

Summary
Despite its widespread use, the underlying mechanism of deep brain stimulation (DBS) remains unknown. Once thought to impart a “functional inactivation”, there is now increasing evidence showing that DBS actually can both inhibit neurons and activate axons, generating a wide range of effects. This implies that the mechanisms that underlie DBS work not only locally but also at the network level. Therefore, not only may DBS induce membrane or synaptic plastic changes in neurons over a wide network, but it may also trigger cellular and molecular changes in other cells, especially astrocytes, where, together, the glial–neuronal interactions may explain effects that are not clearly rationalized by simple activation/inhibition theories alone. Recent studies suggest that (1) high‐frequency stimulation (HFS) activates astrocytes and leads to the release of gliotransmitters that can regulate surrounding neurons at the synapse; (2) activated astrocytes modulate synaptic activity and increase axonal activation; (3) activated astrocytes can signal further astrocytes across large networks, contributing to observed network effects induced by DBS; (4) activated astrocytes can help explain the disparate effects of activation and inhibition induced by HFS at different sites; (5) astrocytes contribute to synaptic plasticity through long‐term potentiation (LTP) and depression (LTD), possibly helping to mediate the long‐term effects of DBS; and (6) DBS may increase delta‐opioid receptor activity in astrcoytes to confer neuroprotection. Together, the plastic changes in these glial–neuronal interactions network‐wide likely underlie the range of effects seen, from the variable temporal latencies to observed effect to global activation patterns. This article reviews recent research progress in the literature on how astrocytes play a key role in DBS efficacy.
Keywords: Astrocytes, Deep brain stimulation, Network, Plasticity, δ‐opioid receptor
Introduction
Ever since its use to refine brain‐lesioning procedures over 50 years ago to the more current practice popularized by Benabid et al. 1, deep brain stimulation (DBS) has become a widely recognized technique for reversible modulation of brain function that is adjunctive to medical management of neurological disorders. The proven efficacy in Parkinson Disease (PD) 1, 2, 3, 4 has led to its worldwide application in a spectrum of hyperkinetic diseases, including essential tremor (ET) 5, 6, 7, 8 dystonia 9, 10, 11, 12, cerebellar outflow tremor 13, 14, 15, 16, Gilles de la Tourette syndrome (GTS) 17, 18, 19, as well as a growing number of other medically intractable disorders, including pain, obsessive‐compulsive disorder (OCD) 20, 21, 22; major depressive disorder (MDD) 23, 24, 25, cluster headache 26, 27, and epilepsy 28, 29. The spectrum of applications continues to grow as new targets are identified and as more treatment‐resistant diseases seek other options for therapy.
Interestingly, despite its widespread use, the underlying mechanism of DBS for its therapeutic efficacy remains unknown. Although there once was a divergence in opinions on the primary action of DBS, ranging from a “functional inactivation” due to local depolarization block to cell activation, there is now increasing evidence that DBS actually can both inhibit neurons and activate axons, generating a wide range of effects. This implies that the mechanisms that underlie DBS work not only locally but at the network level.
Support for fiber activation as a critical mechanism of DBS is provided by the clinical beneficial effects obtained by stimulation of fiber paths in a wide range of disorders, such as the dentate‐rubro‐thalamic (DRT) tract in tremor, as well as the internal capsule 30, 31 in OCD and subgenual cingulate white matter in treatment‐resistant depression (TRD) 23. Yet, stimulation of subcortical nuclear structures such as the subthalamic nucleus and globus pallidus internus in PD clearly cause widespread effect, as illustrated by cerebral blood flow changes in the cortex 32, 33. It seems that the mechanism cannot embody solely one hypothesis or the other, but more of a combination of the two.
