

Abstract
Arsenic (As) is a prevalent environmental toxin; readily accessible for human consumption and has been identified as an endocrine disruptor. However, it is not known what impact As has on female sexual maturation. Therefore, in the present study, we investigated the effects of prepubertal exposure on mammary gland development and pubertal onset in female rats. Results showed that prepubertal exposure to 10mg/kg of arsenite (As(III)) delayed vaginal opening (VO) and prepubertal mammary gland maturation. We determined that As accumulates in the liver, disrupts hepatocyte function and suppresses serum levels of the puberty related hormone insulin-like growth factor 1 (IGF-1) in prepubertal animals. Overall, this is the first study to show that prepubertal exposure to As(III) acts peripherally to suppresses circulating levels of IGF-1 resulting in delayed sexual maturation. Furthermore, this study identifies a critical window of increased susceptibility to As(III) that may have a lasting impact on female reproductive function.
Keywords: Arsenic, Arsenite, Female puberty, pubertal delay, Mammary gland development, Insulin-like growth factor-1 (IGF-1), Reproductive toxicity
1. Introduction
Arsenic (As) is a well known environmental toxin [1] easily accessible to the human population through ingestion of contaminated drinking water [2–6] and food [7–10]. Exposure to As has been linked to various deleterious health conditions including cancers [11–15], neurological conditions [16], skin lesions, cardiovascular disease [17] and diabetes [18–19]. In many cases, these conditions are predominantly diagnosed in adults as a result of life-long exposure to toxic levels of the non-essential element. However there is a growing concern regarding the effects of low-dose (physiological) exposures during childhood development, a critical window of increased susceptibility to environmental toxins [20–22]. It is estimated that 200 million people worldwide are exposed to As, including a large number of children [4]. Several studies have reported deleterious health effects in children exposed to As [23–26]. In the United States, As levels 6-times greater than the EPA safe drinking water limit (10 ppb) have been detected in organic infant milk and high As concentrations were also detected in cereal bars, high energy diets [7], rice [8–9] and in blood taken from children living in rural areas [6]. While conscious efforts have been made to reduce industrial use and the subsequent environmental impact, these recent studies culminate to suggest that exposure to this toxin is prevalent. To date, few studies have focused on childhood low-dose exposure to As and the ramifications this may have on pubertal development. Impairments in normal endocrine processes that regulate thelarche (mammary gland development) and menarche (pubertal onset) could have lasting detrimental effects on reproduction function throughout a women’s life time [27–29]. Thus, understanding the impact low-level As may have on pubertal developmental processes is of vital importance to the well-being of that individual later in life.
While many human [16] and animal studies [30] have focused on elucidating the cognitive effects associated with early As exposure, less attention has been given to the reproductive developmental effects of As. Inorganic trivalent arsenite (As(III)), the most detrimental form of As to human health [31], readily crosses the blood brain barrier (BBB) and accumulates in the brain [32–33]. Thus, it is plausible that As(III) poses a threat to the neuroendocrine pathways that regulate female pubertal development. Supporting this, chronic exposure to As(III) has been shown to suppress serum levels of key reproductive hormones including luteinizing hormone (LH), follicle stimulating hormone (FSH) and estradiol (E2) resulting in prolonged diestrus in adult female rats [33–34]. Additionally, As readily accumulates in the liver [35], the organ responsible for the main production of circulating IGF-1 [36], which has been shown to play a critical role in female pubertal onset [37–39]. Importantly, both E2 and IGF-1 are key reproductive hormones also necessary for normal pubertal mammary gland development and maturation [40–43]. Although epidemiological data is lacking, a delay in menarcheal age was reported in women living in the Bengal Delta Plain (India) exposed to drinking water known to have high levels of inorganic arsenic (iAs) [44]. However, it was unclear if these women suffered from additional developmental/nutritional problems associated with As toxicity that might contribute to the reported delay. Overall, these studies culminate to suggest that As could in fact alter pubertal timing, however no study to date has shown that As can directly alter the onset of female puberty. Thus, we hypothesize that exposure to As during a critical developmental window can disrupt key endocrine processes that regulate female sexual maturation. Therefore, the present studies assessed whether prepubertal short-term exposure to low-dose As(III) would alter puberty-related hormones resulting in altered pubertal mammary gland development and the timing of pubertal onset in female rats. Importantly, we show that As(III) is capable of disrupting both pubertal mammary gland maturation (thelarche) and the timing of pubertal onset (menarche) and identify a critical window of vulnerability to this readily available environmental endocrine disruptor that poses as threat to female reproductive health.
2. Materials and Methods
2.1. Animals and Housing
Adult Sprague Dawley females and males were purchased from Charles River (Wilmington, MA) and housed under controlled conditions of temperature (23°C), lighting (lights on: 7:00 h; light off: 18:00 h) and ad libitum access to food and deionized (d.i.) water. Adult females and males were bred to a 1:1 ratio to generate female offspring used in the study. Both the diet (Harland Teklad 2016 rodent chow; Harlan Laboratories, Indianapolis, IN) and water had no detectable levels of As based on sample analyses performed in the heavy metals analysis laboratory of Dr. Parsons at UTPA. All procedures were approved by University of Texas- Pan American (UTPA) Institutional Animal Care and Use Committee (IACUC) and in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
2.2. Experimental Procedure and Justification of Dose
A minimum of 5 breeding pairs of rats for each experiment were bred and allowed to deliver their pups normally and litters were culled to 10–12 pups with 5–6 females per litter. Half of the female pups from each dam were designated as controls and the other half As-treated. We conducted a preliminary study using doses of either 1mg/kg body weight (b.w.) sodium arsenite NaAs (As(III)), 5mg/kg of As(III), 10mg/kg of As(III) or 20mg/kg of As(III) (data not shown). Animals were treated with various amounts of As(III) based on mg/kg b.w. concentrations. For example, an animal weighing 25g, receiving a 10mg/kg b.w. dose of As(III), was administered .025mg of NaAs(III) diluted in 200μls of deionized water via gastric gavage. Animals were weighted daily and the dose was adjusted based on body weight. Therefore making it possible to administer various amounts of As in small volumes and know exactly how much As(III) each treated animal received. Based on these preliminary studies, the 10mg/kg As(III) was determined to be the lowest effective dose and used in all experiments.
The 10mg/kg dose of As in this study resulted in a average daily intake of 575μg/day per female rat. This resulted in a cumulative (25 days) intake of 14.38 mg of As per animal. The estimated average daily intake of As for adult females is 50μg/day [45]. While less substantiated, the daily As intake for infants and young children has been reported to be 2–3x higher than adults [46–47]. Based on a comprehensive report conducted by the Agency for Toxic Substance and Disease Registry (ATSDR) which presented detailed information on the known toxic effects of As(III), humans were shown to be 10–100 times more sensitive to As(III) [45]. This was based on the lowest adverse effect level (LOAEL) of intermediate (as few a 15 days to as many as 365 days) As(III) exposure (mg/kg exposure a day) to cause similar systemic effects in rats compared to humans. Thus, conservatively, our 10mg/kg dose of As(III) resulted in an average daily intake of 575μg/day, which would be equivalent to a human daily intake of 57.5μg (based on a 10 fold difference); thus resulting in a 25 day cumulative dose of 1.4 mg. Comparatively a cumulative dose in humans for the same duration would result in 1.25mg (50μg/day). Therefore the dose used in our model is relevant to human health concerns.
