubator until cells become confluent (2 or 3 d).
Prepare 1 L L-cell media in a 1 L bottle.
Wash cells with PBS-EDTA (20 ml per each flask) and aspirate.
Add 1 ml trypsin-EDTA per flask. Tap the flask several times to coat the plate surface with the trypsin-containing solution.
Incubate flasks for 3–5 min in the cell culture incubator.
Suspend cells in 12 ml L-cell media per flask. Collect cell suspension in two 50 ml tubes (~39 ml per tube).
Spin down cells at 200 g for 5 min.
Aspirate media and resuspend cells in 12 ml L-cell media per tube. Transfer cell suspension in the L-cell media bottle and mix well.
Pour cell suspension in EasyFill Cell Factory Nunclon (10 trays). Follow manufacturer’s instruction to distribute cell suspension into each tray.
-
Incubate cell factory in the cell culture incubator until cells become over-confluent (3 or 4 d).
▲ CRITICAL STEP Media will be turbid with floating cells.
Discard L-cell media from the cell factory.
Wash cells with 500 ml primary culture media and discard media.
Pour 1,050 ml (50 ml for expected loss due to evaporation) primary culture media into the Cell Factory.
Incubate the Cell Factory in the cell culture incubator for 24 h.
Collect conditioned media in a 1 L bottle and pour 1,050 ml fresh primary culture media in the Cell Factory. Return the Cell Factory to the cell culture incubator.
Decant conditioned media into twenty 50 ml centrifuge tubes (50 ml per tube).
Spin down cells and debris at 2,000 g for 5 min.
Decant supernatant into four 2 L glass media bottles (5 tubes per bottle). Store bottles at 4 °C (1st conditioned media).
Collect 2nd, 3rd and 4th conditioned media every 24 h.
After the 4th collection, dilute conditioned media with 1 L (per bottle) primary culture media (final concentration: 50%), mix well, and aliquot media into 15 ml or 50 ml centrifuge tubes using an autoclaved bottle top dispenser. Store media at −20 °C.
-
Collect 5th – 8th and 9th – 12th media if desired.
■ PAUSE POINT Conditioned media can be stored at −20 °C for at least 3 months without decrease in activity. After thawing, store at 4 °C and use within one week (do not freeze again).
Isolation and culture of mouse gastrointestinal epithelial units ● TIMING 1.5–2 h
-
13|
Euthanize mice using a chemical reagent (e.g. barbiturates, nonexplosive inhalant anesthetics) according to an approved animal protocol and dissect tissues of your interest. For the small intestine and colon, flush the intestinal tube with PBS using a 10 ml syringe with a 19 G blunt needle and open longitudinally. For the stomach and cecum, open the tissue longitudinally and roughly remove contents on the paper towel.
-
14|
Rinse the tissue with ice cold PBS in a 90 mm petri dish.
-
15|
Wash the tissue with ~20 ml ice cold PBS in a 50 ml centrifuge tube by vigorous shaking.
-
16|
Rinse the tissue with ice cold PBS in a 90 mm petri dish.
-
17|
Dissect out fat and connective tissues carefully using fine scissors and forceps under a dissection microscope. For the small intestine, immerse tissues in washing media to inactivate endogenous proteases and remove villi by carefully scraping mucosa with fine forceps or a glass slide under a dissection microscope (Supplementary Fig. 4).
▲ CRITICAL STEP For gastrointestinal tissues, a 0.5–2 cm2 region of tissue is sufficient. Do not process too much tissue at once or the collagenase solution will become too viscous. When handling multiple tissues at one time, keep petri dishes on ice to avoid cell death. Note that processing the small intestine is time sensitive until it is immersed in washing media.
-
18|
Place the tissue in a 35 mm petri dish.
-
19|
Bring the dish to the biological hood.
-
20|
Mince the tissue with fine scissors.
▲ CRITICAL STEP Use sharp scissors and mince into <1 mm2 fragments that allows them to pass through 1,000 μl pipette tips.
-
21|
Add 1 ml collagenase solution using a 1,000 μl pipette and suspend tissue fragments in the solution.