When scouring the mechanisms hypothesized for DBS efficacy, in terms of both local and network effects, one cannot dismiss the fact that a nonneuronal factor, that is, the glial interaction with neurons, is important in mediating cellular signaling. Astrocytes, a major type of glia, contain numerous processes that extend hundreds of millimeters and connect to those of other astrocytes through gap junctions, and surround, and are intertwined in neuronal synapses, forming what is termed the ‘tripartate synapse’ 34, 35, 36. This position makes them well‐poised to function actively in the regulation of neuronal communication. Indeed, they are active players in neuronal signaling. If modulation of axonal flow – and hence, synaptic transmission – is considered a key element underlying the mechanism of DBS, then quite likely, both neurons and astrocytes contribute to the DBS therapeutic effect. In addition, not only may DBS electronically stimulate the brain, but may also chemically regulate the integrative function of the local environment and network via regulation of neurotransmitter/modulator release that may affect both neurons and astrocytes. Therefore, DBS‐induced regulation of astrocytes, especially its interaction with neurons, quite likely, contributes to the beneficial effect of DBS. In fact, the contribution from the astrocytes has been the subject of a few recent reviews 37, 38. However, these reviews have not been inclusive of all mechanisms by which astrocytic – neuronal interactions may contribute to the mechanisms of action of DBS. Our recent investigations on how astrocytes react to applied hypoxia gives us a new insight into their under‐recognized importance in neuroprotection in disease states. We also noticed that applied electrical stimulation to the hypothalamus evokes the release of enkaphalin in the brain, which potentially promotes signaling cascades of the delta‐opioid receptor (DOR) to confer neuroprotection. This is of direct significance to how DBS may influence networks far more than just the local milieu. These findings are discussed below in the context of the role of DOR activation in increasing astrocytic glutamate transport and other neuroprotective effects.
What follows is a unifying discussion highlighting the properties and signaling characteristics of astrocytes as found in the literature, and in the context of activation by DBS how astrocytes may be considered key players in promoting the wide range of observed DBS effects.
Astrocytes and PD
Astrocytes are the predominant glial cell in the central nervous system, and not only serve to regulate the external chemical environment of neurons by removing excess ions and recycle neurotransmitters released during synaptic transmission, but also signal to each other and to neurons. Microglia, like astrocytes, function to protect neurons through phagocytosis and cytokine release, chiefly in infection or inflammatory conditions. Thus, they both react to and communicate changes detected within their immediate external milieu both locally and over large networks.
It has recently been suggested that a glial reaction and inflammatory processes participate in the cascade of events, leading to neuronal degeneration in PD 39, 40. Neuropathological examination of the substantia nigra in PD has shown increased glial fibrillary acidic protein (GFAP), confirming astrocytic reaction 41, and the presence of activated microglia has consistently been reported 42. In PD models and other neurodegenerative disease states, it seems that astrocytes play a conflicting role by releasing toxic substances, leading to neurodegeneration 43, but also by eliminating glutamate from the extracellular space, conferring neuroprotection.
When astrocytes undergo a state of gliosis in response to neuronal injury or toxic insults, they, like the microglia, release cytokines that together with released reactive oxygen species (ROS) and nitrite may be the key mediator of glia‐facilitated (1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine) (MPTP) neurotoxicity 44.
Our recent studies show that astrocytes behave differently depending on their status 45, 46, 47. For example, their release of TNF‐α is very low in normoxic conditions, while largely increases in response to hypoxic stress. Therefore, their influence on neurons is differential in different conditions. Moreover, we found that TNF‐α is very different between neuron‐like cells (PC‐12 cells) and astrocytes in response to hypoxic stress [Wang Q, Chao D, Chen T, Xia Y, unpublished data].
Since TNF‐α dysregulates ionic homeostasis 47, 48, 49; Wang Q, Chao D, Chen T, Xia Y, unpublished data] and causes neural dysfunction and injury, an increased release of TNF‐α from astrocytes may, at least partially, contribute to the pathogenesis of some neurodegenerative disorders including PD, especially those directly or indirectly related to hypoxic/ischemic injury. As PD and some neurodegenerative diseases are mainly found in older populations 50, 51, their pathogenesis might be associated with age‐related vasculopathy and related conditions that result in insufficient blood and/or oxygen supply to the brain 52. Indeed, hypoxic and/or ischemic stress has been recognized as one of pathogenic factors that contribute to the development of PD 53, 54, 55. Many people with ischemic/hypoxic brain injury (e.g., cerebral palsy in children, posthypoxic PD or dystonia in adults) develop movement disorders that can manifest weeks, months or even years after the predisposing event. Therefore, hypoxic/ischemic dysfunction of astrocytes may be one of many factors behind PD and other neurodegenerative diseases.
On the other hand, it has been recently confirmed that astrocyte‐derived glial cell line‐derived neurotrophic factor (GDNF) is a potent inhibitor of microglial activation, suggesting an important and regulatory role of astrocytes 56. If astrocyte‐derived GDNF plays a major role in the control of nigrostriatal microglial activation, then, it appears that astrocyte activation is directly involved in midbrain protection from neurodegeneration through the inhibition of neuroinflammation. In addition, we have recent evidence to suggest that DOR activation on the astrocyte may inhibit inflammatory cytokines and upregulate GDNF, leading to protection [Liang J, Chao D, Chen S, Xia Y, 2010, unpublished data; Liang J, Teng S, Chao D, Kim DH, Xia Y, 2011, unpublished data]. This balance between promoting neurodegeneration and protection seems to be on a fine line, and likely there are other unidentified contributors that help to sway to one side over another. Potentially, it seems that DBS may be such an exogenous contributor, as discussed below.