In the first experiment (figure 1; continuous dose), female offspring were dosed via gastric gavage with either 10mg/kg of As(III) or 0.9% saline solution (control) starting on postnatal day (PND) 12 until pubertal onset (approximately 25 days). Gastric gavage was used given the most common route of As exposure in children is oral [45]. Animals were weighed daily and monitored for acute toxicity to As(III) exposure. Starting on day 27 female offspring from each group were inspected daily for vaginal opening (VO). The day of vaginal opening was recorded and a vaginal smear was used to define the stage of first estrus as described previously [48]. Dosing was continued and vaginal smears were taken until a diestrus smear was recorded indicating first ovulation had occurred. The interval between VO and diestrus was recorded for all rats. It is important to note that vaginal opening and first diestrus, determined by vaginal smears, is a proven method for determining sexual maturation in rats [48]. The mean day of vaginal opening, thus marking sexual maturation, in our Sprague Dawley colony is 35 days of age. The mean vaginal opening and first diestrus occurring significantly later than this was considered a delay is sexual maturation in this study.
Figure 1. Model depicting the experimental dosing protocol.
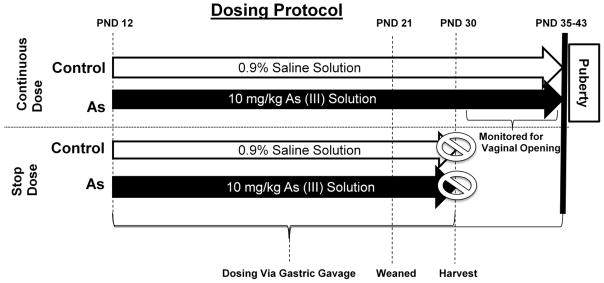
In short, the continous dose group was exposed via gastric gavage to either saline or 10mg/kg of As(III) starting at post natal day (PND) day 12 until vaginal opening. Stop dose group was dosed until PND 30, at which time tissue and serum was collected for analysis.
In the second experiment (figure 1; stop dose), dosing was administered as stated above and continued until PND 29. On PND 30, females were killed by decapitation and trunk blood was collected for serum hormone analysis. All animals were verified to be in the late juvenile phase of development by well established criteria [49]. The hypothalamus, pituitary, and ovaries were harvested and frozen at −80°C until they were analyzed for As accumulation. Livers were collected for transmission electron microscopy and As analysis. Mammary glands were collected for whole mount and As analysis.
2.3. Mammary whole gland morphological analysis
To analyze the effects of As exposure on pubertal mammary gland growth and differentiation, mammary gland whole mounts were processed similarly as previously described [50] with the following modifications. On PND 30, the #4 right abdominal mammary glands from control and treated animals were removed and spread flatly on glass microscope slides and placed in Carnoy’s fixative (75% glacial acetic acid, 25% absolute ethanol) at RT for 3 days. Next slides were washed in 70% ethanol (EtOH) for 1 hour, distilled water for 30 minutes, and placed in Carmine Alum stain for 4 days at RT. Then, mammary glands were destained as follows: 70% EtOH for 1hr.; 95% EtOH for 1hr.; 100% EtOH for 1hr.; and cleared in xylene for 2 days at RT. Finally tissues were permanently stored in methylsalicylate until analyzed under an Olympus CKX41 microscope.
Under the microscope, the number of mammary gland terminal end buds (TEB), alveolar buds (Ab), lobular type 1 (Lob1) and terminal ducts (TD) were counted in a localized exterior area (opposite of the nipple), the most actively growing area in the mammary gland, previously described as Zone C [51]. The number of each of the above ductal structures was counted in each mammary gland analyzed. There were a total of 4 mammary glands per group (one per animal) analyzed. The mean number of each of these structures per group was calculated and statistical analysis between groups was conducted.
2.4. Arsenic (As) Analysis
Arsenic was measured by the heavy metal analysis laboratory of Dr. Parsons in the Department of Chemistry, College of Science and Mathematics, University of Texas-Pan American. The samples: rat tissues (liver, ovaries, pituitary glands, mammary glands, and hypothalamus) food, and bedding were dried in a Labconco Freezezone 6 freeze dry system (Kansas City, MO). The dried tissue, food and bedding samples were digested using a nitric acid digestion method. Samples were weighed and 10 mL of concentrated trace pure nitric acid was added to each sample. Samples with nitric acid were then heated on a hot plate until the solution turned clear. Subsequently, the samples were evaporated almost to dryness and cooled to room temperature. To the digested samples 5 mL of 30 % H2O2 was added to aid in the decomposition of materials. The samples were then cooled to room temperature and diluted using ultrapure water (water with a resistance of 18MΩ). Additionally water samples were tested for arsenic content without pretreatment. All samples were analyzed using a Graphite Furnace atomic absorption spectroscopy Perkin Elmer AAnalyst 800 Atomic absorption Spectrometer (Shelton Connecticut). The Furnace program was optimized by varying the atomization temperatures to obtain the optimum sensitivity and the characteristic mass was found to be 20 pg/0.0044 A-s at a wavelength of 197.3 nm. To obtain the data, matrix modifiers consisting of Pd 5μg and Mg 3μg were also used. For each analysis 20 μL of sample was injected with the matrix modifier into the graphite furnace atomic absorption spectrometer at 20°C. Additionally, a correlation coefficient of 0.99 or better was obtained for all analyses performed.
2.5. Transmission Electron Microscopy
Liver was harvested from control and treated PND 30 day animals and fixed for transmission electron microscopy (TEM) following a procedure previously described [52] with the following modifications. Tissues were diced into 1 mm3 in 2% formalin/0.1M phosphate buffer (7.2 pH). After dicing, tissues were placed in fixative containing 4% formalin, 2% glutaraldehyde in a 0.1M phosphate buffer (7.2 pH) for 24 hrs at 4°C. The next day tissues were fixed with 2% osmium tetroxide in 0.1M phosphate buffer (7.2 pH) for 1 hr at RT and placed in a 2% uranyl acetate solution (in H2O) for 1hr at RT. Fixed tissues were then dehydrated at RT as follows: 30% EtOH for 15min, 50% EtOH for 15min, 70% EtOH for 15min, 95% EtOH for 15min, 95% EtOH for 30 min, 100% EtOH for 1h, and acetone for 15min. Tissues were embedded into epoxy resin blocks (0.5 × 1 cm) using 46% araldite 502 resin (Ted Pella Incorporated, Redding, CA), 50% dodecenyl succinic anhydride (DDSA; Ted Pella Incorporated) and 4% benzyldimethylamine (BDMA; Ted Pella Incorporated). Propylene oxide was used as a transitional buffer as follows: 1:1 propylene oxide (250μl)/epoxy resin (250μl) for 1hr.; 1:2 propylene oxide (250μl)/epoxy resin (500μl) for 2hrs; 1:4 propylene oxide (250μl)/epoxy resin (1ml) for 2hrs. at RT then incubated at 60° C for 12 hours. Tissues were sectioned (70nm), placed on copper grids (75 mesh count), treated with 2% uranyl acetate for 2 mins, washed in ddH2O, treated with lead citrate for 90 secs, washed with ddH2O and immediately analyzed using the EVO LS 10 electron microscope. Three different tissue samples were fixed from each liver (4 livers per group).