-
22|
Incubate the petri dish in the cell culture incubator and pipette tissue mixture vigorously every 5–10 min using a 1,000 μl pipette. After pipetting, check if single epithelial units (crypts/pits) have separated from the larger tissue fragments by using a phase or dissection microscope (Fig. 1a).
▲ CRITICAL STEP This step will take 30–60 min. Digest tissue fragments until 50–80% of epithelial units have separated from the larger tissue fragments.
-
23|
Set a 70 μm (or 40 μm for small intestine) cell strainer in a 50 ml centrifuge tube. Filtrate tissue mixture through the cell strainer using a 1,000 μl pipette.
-
24|
Wash the strainer with 9 ml washing media.
-
25|
Transfer filtrated cell solution to a 15 ml centrifuge tube.
-
26|
Centrifuge the tube at 20 g for 5 min.
▲ CRITICAL STEP If there is little or no precipitant, increase the centrifugation speed to 50–150 g. Low centrifugation speed promotes the collection of the heavier epithelial units over lighter single cells. A significant number of epithelial units may stay in the supernatant, but loss of some epithelial units in this step is acceptable because, in general, a relatively small number of epithelial units are required to start the culture.
-
27|
Aspirate supernatant with ~100 μl leftover using the vacuum aspirator.
-
28|
Aspirate remaining supernatant completely using a 200 μl pipette.
▲ CRITICAL STEP Aspirate carefully because precipitants are fragile.
-
29|
Add 500 μl–1 ml washing media and suspend well.
-
30|
Take a drop of suspension and check the density and viability of epithelial units (Fig. 1b).
? TROUBLESHOOTING
-
31|
Transfer appropriate volume of suspension to a 1.5 ml tube (1,000–3,000 intact epithelial units per well).
-
32|
Centrifuge the tube at 200 g for 5 min.
-
33|
Aspirate supernatant completely.
▲ CRITICAL STEP First aspirate supernatant with ~100 μl leftover using the vacuum aspirator and then aspirate remaining supernatant completely with a 200 μl pipette.
-
34|
Place the tube on ice. Suspend epithelial units in Matrigel (15 μl per well).
▲ CRITICAL STEP Gently pipette until cell aggregates are completely dispersed. Do not generate many air bubbles in the Matrigel. Do not warm the tube.
-
35|
Place the tube and a 24-well plate on ice. Place 15 μl of cell-Matrigel suspension in the center of each well using a 20 μl pipette and spread it carefully with a pipette tip (Fig. 3).
-
36|
Incubate plates in the cell culture incubator to polymerize the Matrigel. Turn the plates upside down to avoid the attachment of epithelial units to the surface of the plate.
-
37|
Add 500 μl 50% conditioned media to the well. For the small intestine and cecum, add 500 μl 50% conditioned media containing 10 μM Y27632 and 10 μM SB431542. Change media at least every 2 d.
▲ CRITICAL STEP Developing primary organoids may take 2–4 d. Observe daily and passage organoids when spherical organoids begin to collapse. Generally mesenchymal cell population is diluted out after several passages. If mesenchymal cells dominate primary culture, separate epithelial organoids according Box 1.
? TROUBLESHOOTING
Passage for maintenance ● TIMING 30–40 min
-
38|
Grow organoids in the 24-well plate for 3 d.
▲ CRITICAL STEP The best interval between passages is 3 d for gastrointestinal organoids. The interval may differ slightly depending on initial density of organoids.
-
39|
Wash the well with 500 μl PBS-EDTA and aspirate.
-
40|
Add 200 μl trypsin-EDTA to the well. Scratch and suspend Matrigel with a 1,000 μl pipette.
-
41|
Incubate the plates in the cell culture incubator for 2–5 min.
▲ CRITICAL STEP Too much trypsinization may cause poor survival of cells. Optimal incubation time should be determined for each origin of organoids. Generally, small intestinal organoids are more sensitive to trypsinization than other organoids.
-
42|
Add 600–800 μl washing media and dissociate organoids by vigorous pipetting.
▲ CRITICAL STEP Too much pipetting may cause poor survival of cells. Leave large aggregates (>100 cells) for the routine passage.
-
43|
Transfer organoid suspension into a 15 ml centrifuge tube and add 5 ml washing media.
-
44|
Centrifuge the tube at 200 g for 5 min.