Potential Signaling between Astrocytes and Neurons in PD
Astrocytes, once activated in disease states such as PD, are known to display excitability due to a change in intracellular Ca2+ concentration, which can cause the extracellular release of gliotransmitters – glutamate, adenosine (from adenosine triphosphate (ATP) and D‐serine – that act on neighboring neuronal pre‐and postsynaptic receptors and can modulate synaptic transmission 57. Increased Ca2+ in astrocytes evokes postsynaptic slow inward currents (SICs) in nearby neurons through astrocytic glutamate release, which binds to the NR2B subunit of extrasynaptic N‐methyl‐D‐aspartate receptors (NMDA‐R) 57, 58, 59, 60, 61, 62, 63, 64, 65, 66; this increases neuronal excitability and increases probability of neurotransmitter release from neuronal presynaptic terminals at excitatory 63, 64 and inhibitory synapses 60, 67. Such an SIC is generated in many nearby neurons, and can promote synchrony of neuronal firing 57.
Astrocytes are also known to communicate with each other through Ca2+ waves. Possible mechanisms of Ca2+ wave propagation include the presence of hemichannels on astrocytes which are permeable to ATP, glutamate, and glucose 68, 69, 70, 71 and can mediate nonsynaptic neuronal synchronization via glutamate release 61, 66 or gap junctions which can also mediate glucose uptake and trafficking from blood vessels and clear neuronally released extracellular K+ and glutamate 71, 72, 73. In the seizure model, increased astrocytic coupling through these structures may facilitate the propagation of Ca2+ waves, leading to hypersynchronization and spread of ictal activity 74. It is unclear how these may function to cause widespread communication in other disease states or due to exogenous stimulation. Such astrocytic Ca2+ waves can either be initiated in response to neuronal activation via Gq G protein‐coupled receptor signaling on astrocytic micro‐domains 75, forming networks of cells, or can occur spontaneously even independent of neuronal activity 67, 76, 77, exhibiting so‐called ‘pairwise synchronization’ with a nearly zero time lag in neighboring astrocyte activity 65, 78, 79. Even sensory stimulation, as reported by Wang et al. 80 via whisker stimulation in vivo can evoke calcium activity in the astrocytes in the barrel cortex.
Sasaki et al. 81 employed in vivo calcium imaging to reveal that neighboring astrocytes spontaneously exhibited synchronous Ca2+ elevations and formed locally correlated cell groups of 2–5 cells, which is believed to be due to release of glutamate upon astrocyte activation and subsequent activation of other astrocytes through metabotropic glutamate receptor type 1 activity, independent of neuronal activity 65. These clusters were found to enhance the local excitability of neuronal networks by inducing tonic depolarization of neurons, whereas sporadic activity of single astrocytes – as elicited by Ca2+ uncaging by photostimulation – could not induce an action potential (AP) and was speculated to lower the threshold of synaptic plasticity by enhancing NMDA‐R activity and SICs.
Kuga et al. 82 monitored spontaneous calcium activity simultaneously from hundreds of mouse hippocampal and neocortical astrocytes in vivo and found participation of most in the propagation of regenerative waves from cell to cell – referred to as glissandi – which, as they were blocked by sodium channel blocker tetrodotoxin (TTX), are believed to emerge in response to changes in coordinated intrinsic neuronal network oscillations, or state changes. Neuronal activity, ATP, and gap junctions were found to mediate such waves 82. As the generation of such waves were associated with a decrease in the power of infraslow field oscillations, which is correlated with the blood oxygen level‐dependent (BOLD) signal in functional magnetic resonance imaging (fMRI), 83, 84, 85, 86, it is speculated that these waves mediate the activity‐dependent large scale modulations of cerebral blood flow (CBF). Astrocytes extend several processes, or endfeet, around the cerebral microvasculature 35. In response to intracellular Ca2+ influxes that cause astrocyte activation, Ca2+‐gated K+ channels in these endfeet are also activated, resulting in increased extracellular K+. It is this increased extracellular K+ that causes the dilation of arterioles 57, 87. By being strategically positioned between synapses and blood vessels, astrocytes can indeed act as mediators of neurovascular coupling and may modulate CBF through such highly organized large‐scale astrocyte dynamics 35, 88, 89, 90, 91.