2.6. Hormone and Statistical Analysis
Blood was collected from controls and treated animals, placed in 4°C overnight, centrifuged at 4,000×G for 45 mins, and serum was used for hormonal assays. LH and FSH were measured using a rat MILLIPLEX rat pituitary fluorescent-coded bead kit purchased from Millipore Corporation (Billerica, MA). The assay sensitivity was 4.9 pg/ml and 47.7 pg/ml for LH and FSH, respectively. IGF-1 levels were measured in serum using a MILLIPLEX rat/mouse IGF-I fluorescent-coded bead assay purchased from the Millipore Corporation. The assay sensitivity was 3.2 pg/ml. LH, FSH and IGF-1 assays were all run in the Biomarkers Core Laboratory, at the University of Texas Health Science Center San Antonio (UTHSCSA) Edinburg Regional Health Center (E-RAHC). E2 levels were measured using a mouse/rat estradiol ELISA assay from Calbiotech (Spring Valley, CA). The assay sensitivity was 3pg/ml.
Differences between control and treated groups were analyzed by unpaired Student’s t test assuming random sampling. Probability values <0.05 were considered to be statistically significant. The IBM PC programs INSTAT and PRISM software (GraphPad, San Diego, CA, USA) were used to calculate and graph the results.
3. Results
3.1. Effects of prepubertal exposure to As(III) (arsenite) on female pubertal onset
Figure 2A depicts the growth curves of female pups exposed to either saline (controls) or 10mg/kg As(III) starting at PND 12 through pubertal onset. Importantly, there were no weight differences in controls compared to the arsenite-treated females during the dosing period and both groups had similar daily weight gains (Figure 2B). In addition, there were no observable signs of dehydration, loss of activity, or abnormal behavior between groups. The 10mg/kg As(III) dose did not result in acute toxicity or physically debilitate the animals (data not shown).
Figure 2. Prepubertal exposure to As(III) did not effect somatic growth.
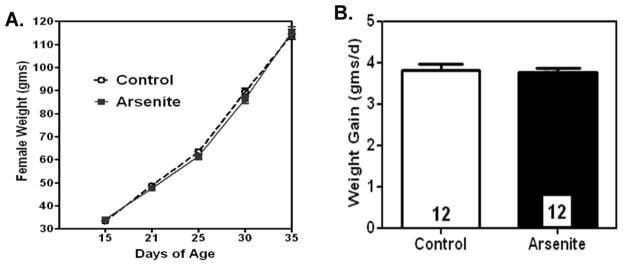
Overall, 10mg/kg of As(III) did not alter developmental growth throughout the study, nor did it have any effect on daily weight gain compared to saline treated females. A.) Line graph represents the mean (±SEM) weight in grams (g) at specific days of pubertal growth in arsenite (As(III)) and control (saline) treated females. Control; N=20 for each day. Arsenite; N= 15 for each day. B.) Bar graph depicts the mean (±SEM) daily weight in grams in arsenite and control treated females. N=12 in each group.
Figure 3 demonstrates that prepubertal females exposed to As(III) had almost a 2 day delay in the timing of pubertal onset as determined by the day of VO and the day of first diestrus (D). Specifically, prepubertal As(III) exposure delayed both VO (37.266 ± 0.613) and D (38.400 ± 0.615) compared to rats treated with saline (35.450 ± 0.413; 36.450 ± 0.413 days of age, respectively). However, there were no differences in length of estrus (number of days between VO and D) between groups.
Figure 3. Prepubertal exposure to As(III) delays pubertal onset.
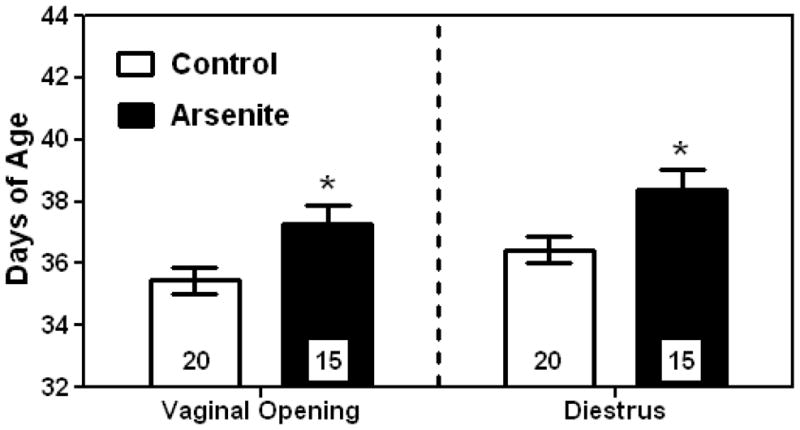
Exposure to 10mg/kg of As(III) significantly (p <0.05) delayed the onset of female puberty indicated by a delay in vaginal opening (VO) and first diestrus (D1). The panel on the left represents the mean (±SEM) age in days at VO for the two treatment groups. The panel on the right represents the mean (±SEM) age in days at D1 for each group. *= P< 0.05; Number of females per group are within bars.
3.2 Effects of prepubertal As(III) (arsenite) exposure on pubertal mammary gland morphology
To determine the effects of prepubertal As(III) exposure on pubertal mammary gland maturation, the number of terminal mammary gland structures were counted in PND 30 animals (figure 4). Figure 4A shows that the mean number of terminal end buds (TEB), undifferentiated progenitor cells, were significantly higher (p < 0.01) in prepubertal animals exposed to As(III) (68.0 ± 10.654) compared to saline treated controls (30.0 ± 3.342). Prepubertal As(III) exposure also resulted in a significantly (p< 0.01) higher presence of alveolar buds (AB; 12.80 ± 2.198) compared to controls (4.50 ± 0.645). Figure 4C shows that there were significantly (p <0.01) less mean number of terminal ducts in As(III)-treated females (TD; 30.25 ± 4.029) compared to the saline treated group (53.5 ± 4.907). Although fewer mean number of lobular type 1 (Lob1) structures were observed in the arsenite treated group (1.00 ± 0.707) compared to controls (3.5 ± 1.190), the difference was not significant (figure 4C). Importantly, no Lob1 structures were observed in 50% (2 of 4) of the mammary glands analyzed in the As(III)-treated group, comparatively Lob1 structures were present in all of the control mammary glands (N=4).
Figure 4. Morphological assessment of altered mammary gland growth due to prepubertal As(III) exposure at 30 days of age.

A.) As(III) -treated females had a significantly higher (p<0.01) mean (±SEM) number of TEB compared to saline treated controls. B.) As(III) exposed prepubertal mammary glands had significantly more (p<0.01) mean (±SEM) AB ductal structures vs. controls. C.) As(III) exposure resulted in significantly lower (p <0.01) number of TD (±SEM) structures vs. controls. D.) As(III)-treated females had a reduced mean (±SEM) number of Lob1 structures in prepubertal mammary gland vs. controls. Representive images of each structure are included within each graph. N= 4 per group, same animals were assessed for all 4 structures. **p<0.01.