-
45|
Aspirate supernatant with ~100 μl leftover.
-
46|
Add 500 μl–1 ml washing media and re-suspend cells.
-
47|
Transfer appropriate volume of suspension to a 1.5 ml test tube.
▲ CRITICAL STEP Dilution should be determined based on the cell density and growth rate (usually between 1:4 and 1:8 for gastrointestinal organoids).
-
48|
Centrifuge the tube at 200 g for 5 min.
-
49|
Aspirate supernatant completely.
▲ CRITICAL STEP First aspirate supernatant with ~100 μl leftover using the vacuum aspirator and then aspirate remaining supernatant completely with a 200 μl pipette.
-
50|
Place the tube on ice. Suspend organoids in Matrigel (15 μl per well).
▲ CRITICAL STEP Gently pipette until cell aggregates are completely dispersed. Do not generate many air bubbles in the Matrigel. Do not warm the tube.
-
51|
Place the tube and a 24 well plate on ice. Place 15 μl of cell-Matrigel suspension in the center of each well using a 20 μl pipette and spread it carefully with a pipette tip (Fig. 3).
-
52|
Incubate the plates in the cell culture incubator to polymerize the Matrigel. Turn the plates upside down to avoid attachment of cells to the surface of the plate.
-
53|
Add 500 μl 50% conditioned media to the well. Change media at least every 2 d.
? TROUBLESHOOTING
-
54|
Organoids can now be passaged in 96-well plates for assays (option A), cryopreserved (option B) or transfected with lentiviruses (option C) according to specific experiment.
A) Passage for assays ● TIMING 40 min-60 min
-
Grow organoids in the 24-well plate for 2–3 d.
▲ CRITICAL STEP The majority of the cells in the organoids should be in growth phase to maximize viability after passage. Do not overgrow organoids (i.e., until the media turns yellow). Make sure that the majority of organoids have a spherical shape and that there are few dead cells and debris in organoids.
Scratch and suspend Matrigel in culture media with a 1,000 μl pipette. Transfer organoid suspension to a 15 ml centrifuge tube. Combine suspension from multiple wells depending on the assay scale.
Centrifuge the tube at 200 g for 5 min.
Aspirate supernatant with ~100 μl leftover.
Add 5 ml PBS-EDTA.
Centrifuge at 200 g for 5 min.
Aspirate supernatant with ~100 μl leftover.
Disperse organoids in 200 μl trypsin-EDTA.
-
Incubate the tube in the 37 °C water bath for 2–5 min.
▲ CRITICAL STEP Too much trypsinization results in poor survival.
-
Add 1 ml washing media and dissociate organoids by vigorous pipetting.
▲ CRITICAL STEP The average size of cell aggregates depends on the number of times that they are pipetted. Make sure that the size of cell aggregates is homogeneous. Irregularly large cell aggregates can be removed by passing cells through a 40 μm cell strainer
Dilute cell suspension with 9 ml washing media.
Centrifuge the tube at 200 g for 5 min.
Aspirate supernatant with ~100 μl leftover.
Add 500 μl–1 ml washing media and re-suspend cells.
Take a drop of suspension and check the density and viability of epithelial aggregates.
-
Transfer appropriate volume of suspension to a 1.5 ml test tube.
▲ CRITICAL STEP The optimal cell number to plate should be determined by preliminary experiments. As a guideline, 1,000–3,000 cell aggregates (excluding single cells and doublets) can be seeded per well in 96-well plates.
Centrifuge the tube at 200 g for 5 min.
-
Aspirate supernatant completely.
▲ CRITICAL STEP First aspirate supernatant with ~100 μl leftover using the vacuum aspirator and then aspirate remaining supernatant completely with a 200 μl pipette.
-
Place the tube on ice. Suspend cell aggregates in Matrigel (4 μl per well).
▲ CRITICAL STEP Gently pipette until cell aggregates are completely dispersed. Try not to generate many air bubbles in the Matrigel.
Place the tube and a 96-well plate on ice. Place 4 μl of cell-Matrigel suspension in the center of each well using a 10 μl pipette (Matrigel should not touch the wall of the well) (Fig. 3).