3.3 Effects of prepubertal exposure of As(III) (arsenite) on key puberty-related hormones
To determine how arsenite delayed pubertal sexual maturation, serum levels of key puberty-related hormones were assessed on PND 30, just prior to entering the peripubertal phase of female development. Figure 5A depicts that daily prepubertal exposure to low-level As(III) (arsenite) significantly decreased (p< 0.01) serum levels of IGF-1 (629.605 ± 46.018 ng/ml) compared to controls (802.914 ± 35.440 mg/ml). Surprisingly, exposure to arsenite had no effect on serum levels of growth hormone (GH; 9.235 ± 3.138 ng/ml; arsenite vs. 9.648 ± 3.383 ng/ml; controls), LH (0.496 ± 0.174 ng/ml; arsenite vs. 0.791 ± 0.262 ng/ml; controls), and E2 (14.336 ± 0.618 pg/ml; arsenite vs. 13.478 ± 0.924 pg/ml; controls) compared to saline treated females (Figure 5 B, C and D). In addition, no differences were observed in serum levels of FSH in As(III) treated (12.225 ± 1.194 ng/ml; N= 10) vs. saline treated (11.152 ± 2.077 ng/ml; N=9) animals (data not shown). Supporting this, Table 1 shows that As concentrations in the liver (29.405 ± 1.146 μg/g), the organ responsible for producing and secreting the majority of circulating levels of IGF-1, were more than 3x higher than As levels measured in the hypothalamus (7.402 ± 0.766 μg/g), pituitary (10.997 ± 1.202 μg/g), ovaries (2.940 ± 0.445 μg/g) and mammary gland (0.923 ± 0.179 μg/g). As expected, As levels in controls tissues were non-detectable (ND) with the liver (0.0449 ± 0.008 μg/g) being the only exception. The likely source was from trace levels (under detection limit) of As in food and water that accumulated in the liver over the experimental period. As-treatment did not affect tissue growth given there were no measurable differences in organ wet weight between groups at PND 30 in these respective tissues (data not shown).
Figure 5. The effects of.
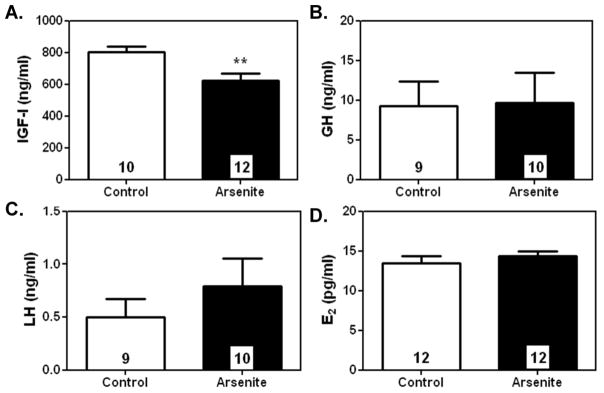
As(III) exposure on reproductive hormonal secretion at 30-days of age.
A.) Prepubertal exposure to 10mg/kg of As(III) significantly (p <0.01) suppressed mean (±SEM) serum levels of IGF-1 at 30 days of age compared to controls. However, AsIII exposure did not alter mean (±SEM) serum levels of B.) GH, C.) LH or D.) E2 compared to saline treated controls. **p<0.01. Number of animals per group is within each bar.
Table 1.
Accumulation of Arsenic (As) in Prepubertal Female Rats at 30 Days of Age
| Tissue | Saline Treated (μg/g) | NaAs(III) Treated (μg/g) |
|---|---|---|
| Liver | 0.0449 ± 0.008 | 29.405 ± 1.146 |
| Hypothalamus | ND | 7.402 ± 0.766 |
| Pituitary | ND | 10.997 ± 1.202 |
| Ovaries | ND | 2.940 ± 0.445 |
| Mammary Gland | ND | 0.923 ± 0.179 |
This table shows the mean (±SEM) microgram per gram (μg/g) of total As measured in tissue harvested at 30 days of age from female rats prepubertally exposed to either saline or 10mg/kg of As(III) N=4 for each tissue assessed for total As accumulation in each treatment group.
3.2. Effects of prepubertal exposure of As(III) (arsenite) on liver histology and function
Given the high accumulation of As in the liver and subsequent suppression of circulating IGF-1 due to prepubertal As(III) exposure TEM was used to observe histological changes in the liver previously shown to occur due to As treatment [35]. Figure 6 shows representative TEM images of hepatocytes at PND 30 from saline (A, C and G) and As(III) (arsenite) treated animals (B, D, E, F and H). Arsenite treated hepatocytes exhibited irregularity in their nuclear shape (figure 6F), increased heterochromatin condensation (figure 6B, D, E and F) and an increased presence of lipid (droplets) vacuolization (figure 6B and D); structures previously used as markers to determine cell stress and toxicity [35]. In addition, livers from arsenite treated animals expressed a higher observable concentration of engorged Kupffer cells containing lipid vacuoles (figure 6H) compared to normal nonreactive cells in controls (figure 6G). This is indicative of an immune response to As in these animals. A large majority of hepatocytes (2 out of three hepatocytes) in the arsenite treated tissue samples exhibited these cellular changes compared to sporadic and minute expression of these cell stress determinates in control samples. This result was confirmed on multiple serial sections taken from 3 different tissue samples per liver (4 animals per group).
Figure 6. Histological differences observed in hepatocytes at 30 days of age.

Representative TEM images showing hepatocytes in livers from prepubertal saline treated (A, C and G) and As (III)- treated females (B, D, E, F and H). Observable differences were noted in hepatocytes from As(III)–treated females including: increased intracellular lipid concentration (B and D); heterochromatin condensation (B, D and E) seen scattered throughout and densely along the periphery of the nucleus; and irregularity in their nuclear shape (F). In addition, liver of As(III)-treated animals contained a high concentration of engourged Kupffer cells containing lipid vacuoles (H) compared to controls (G). Number of livers was 4 in each group. N= Nucleus, n= nucleolus, L= Lipid droplet, M= Mitochondria, rER= Rough endoplasmic reticulum, K = Kupffer cell; arrows indicate irregularity in nuclear membrane, small arrow heads indicated heterochromatin.
4. Discussion
In the present study we show that prepubertal exposure to As(III) delays sexual maturation in female rats as determined by a delay in pubertal onset and suppression of prepubertal mammary gland development. The dose used was determined to be nontoxic given it did not disrupt overall somatic growth, limit food and water intake, or alter the wet weight of key reproductive organs, all signs of As toxicity. Although no study to date has assessed the effects of prepubertal As exposure on mechanisms involved in reproductive pubertal onset, the dose of As used in this study is low compared to previous studies showing As(III) effects of physiological processes in rats [35, 53–55]. In the most comprehensive studies to date, with regards to As(III) effects on reproduction, adult females exposed to 0.4ppm/100 g body weight of As(III) for 28 days resulted in suppressed circulating levels of reproductive hormones, ovarian function and disrupted estrous [33–34]. While this data is compelling, the mode of exposure (drinking water vs. gavage in our study), days exposed (28 days vs. 19 days), and age exposed (adult vs. prepubertal) make it difficult to compare. However, the As dosed used significantly lowered ovarian wet weight [34], which we did not observe in our current study. Additionally, our dose of 10mg/kg resulted in a considerably lower accumulated amount of As in reproductive tissues. Furthermore, as described above (see methods), humans are known to be considerably more sensitive to As toxicity than rats [45]. The 10mg/kg dose of As used in this study is equivalent to a human daily intake of 57.5μg/day. Similarly, the known average daily intake of As for adult females is 50μg/day [45] and As daily intake has been reported to be 2–3x higher in young children and infants [46–47]. This combined with epidemiological data reporting a delay in menarcheal age in women exposed to high levels of iAs in drinking water [44] support that As-induced effects we have described are relevant to human health concerns.
The initiation of female puberty (first ovulation) is a highly intricate and succinct physiological process directed by neuroendocrine regulatory pathways and timing in of which these events occur is critical. Our study suggests that prepubertal As exposure disrupts this timing by suppressing the peripheral release of circulating levels of IGF-1. There are several lines of evidence that support this hypothesis. Circulating IGF-1 plays a significant role in the pubertal process by linking somatic development to the activation of the LH releasing system. IGF-1 has been shown to act centrally to induce luteinizing hormone releasing hormone (LHRH) from immature female median eminence (ME) in vitro [56] and LH release during first proestrus and estrus in vivo [37]. Furthermore, IGF-1 administration during the late juvenile period (just prior to first proestrus) has also been shown to advance puberty in rats [37] and prepubertal treatment of IGF-1 advanced puberty in primates [38]. Conversely, suppressed IGF-1 levels have been previously shown to be associated with delayed puberty in female rats maternally exposed to low levels of lead (Pb) [57]. This effect was negated by IGF-1 replacement (during the late juvenile phase of development) which stimulated LH release and restored normal pubertal onset in these animals. These studies culminate to demonstrate that circulating IGF-1 plays an important role in the timing of pubertal onset.
Our results demonstrated that prepubertal exposure to As suppressed circulating levels of IGF-1, without affecting systemic levels of LH or E2. This is intriguing, but could be explained through the timing at which these hormones were assessed and/or the site of As actions. IGF-1 significantly increases just prior to puberty [58–60] which is marked by a change in the physiological release pattern of LH from the pituitary during first proestrus [61–62]. It is likely that the decreased levels of IGF-1 measured during late juvenile development in our study had no effect on basal levels of these hormones but delayed the proestrus rise in LH required to stimulate the release of E2, which subsequently results in the large preovulatory surge of LH that causes first ovulation [61–63]. As previously stated, IGF-1 replacement in late juveniles can stimulate proestrus LH release and restore pubertal onset in females maternally exposed to Pb [57]. Furthermore, studies in ovariectomized (OVEX) prepubertal rats [58] and primates [64] have shown that the prepubertal rise in IGF-1 shown to regulate LH release, is E2 independent. The prepubertal rise in circulating IGF-1 is centrally regulated by GH [65], but we showed that circulating levels of GH were not suppressed due to As(III) exposure, signifying that the central mechanisms involved in GH regulation were still intact, yet IGF-1 levels were suppressed. This coupled with our observed low levels of As accumulation (relative to the liver) in the ovary, hypothalamus and pituitary and unaltered levels of another centrally regulated pituitary hormone (FSH), supports the idea that As does not directly affect central or ovarian regulation of these hormones. Rather, it is more likely that As disrupts the peripheral rise of IGF-1 just prior to first proestrus, thus disrupting the timing of LH/E2 secretory patterns responsible for normal pubertal onset.
Supporting our observed effect of As(III) on pubertal onset we also demonstrated that As could impede another indicator of sexual maturity, mammary gland prepubertal growth. Morphological assessment confirmed that As treated females had a significantly higher number of undifferentiated epithelial structures (TEB and AB) in the most actively growing area in the mammary gland [51]. This would suggest that prepubertal As(III) exposure delayed the differentiation of terminal mammary gland structures, signifying a delay in the normal development of the gland. This is important given the TEB is the most susceptible ductal structure to malignant transformation [27] and impaired lactation in humans [28–29] and rodents [66–68] due to gestational exogenous chemical exposure has been associated with altered mammary gland development. The mechanism that As alters mammary gland development is not known, however it could act indirectly or directly by altering hormonal regulation of the gland. The combination of IGF-1 and E2 is critical for pubertal MG development [40]. Supporting this, IGF-1-null mice have severely stunted mammary gland growth and ductal branching [69], which can be reversed with IGF-1 and E2 treatment [41]. Furthermore, increased circulating levels of IGF-1 have been shown to increase mammary gland proliferation and ductal branching at 28 days in liver-specific IGF-1 producing transgenic mice [70]. Thus, the As-induced decrease in serum IGF-1 levels demonstrated in our study suggest that As(III) possibly acts indirectly to alter the development of undifferentiated epithelial ductal structures stunting normal pubertal mammary gland growth. Yet, it is plausible that As(III) directly alters hormonally regulated mechanisms within the mammary gland. While there is a lack of studies that have addressed this hypothesis, in vitro studies using the breast cancer cell line MCF-7, have shown that As(III) has complex dose- dependent effects on estrogen receptor α (EsR1). At very low doses As(III) has been shown to upregulate EsR1–regulated gene expression [71] and, at higher yet nontoxic doses, suppress E2 dependent EsR1-regulated gene expression [72]. EsR1- null mice do not go through pubertal mammary gland ductal elongation [42–43]. So, As might directly alter EsR1 function, disrupting E2-regulated mammary gland development. Given the low amounts of As(III) measured in pubertal mammary glands and normal levels of serum E2 in As(III) treated females in our study, it is unlikely that As disrupted E2 regulated mechanisms controlling mammary gland growth. However we cannot rule out that As(III) may also have a direct effect on the mammary gland through some other hormonal independent mechanism. Therefore, further studies are warranted to conclusively explain the mechanism that As(III) delays normal mammary gland growth.
The present study suggests that the liver is the site of endocrine disruption responsible for delaying female sexual maturation. The direct mechanism in which As alters liver IGF-1 production and/or secretion is not known, however several studies demonstrate the ability of As to disrupt hepatocyte viability. Arsenic at various concentrations and experimental models can cause oxidative stress [35, 55, 73], apoptosis [35], disrupt DNA methylation causing genomic instability [74–76] and impair DNA repair enzymes [77–78] in the liver. Bashir et al. [35] showed that livers from male rats chronically exposed to 5mg/L of As(III) had increased heterochromatin condensation, shrunken nuclear membranes, increased fatty vacuolization (lipid droplets) and altered antioxidant activity. The authors concluded that these marked histological and biochemical changes due to As resulted in hepatocyte oxidative stress and cell death. Similarly, our current study demonstrated that prepubertal As(III) exposure increased fatty vacuolization, as well as increased heterochromatin condensation and augmented nuclear membrane shape. However, we observed no signs of apoptosis, which is likely due to the length of exposure. In the above mentioned study [35], adult rats were exposed to the toxin 3 times longer and exhibited signs of toxicity indicated by decrease body weight and liver wet weight, both not observed in our current study. In addition, As has been shown to block ubiquitination of Nrf2 (nuclear factor erythroid 2-related factor 2) increasing its binding to the antioxidant response element (ARE) in the nucleus of hepatocytes resulting in the induction of the detoxification gene NAD(P)H-quinone oxidoreductases (Nqo1) [79]. Nqo1 combats against oxidative stress. This would allow for cells to compensate under As-induced stress. However, if As liver toxicity were to continue (due to a longer exposure time and/or at a higher dose) it is likely that the resulting DNA [74–75] damage would be sufficient enough to turn off this pathway resulting in cell death. It is plausible that the abbreviated As(III) exposure in our study was sufficient to limit certain aspects of hepatocyte function but not toxic enough to induce robust cell death detrimental to the animals well being. Thus, it is likely that complete hepatocyte function is restored after As treatment. Collectively, it is conceivable that As(III) reduces the number of properly functioning hepatocytes impairing the ability to produce the necessary levels of IGF-1 during puberty. However, future studies are necessary to elucidate the direct effects of low-level As(III)on IGF-1 production and secretion in the liver.
In summary, our results are the first to show that prepubertal exposure to As(III) delays the timing of pubertal onset and suppresses morphological differentiation in prepubertal mammary gland epithelial structures. We conclude that As(III) acts peripherally to disrupt liver function, suppressing circulating levels of IGF-1 resulting in the overall delay in sexual maturation. Importantly, the As-induced modifications in mammary gland morphology suggest that normal pubertal mammary gland development is delayed. Thus, making the gland more susceptible to dysfunction [28–29, 66–67] and/or breast cancer [27]. Our data suggests that, if exposed during a critical developmental window, relatively low levels of As(III) are sufficient to disrupt sexual maturity with no other observable effects. This raises concern over the amount young females could be unknowingly exposed to daily from multiple food sources. Notably, we have identified As(III) as a pubertal endocrine disruptor capable of causing detrimental health effects. Further epidemiological and experimental research in rodent and primate models is necessary to understand the impact childhood exposure to As(III) could have on a female’s reproductive health.