Incubate the plates in the cell culture incubator to polymerize Matrigel. Turn the plates upside down to avoid attachment of cells to the plate surface.
-
Culture organoids with your preferred conditions.
▲ CRITICAL STEP It should be noted that conditioned media may contain undefined factors secreted from L-WRN or L-WR cells. To avoid potential undesired effects of conditioned media, primary culture media supplemented with recombinant R-spondin 1 and Wnt3a (CM-free organoid culture media) or reconstituted non-serum media can be used instead.
? TROUBLESHOOTING
B) Cryopreservation ● TIMING steps i–vi, 15–20 min; steps vii–iX, 20–30 min
Freezing organoids. Grow organoids in a 24-well plate for 2–3 d.
Scratch and suspend Matrigel in the culture media by vigorous pipetting.
Transfer the suspension to a 1.5 ml test tube (up to 2 wells) or a 15 ml centrifuge tube (>2 wells).
Centrifuge the tube at 200 g for 5 min.
Aspirate supernatant with ~100 μl leftover and re-suspend organoids gently in 500 μl freezing media per well.
-
Transfer cell suspension to cryotubes and freeze at −80 °C using an appropriate freezing device.
■ PAUSE POINT Frozen cells can be stored for >1 year in a −80 °C deep freezer. For more secure storage, store cryotubes in a liquid nitrogen tank or a cryogenic freezer (−150 °C).
Thawing organoids. Incubate frozen cryotubes in a 37 °C water bath.
Transfer organoids to pre-warmed 5 ml washing media in a 15 ml centrifuge tube.
Culture recovered organoids in 2 wells of a 24-well plate according to Steps 44–53.
C) Transfection with lentivirus ● TIMING steps i–xix, 5 d; steps xx –xlv, 12–16 d
Generation of lentiviral particles. Plate ~2,000,000 293FT cells in a 60 mm dish with 5 ml antibiotics-free L-cell media and incubate cells in the cell culture incubator for 1 d.
-
Aspirate media and carefully add 5 ml pre-warmed antibiotics-free L-cell media (cells easily detach). Incubate cells in the cell culture incubator until transfection mixture is ready.
▲ CRITICAL STEP Start transfection in the late afternoon or evening.
Add 4 μg lentiviral vector plasmid, 2.6 μg psPAX2 packaging plasmids and 1.4 μg pMD2.G envelope plasmids to 500 μl Opti-MEM in a 1.5 ml test tube.
Add 20 μl Lipofectamine 2000 to 500 μl Opti-MEM in another 1.5 ml test tube. Incubate mixture at room temperature (20–28 °C) for 5 min.
Combine plasmid solution with Lipofectamine solution into one tube and mix gently.
Incubate the Lipofectamine-DNA complex at room temperature for 20 min.
-
Add the transfection complex to cells. Incubate cells in the cell culture incubator for over night.
! CAUTION In the following steps, handle and dispose of infectious materials appropriately according to the regulations of individual facilities.
Aspirate media with 5 ml pipettes and add 2.5 ml pre-warmed L-cell media.
-
Recover media in a 15 ml centrifuge tube 48 h after infection and add 2.5 ml pre-warmed L-cell media. Store 1st virus solution at 4 °C.
▲ CRITICAL STEP Check transfection efficiency under a fluorescence microscope. GFP signal should be observed in nearly 100% cells. If transfection efficiency is low, discard the dish and redo the transfection.
Recover media 72 h after infection and combine with 1st viral solution.
Filtrate virus solution using a 0.45 μm syringe filter.
Add 1/4 volume of PEG-it Virus Precipitation Solution (5X) to virus solution and mix in a 15 ml centrifuge tube.
Incubate the tube at 4 °C over night.
Centrifuge the tube at 1,500 g for 30 min at 4 °C.
Aspirate supernatant with a 5 ml pipette with ~100 μl leftover.
Centrifuge the tube at 1,500 g for 5 min at 4 °C.
Aspirate supernatant completely with a 200 μl pipette.
Re-suspend the pellet in 410 μl washing media.
-
Aliquot supernatant in four 1.5 ml test tubes (100 μl per tube) and store at −80 °C
■ PAUSE POINT Aliquots can be stored for 6 months without decrease in infection efficiency.