Research Highlights.
Prepubertal exposure to Arsenic(III) delayed vaginal opening in female rats.
As(III) exposure delays differentiation of the prepubertal mammary gland.
Prepubertal exposure to As(III) significantly decreases circulating levels of IGF-1
Acknowledgments
This research was supported by a University of Texas-Pan American (UTPA) Facility Research Council (FRC) grant (RKD), NIH/NIGMS 1R25GM100866-01 (RKD and JGP) and the Howard Hughes Medical Institute Precollege and Undergraduate Science Education Program (MPR and RFS). The authors would like to thank Dr. Anxiu Kuang and Dr. Karen Lozano for assisting in the utilization of the environmental scanning electron microscope which is supported by a grant from the NSF (DMR 1040419) (KL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael P. Reilly, Email: mreilly@mail.utexas.edu.
James C. Saca, Email: sacaj@livemail.uthscsa.edu.
Alina Hamilton, Email: ahamilton@broncs.utpa.edu.
Rene F. Solano, Email: solanofonsec@livemail.uthscsa.edu.
Jesse R. Rivera, Email: jrriveray@broncs.utpa.edu.
Wendy Whitehouse-Innis, Email: innis@uthscsa.edu.
Jason G. Parsons, Email: parsonsjg@utpa.edu.
Robert K. Dearth, Email: dearthrk@utpa.edu.
References
- 1.Tchounwou PB, Wilson B, Ishaque A. Important considerations in the development of public health advisories for arsenic and arsenic-containing compounds in drinking water. Rev Environ Health. 1999;14:211–29. doi: 10.1515/reveh.1999.14.4.211. [DOI] [PubMed] [Google Scholar]
- 2.USEPA. Technical Fact Sheet: Final Rule for Arsenic in Drinking Water. EPA; 2001. 815-F-00-016. [Google Scholar]
- 3.Focazio MJ, Welch AH, Watkins SA, Helsel DR, Horn DR, Horn MA. A Retrospective Analysis on the Occuurence of Arsenic in Ground-water Resources of the Uninted States and Limitations in Drinking-water Resources of the United States and Limitations in Drinking-water-supply Characterizations. US Geological Survey Water-Resources Investigation Report. 1999:21. 99-4279. [Google Scholar]
- 4.NRC. Arsenic in Drinking Water: 2001 Update. National Academy Press; Washington DC: 2001. [Google Scholar]
- 5.Smith AH, Lopipero PA, Bates MN, Steinmaus CM. Public health. Arsenic epidemiology and drinking water standards. Science. 2002;296:2145–6. doi: 10.1126/science.1072896. [DOI] [PubMed] [Google Scholar]
- 6.Postma J, Butterfield PW, Odom-Maryon T, Hill W, Butterfield PG. Rural children’s exposure to well water contaminants: implications in light of the American Academy of Pediatrics’ recent policy statement. J Am Acad Nurse Pract. 2011;23:258–65. doi: 10.1111/j.1745-7599.2011.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson BP, Taylor VF, Karagas MR, Punshon T, Cottingham KL. Arsenic, organic foods, and brown rice syrup. Environ Health Perspect. 2012;120:623–6. doi: 10.1289/ehp.1104619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone R. Food safety. Arsenic and paddy rice: a neglected cancer risk? Science. 2008;321:184–5. doi: 10.1126/science.321.5886.184. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert-Diamond D, Cottingham KL, Gruber JF, Punshon T, Sayarath V, Gandolfi AJ, et al. Rice consumption contributes to arsenic exposure in US women. Proc Natl Acad Sci U S A. 2011;108:20656–60. doi: 10.1073/pnas.1109127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akter KF, Owens G, Davey DE, Naidu R. Arsenic speciation and toxicity in biological systems. Rev Environ Contam Toxicol. 2005;184:97–149. doi: 10.1007/0-387-27565-7_3. [DOI] [PubMed] [Google Scholar]
- 11.Chiou HY, Chiou ST, Hsu YH, Chou YL, Tseng CH, Wei ML, et al. Incidence of transitional cell carcinoma and arsenic in drinking water: a follow-up study of 8,102 residents in an arseniasis-endemic area in northeastern Taiwan. Am J Epidemiol. 2001;153:411–8. doi: 10.1093/aje/153.5.411. [DOI] [PubMed] [Google Scholar]
- 12.Smith AH, Goycolea M, Haque R, Biggs ML. Marked increase in bladder and lung cancer mortality in a region of Northern Chile due to arsenic in drinking water. Am J Epidemiol. 1998;147:660–9. doi: 10.1093/oxfordjournals.aje.a009507. [DOI] [PubMed] [Google Scholar]
- 13.Steinmaus C, Moore L, Hopenhayn-Rich C, Biggs ML, Smith AH. Arsenic in drinking water and bladder cancer. Cancer Invest. 2000;18:174–82. doi: 10.3109/07357900009038249. [DOI] [PubMed] [Google Scholar]
- 14.Chen CJ, Chen CW, Wu MM, Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992;66:888–92. doi: 10.1038/bjc.1992.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morales KH, Ryan L, Kuo TL, Wu MM, Chen CJ. Risk of internal cancers from arsenic in drinking water. Environ Health Perspect. 2000;108:655–61. doi: 10.1289/ehp.00108655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinkel J, Khan MH, Kraemer A. A systematic review of arsenic exposure and its social and mental health effects with special reference to Bangladesh. Int J Environ Res Public Health. 2009;6:1609–19. doi: 10.3390/ijerph6051609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Parvez F, Gamble M, Islam T, Ahmed A, Argos M, et al. Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol Appl Pharmacol. 2009;239:184–92. doi: 10.1016/j.taap.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tseng CH, Tai TY, Chong CK, Tseng CP, Lai MS, Lin BJ, et al. Long-term arsenic exposure and incidence of non-insulin-dependent diabetes mellitus: a cohort study in arseniasis-hyperendemic villages in Taiwan. Environ Health Perspect. 2000;108:847–51. doi: 10.1289/ehp.00108847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai MS, Hsueh YM, Chen CJ, Shyu MP, Chen SY, Kuo TL, et al. Ingested inorganic arsenic and prevalence of diabetes mellitus. Am J Epidemiol. 1994;139:484–92. doi: 10.1093/oxfordjournals.aje.a117031. [DOI] [PubMed] [Google Scholar]
- 20.Yorifuji T, Tsuda T, Doi H, Grandjean P. Cancer excess after arsenic exposure from contaminated milk powder. Environ Health Prev Med. 2011;16:164–70. doi: 10.1007/s12199-010-0182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: new data on the significance of age at exposure. Environ Health Perspect. 2007;115:1406–14. doi: 10.1289/ehp.10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cory-Slechta DA, Virgolini MB, Rossi-George A, Thiruchelvam M, Lisek R, Weston D. Lifetime consequences of combined maternal lead and stress. Basic Clin Pharmacol Toxicol. 2008;102:218–27. doi: 10.1111/j.1742-7843.2007.00189.x. [DOI] [PubMed] [Google Scholar]
- 23.Luna AL, Acosta-Saavedra LC, Lopez-Carrillo L, Conde P, Vera E, De Vizcaya-Ruiz A, et al. Arsenic alters monocyte superoxide anion and nitric oxide production in environmentally exposed children. Toxicol Appl Pharmacol. 2010;245:244–51. doi: 10.1016/j.taap.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Calderon J, Navarro ME, Jimenez-Capdeville ME, Santos-Diaz MA, Golden A, Rodriguez-Leyva I, et al. Exposure to arsenic and lead and neuropsychological development in Mexican children. Environ Res. 2001;85:69–76. doi: 10.1006/enrs.2000.4106. [DOI] [PubMed] [Google Scholar]
- 25.Rosado JL, Ronquillo D, Kordas K, Rojas O, Alatorre J, Lopez P, et al. Arsenic exposure and cognitive performance in Mexican schoolchildren. Environ Health Perspect. 2007;115:1371–5. doi: 10.1289/ehp.9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore LE, Lu M, Smith AH. Childhood cancer incidence and arsenic exposure in drinking water in Nevada. Arch Environ Health. 2002;57:201–6. doi: 10.1080/00039890209602937. [DOI] [PubMed] [Google Scholar]
- 27.Russo J, Russo IH. DNA labeling index and structure of the rat mammary gland as determinants of its susceptibility to carcinogenesis. J Natl Cancer Inst. 1978;61:1451–9. [PubMed] [Google Scholar]
- 28.Cupul-Uicab LA, Gladen BC, Hernandez-Avila M, Weber JP, Longnecker MP. DDE, a degradation product of DDT, and duration of lactation in a highly exposed area of Mexico. Environ Health Perspect. 2008;116:179–83. doi: 10.1289/ehp.10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gladen BC, Rogan WJ. DDE and shortened duration of lactation in a northern Mexican town. Am J Public Health. 1995;85:504–8. doi: 10.2105/ajph.85.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez VM, Carrizales L, Jimenez-Capdeville ME, Dufour L, Giordano M. The effects of sodium arsenite exposure on behavioral parameters in the rat. Brain Res Bull. 2001;55:301–8. doi: 10.1016/s0361-9230(01)00477-4. [DOI] [PubMed] [Google Scholar]
- 31.Mass MJ, Tennant A, Roop BC, Cullen WR, Styblo M, Thomas DJ, et al. Methylated trivalent arsenic species are genotoxic. Chem Res Toxicol. 2001;14:355–61. doi: 10.1021/tx000251l. [DOI] [PubMed] [Google Scholar]
- 32.Zheng W. Toxicology of choroid plexus: special reference to metal-induced neurotoxicities. Microsc Res Tech. 2001;52:89–103. doi: 10.1002/1097-0029(20010101)52:1<89::AID-JEMT11>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chattopadhyay S, Ghosh S, Debnath J, Ghosh D. Protection of sodium arsenite-induced ovarian toxicity by coadministration of L-ascorbate (vitamin C) in mature wistar strain rat. Arch Environ Contam Toxicol. 2001;41:83–9. doi: 10.1007/s002440010223. [DOI] [PubMed] [Google Scholar]
- 34.Chattopadhyay S, Pal Ghosh S, Ghosh D, Debnath J. Effect of dietary co-administration of sodium selenite on sodium arsenite-induced ovarian and uterine disorders in mature albino rats. Toxicol Sci. 2003;75:412–22. doi: 10.1093/toxsci/kfg194. [DOI] [PubMed] [Google Scholar]
- 35.Bashir S, Sharma Y, Irshad M, Nag TC, Tiwari M, Kabra M, et al. Arsenic induced apoptosis in rat liver following repeated 60 days exposure. Toxicology. 2006;217:63–70. doi: 10.1016/j.tox.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 36.Ohlsson C, Mohan S, Sjogren K, Tivesten A, Isgaard J, Isaksson O, et al. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009;30:494–535. doi: 10.1210/er.2009-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiney JK, Srivastava V, Nyberg CL, Ojeda SR, Dees WL. Insulin-like growth factor I of peripheral origin acts centrally to accelerate the initiation of female puberty. Endocrinology. 1996;137:3717–28. doi: 10.1210/endo.137.9.8756538. [DOI] [PubMed] [Google Scholar]
- 38.Wilson ME. Premature elevation in serum insulin-like growth factor-I advances first ovulation in rhesus monkeys. J Endocrinol. 1998;158:247–57. doi: 10.1677/joe.0.1580247. [DOI] [PubMed] [Google Scholar]
- 39.Danilovich N, Wernsing D, Coschigano KT, Kopchick JJ, Bartke A. Deficits in female reproductive function in GH-R-KO mice; role of IGF-I. Endocrinology. 1999;140:2637–40. doi: 10.1210/endo.140.6.6992. [DOI] [PubMed] [Google Scholar]
- 40.Ruan W, Catanese V, Wieczorek R, Feldman M, Kleinberg DL. Estradiol enhances the stimulatory effect of insulin-like growth factor-I (IGF-I) on mammary development and growth hormone-induced IGF-I messenger ribonucleic acid. Endocrinology. 1995;136:1296–302. doi: 10.1210/endo.136.3.7867584. [DOI] [PubMed] [Google Scholar]
- 41.Ruan W, Kleinberg DL. Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development. Endocrinology. 1999;140:5075–81. doi: 10.1210/endo.140.11.7095. [DOI] [PubMed] [Google Scholar]
- 42.Bocchinfuso WP, Korach KS. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J Mammary Gland Biol Neoplasia. 1997;2:323–34. doi: 10.1023/a:1026339111278. [DOI] [PubMed] [Google Scholar]
- 43.Bocchinfuso WP, Lindzey JK, Hewitt SC, Clark JA, Myers PH, Cooper R, et al. Induction of mammary gland development in estrogen receptor-alpha knockout mice. Endocrinology. 2000;141:2982–94. doi: 10.1210/endo.141.8.7609. [DOI] [PubMed] [Google Scholar]
- 44.Sen J, Chaudhuri AB. Effect of arsenic on the onset of menarcheal age. Bull Environ Contam Toxicol. 2007;79:293–6. doi: 10.1007/s00128-007-9206-7. [DOI] [PubMed] [Google Scholar]
- 45.ATSDR. Toxicological Profile for Arsenic. Public Health Service USDoHaHS; Atlanta Georgia: 2007. [Google Scholar]
- 46.EFSA. Scientific Opinion on Arsenic in Food. 2009. Panel on Contaminants in the Food Chain (CONTAM) [Google Scholar]
- 47.Meharg AA, Sun G, Williams PN, Adomako E, Deacon C, Zhu YG, et al. Inorganic arsenic levels in baby rice are of concern. Environ Pollut. 2008;152:746–9. doi: 10.1016/j.envpol.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 48.Dearth RK, Hiney JK, Srivastava V, Burdick SB, Bratton GR, Dees WL. Effects of lead (Pb) exposure during gestation and lactation on female pubertal development in the rat. Reprod Toxicol. 2002;16:343–52. doi: 10.1016/s0890-6238(02)00037-0. [DOI] [PubMed] [Google Scholar]
- 49.Dees WL, Skelley CW. Effects of ethanol during the onset of female puberty. Neuroendocrinology. 1990;51:64–9. doi: 10.1159/000125317. [DOI] [PubMed] [Google Scholar]
- 50.Williams JM, Daniel CW. Mammary ductal elongation: differentiation of myoepithelium and basal lamina during branching morphogenesis. Dev Biol. 1983;97:274–90. doi: 10.1016/0012-1606(83)90086-6. [DOI] [PubMed] [Google Scholar]