Stable gene transfection of organoids with lentiviruses. Trypsinize organoids in one well (24-well plate) according Steps 54A(i)-(xviii). Use 1/4 – 1/10 volume of cell suspension for each infection.
Re-suspend cells in 100 μl lentiviral solution.
Add hexadimethrine bromide (10 μg/ml) and Y27632 (10 μM) to the tube.
Place the tube (lying on the side) in the cell culture incubator and incubate cells for 6 h.
Centrifuge the tube at 200 g for 5 min.
Aspirate supernatant carefully and completely with a 200 μl pipette.
Suspend cells in 15 μl Matrigel.
Place Matrigel-cell mixture in the center of well in a 24-well plate.
Incubate the plates at 37 °C to polymerize Matrigel (Turn the plates upside down).
-
Add 500 μl 50% conditioned media containing 10 μM Y27632 and 10 μM SB431542. Incubate the plates in the cell culture incubator for 4 d (change media once on day 2). Monitor the GFP fluorescence under a fluorescence microscope (Fig. 4a, b and Supplementary Fig. 5).
? TROUBLESHOOTING
Scratch and suspend Matrigel with a 1,000 μl pipette.
Disperse organoids into 5 ml washing media in a 6 cm dish.
Look for partially GFP-positive organoids under the fluorescence microscope. Using a 20 μl pipette, collect organoids in a 1.5 ml test tube with ~100 μl washing media (10–20 organoids).
Centrifuge the tube at 200 g for 5 min.
Aspirate media carefully with a 200 μl pipette. Add 200 μl PBS-EDTA.
Centrifuge the tube at 200 g for 5 min.
Aspirate supernatant carefully with a 200 μl pipette. Add 10 μl trypsin EDTA.
Incubate the tube in 37 °C water bath for 3 min.
Add 200 μl washing media and pipette vigorously with a 200 μl pipette.
Add 500 μl washing media and centrifuge the tube at 200 g for 5 min.
Aspirate media carefully and completely with 1,000 μl and 20 μl pipettes. Suspend cells in 15 μl Matrigel.
Place Matrigel-cell mixture in the center of well in a 24-well plate.
Incubate the plate in the cell culture incubator until Matrigel polymerizes (Turn the plates upside down).
Add 500 μl 50% conditioned media containing 10 μM Y27632 and 10 μM SB431542 to the well. Incubate the plate in the cell culture incubator for 4 d (change media once on day 2).
Pick up 100% GFP-positive organoids (Fig. 4c) according Steps 54C(xxx)-(xliii).
Repeat the selection step unless all cells express GFP.
TIMING
Steps 1–12, Preparation of the conditioned media: 10–23 d.
Steps 13–37, Isolation and culture of mouse epithelial units: 1.5–2 h.
Steps 38–53, Passage for maintenance: 30–40 min.
Steps 54A, Passage for assays: 40–60 min.
Steps 54B (i)–(vi), Cryopreservation: 15–20 min.
Steps 54B (vii)–(ix), Thawing organoids: 20–30 min.
Steps 54C (i)–(xix), Generation of lentiviral particles: 5 d.
Steps 54C (xx)-(xlv), Stable gene transfection of organoids with lentiviruses: 12–16 d.
Box 1, Purifying organoids from stromal cell contamination: 40–60 min.