- 51.Russo J, Russo IH. Susceptibility of the mammary gland to carcinogenesis. II. Pregnancy interruption as a risk factor in tumor incidence. Am J Pathol. 1980;100:497–512. [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng J, Nagashima K, Parmiter D, de la Cruz J, Patri AK. SEM X-ray microanalysis of nanoparticles present in tissue or cultured cell thin sections. Methods Mol Biol. 2011;697:93–9. doi: 10.1007/978-1-60327-198-1_9. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez VM, Limon-Pacheco JH, Carrizales L, Mendoza-Trejo MS, Giordano M. Chronic exposure to low levels of inorganic arsenic causes alterations in locomotor activity and in the expression of dopaminergic and antioxidant systems in the albino rat. Neurotoxicol Teratol. 2010;32:640–7. doi: 10.1016/j.ntt.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Bardullas U, Limon-Pacheco JH, Giordano M, Carrizales L, Mendoza-Trejo MS, Rodriguez VM. Chronic low-level arsenic exposure causes gender-specific alterations in locomotor activity, dopaminergic systems, and thioredoxin expression in mice. Toxicol Appl Pharmacol. 2009;239:169–77. doi: 10.1016/j.taap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 55.Nain S, Smits JE. Pathological, immunological and biochemical markers of subchronic arsenic toxicity in rats. Environ Toxicol. 2012;27:244–54. doi: 10.1002/tox.20635. [DOI] [PubMed] [Google Scholar]
- 56.Hiney JK, Ojeda SR, Dees WL. Insulin-like growth factor I: a possible metabolic signal involved in the regulation of female puberty. Neuroendocrinology. 1991;54:420–3. doi: 10.1159/000125924. [DOI] [PubMed] [Google Scholar]
- 57.Pine MD, Hiney JK, Dearth RK, Bratton GR, Dees WL. IGF-1 administration to prepubertal female rats can overcome delayed puberty caused by maternal Pb exposure. Reprod Toxicol. 2006;21:104–9. doi: 10.1016/j.reprotox.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Handelsman DJ, Spaliviero JA, Scott CD, Baxter RC. Hormonal regulation of the peripubertal surge of insulin-like growth factor-I in the rat. Endocrinology. 1987;120:491–6. doi: 10.1210/endo-120-2-491. [DOI] [PubMed] [Google Scholar]
- 59.Crawford BA, Singh J, Simpson JM, Handelsman DJ. Androgen regulation of circulating insulin-like growth factor-I during puberty in male hypogonadal mice. J Endocrinol. 1993;139:57–65. doi: 10.1677/joe.0.1390057. [DOI] [PubMed] [Google Scholar]
- 60.Copeland KC, Kuehl TJ, Castracane VD. Pubertal endocrinology of the baboon: elevated somatomedin-C/insulin-like growth factor I at puberty. J Clin Endocrinol Metab. 1982;55:1198–201. doi: 10.1210/jcem-55-6-1198. [DOI] [PubMed] [Google Scholar]
- 61.Delemarre-Van De Waal HA, Wennink JM, Odink RJ. Gonadotrophin and growth hormone secretion throughout puberty. Acta Paediatr Scand Suppl. 1991;372:26–31. doi: 10.1111/j.1651-2227.1991.tb17964.x. discussion 2. [DOI] [PubMed] [Google Scholar]
- 62.Urbanski HF, Ojeda SR. The juvenile-peripubertal transition period in the female rat: establishment of a diurnal pattern of pulsatile luteinizing hormone secretion. Endocrinology. 1985;117:644–9. doi: 10.1210/endo-117-2-644. [DOI] [PubMed] [Google Scholar]
- 63.Urbanski HF, Ojeda SR. The development of afternoon minisurges of luteinizing hormone secretion in prepubertal female rats is ovary dependent. Endocrinology. 1986;118:1187–93. doi: 10.1210/endo-118-3-1187. [DOI] [PubMed] [Google Scholar]
- 64.Wilson ME. Regulation of the growth hormone-insulin-like growth factor I axis in developing and adult monkeys is affected by estradiol replacement and supplementation with insulin-like growth factor I. J Clin Endocrinol Metab. 1998;83:2018–28. doi: 10.1210/jcem.83.6.4872. [DOI] [PubMed] [Google Scholar]
- 65.Veldhuis JD, Roemmich JN, Richmond EJ, Bowers CY. Somatotropic and gonadotropic axes linkages in infancy, childhood, and the puberty-adult transition. Endocr Rev. 2006;27:101–40. doi: 10.1210/er.2005-0006. [DOI] [PubMed] [Google Scholar]
- 66.Vorderstrasse BA, Fenton SE, Bohn AA, Cundiff JA, Lawrence BP. A novel effect of dioxin: exposure during pregnancy severely impairs mammary gland differentiation. Toxicol Sci. 2004;78:248–57. doi: 10.1093/toxsci/kfh062. [DOI] [PubMed] [Google Scholar]
- 67.White SS, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L, et al. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007;96:133–44. doi: 10.1093/toxsci/kfl177. [DOI] [PubMed] [Google Scholar]
- 68.Matsumoto C, Miyaura C, Ito A. Bishenol-A suppresses the growth of newborn pubs through insufficient supply of materanl milk in mice. J Health Sci. 2004;50(3):315–8. [Google Scholar]
- 69.Kleinberg DL, Feldman M, Ruan W. IGF-I: an essential factor in terminal end bud formation and ductal morphogenesis. J Mammary Gland Biol Neoplasia. 2000;5:7–17. doi: 10.1023/a:1009507030633. [DOI] [PubMed] [Google Scholar]
- 70.Cannata D, Lann D, Wu Y, Elis S, Sun H, Yakar S, et al. Elevated circulating IGF-I promotes mammary gland development and proliferation. Endocrinology. 2010;151:5751–61. doi: 10.1210/en.2010-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stoica A, Pentecost E, Martin MB. Effects of arsenite on estrogen receptor-alpha expression and activity in MCF-7 breast cancer cells. Endocrinology. 2000;141:3595–602. doi: 10.1210/endo.141.10.7704. [DOI] [PubMed] [Google Scholar]
- 72.Davey JC, Bodwell JE, Gosse JA, Hamilton JW. Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol Sci. 2007;98:75–86. doi: 10.1093/toxsci/kfm013. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Kadiiska MB, Liu Y, Lu T, Qu W, Waalkes MP. Stress-related gene expression in mice treated with inorganic arsenicals. Toxicol Sci. 2001;61:314–20. doi: 10.1093/toxsci/61.2.314. [DOI] [PubMed] [Google Scholar]
- 74.Sciandrello G, Caradonna F, Mauro M, Barbata G. Arsenic-induced DNA hypomethylation affects chromosomal instability in mammalian cells. Carcinogenesis. 2004;25:413–7. doi: 10.1093/carcin/bgh029. [DOI] [PubMed] [Google Scholar]
- 75.Okoji RS, Yu RC, Maronpot RR, Froines JR. Sodium arsenite administration via drinking water increases genome-wide and Ha-ras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis. 2002;23:777–85. doi: 10.1093/carcin/23.5.777. [DOI] [PubMed] [Google Scholar]
- 76.Liu J, Benbrahim-Tallaa L, Qian X, Yu L, Xie Y, Boos J, et al. Further studies on aberrant gene expression associated with arsenic-induced malignant transformation in rat liver TRL1215 cells. Toxicol Appl Pharmacol. 2006;216:407–15. doi: 10.1016/j.taap.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 77.Hartwig A, Blessing H, Schwerdtle T, Walter I. Modulation of DNA repair processes by arsenic and selenium compounds. Toxicology. 2003;193:161–9. doi: 10.1016/j.tox.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 78.Rossman TG. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 79.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–31. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]