TROUBLE SHOOTING
For troubleshooting guidance see Table 1.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 30 | Poor yield of epithelial units. | Incomplete collagenase digestion and pipetting. | Incubate tissues longer in collagenase solution. Pipette more vigorously to dissociate epithelial units. |
| 30 | Low viability of epithelial units after isolation | Some tissues (e.g. small intestine, pancreas) digest themselves with endogenous enzymes. | Dissect tissues quickly and immerse them in washing media as soon as possible. Reduce the number of mincing. Reduce the amount of tissues in the collagenase solution. |
| 30 | Contamination of villi with small intestinal crypts | Villi come off of tissue fragments first and can clog up the isolation system. | During collagenase digestion, separate villi (that come off of tissue fragments first) from crypts that come off from tissue fragments. Wash tissue fragments with washing media after villi come off and re-suspend them in collagenase solution. |
| 30 | Contamination of single cells | A number of single cells may precipitate at centrifugation step. | Resuspend cells in ~10 ml washing media and repeat Steps 26–29. |
| 37 | Contamination of yeasts | Live yeasts may be present in the mouse stomach and trachea. | Start culture in multiple wells of 96 well plates and pick up uncontaminated wells for the next passage. |
| 53 | Low cell viability after passage. | Low stem cell number in organoids. | Harvest organoids in growth phase. Avoid overcrowded culture. |
| 53 | Poor organoid growth | Low quality of Matrigel. | Test another lot of Matrigel. |
| 54A (xxii) | Low viability of strongly dissociated cells | Single cells and small cell aggregates are sensitive to anoikis. | Culture cells in presence of 10 μM Y27632. |
| 54C (xxix) | Low infection rate with lentiviruses. | Low titer of lentiviral solution. | Make sure that transfection efficiency is 100% in virus generation steps. |
ANTICIPATED RESULTS
The timing of processes is not critical (except early steps for isolating small intestinal crypts), thus epithelial units that are isolated using this protocol should be in good condition even without applying strict timing to processes (Fig. 1a, b). Longer incubation (~1 h) with collagenase or delay of processing dose not affect cell viability due to the protective effect of serum, which enables the processing of multiple tissues at one time. Properly isolated gastrointestinal units forms hollow spheres a few hours after plating that go on to develop spherical organoids. Primary gastrointestinal organoids grow quickly enough for passage generally in 3 days (Fig. 1c). At early passages, some organoids undergo budding. After additional passages, most organoids will form a spherical shape (Fig. 2). The small population of mesenchymal cells that are typically isolated with individual crypts should be completely eliminated during early serial passages without separation procedures6. High-density culture results in an increase in budded organoids and accumulation of dead cells in spheres. Such organoids lose their stem cell population and are not suitable for passage. To date, we have maintained gastric and intestinal organioids for over 3 months. The capacity of gastrointestinal organoids for long-term cell division enables permanent genetic modification on tissue stem cells. 293FT cells in a 60 mm dish generate sufficient lentiviral solution for 4 infections of organoids. Concentration of virus particles by polyethylene glycol improves cell viability and transduction efficiency (Supplementary Fig. 5). Titration of virus particles is not necessary to isolate the stably transduced population. In the initial organoid spheres formed after infection, mosaic GFP expression is observed (Fig. 4a, b and Supplementary Fig. 5). Following trypsinization, small cell aggregates composed of only GFP-positive cells are generated. At this stage, the infected population can be enriched by hand-selecting the 100% GFP-positive organoids under the fluorescence microscope (Fig. 4c).
Supplementary Material
Acknowledgments
We thank K. L. VanDussen and K. K. Patel for comments on the manuscript. This work was funded by the NIH (DK071619), the Pew Scholars Foundation, and the Washington University Digestive Disease Research Core (NIH P30-DK52574).
Footnotes
AUTHOR CONTRIBUTIONS H.M. performed the experiments. H.M. and T.S.S. wrote the manuscript.
COMPETING FINANTIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–266. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 2.Barker N, et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338:108–113. doi: 10.1126/science.1223821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nusse R, et al. Wnt Signaling and Stem Cell Control. Cold Spring Harb Symp Quant Biol. 2008;73:59–66. doi: 10.1101/sqb.2008.73.035. [DOI] [PubMed] [Google Scholar]
- 5.Haegebarth A, Clevers H. Wnt Signaling, Lgr5, and Stem Cells in the Intestine and Skin. Am J Pathol. 2009;174:715–721. doi: 10.2353/ajpath.2009.080758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim KA, et al. Mitogenic influence of human R-Spondin1 on the intestinal epithelium. Science. 2005;309:1256–1259. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 7.Willert K, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 8.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 10.Koo BK, et al. Controlled gene expression in primary Lgr5 organoid cultures. Nat Methods. 2011;9:81–83. doi: 10.1038/nmeth.1802. [DOI] [PubMed] [Google Scholar]
- 11.Nagy A. Manipulating the mouse embryo: a laboratory manual. Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- 12.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 13.Sato T. Novel stem cell culture system. Inflamm Regen. 2012;32:43–47. [Google Scholar]
- 14.Snippert HJ, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.