Interestingly, although short electrical microstimulation did activate astrocytes at the tip of the electrode, it did not induce such broad glissandi, which is expected in keeping with our network theory of cellular modulation. Such artificial activation likely disrupts the intrinsic nature of ongoing network activity, and when applied transiently, cannot induce a persistent shift in the global network state 92.
The ability of astrocytes to modulate synaptic transmission – directly on the AP – has been evidenced in vitro in hippocampal slices via the use of calcium imaging again, where uncaging of Ca2+ in peri‐neuronal astrocytes was found to cause AP broadening through ionotropic axonal glutamate (2‐amino‐3‐(5‐methyl‐3‐oxo‐1,2‐oxazol‐4‐yl)propanoic acid) (AMPA) receptor activation 81. Broadened APs triggered larger Ca2+ elevations in presynaptic boutons and facilitated synaptic transmission to postsynaptic neurons. Such broadening of APs occurred in response to the local application of glutamate and adenosine A1 antagonist to axon shafts within 400 μm of the axon hillock, making the suggestion that adenosine tonically inhibits AP (via A1 receptors) and that glutamate released from astrocytes mediates neuronal axonal effects – AP – via extra‐synaptic modulation at non‐NMDA receptors 81.
These are yet other ways astrocytes are believed to influence neuronal signaling, and although this was only seen in vitro on unmyelinated axons in hippocampal slices, the potential for these mechanisms to exist elsewhere in the brain at subcortical or cortical locations is highly possible and exciting, as it build yet new layers of complexity into circuit modulation. In disease states such as PD, the astroglial contribution has a contradictory role of both promoting neurodegenration as well as conferring neuroprotection. The balance is fine, and can be modulated by exogenous stimuli such as HFS. Specifically, this above work could give explanations for the mechanisms for synaptic modulation underlying the beneficial effects seen upon stimulating specific nodes in a dysregulated circuit in certain neurodegenerative diseases, such as PD, or circuits in network disorders, such as in TRD, OCD, and essential tremor (ET). So, there is evidence in vivo that neurons can activate astrocytes, as well as conversely that astrocytes can activate neurons. This could fit into the DBS mechanism theory a bit more readily, as quite possibly, HFS can activate astrocytes locally, which can then cause multiple effects to modulate neuronal activity, leading to the ‘summed final’ network effect seen. Evidence of HFS activation of astrocytic activity is detailed below to help gain focus on this still blurry concept.
Astrocytes and DBS
High‐frequency stimulation, a popular strategy of DBS, induces a sudden change, or interruption of a network status – either physiological or pathological. As detailed above, it has been observed that HFS modulates CBF 93, 94, 95 associated with an increase or decrease in neuronal activity 96. Possibly, after a neuronal network, re‐organization occurs, such as in dystonia or PD after years of HFS, such glissandi are representative of the new current steady‐state of neuronal activity. It is thus also suspected that the observed DBS‐induced modulation in CBF is due to large scale network astrocytic activation representative of such a new global status.
HFS has been known to directly activate astrocytes, and their impact on neuronal signaling has been observed for over a decade 60. Bekar et al. 97 applied HFS to the ventrolateral thalamus to the mouse cortex in a tremor model in vivo and observed increased adenosine levels which they showed were Ca2+‐ independent, and thus believed to be from astrocyte origin. ATP released from astrocytes is broken down in extracellular space by ecto‐ATPase to adenosine; adenosine was seen to increase in the extracellular space around the electrode and is thought to be a key mediator in promoting the efficacy of HFS in suppressing tremor. When an adenosine A1‐receptor antagonist or ecto‐ATPase inhibitor was used, HFS‐induced suppression of thalamic neuronal activity was abolished. Also, HFS induced was seen to induce astrocytic Ca2+ waves that propagated away from the site of stimulation in a frequency and amplitude‐dependent manner.
These findings corroborate the later study of Tawfik et al. 98, where the application of HFS to the thalamic slice resulted in abolishment of oscillations concomitant with the release of glutamate and adenosine. This was interpreted to be due to astrocyte activation, as the glutamate persisted despite TTX application – which would inhibit axonal synaptic release – but was eliminated with a vesicular H+‐ATPase inhibitor. This group also saw HFS‐induced triggering of Ca2+ waves and release of glutamate in astrocyte cultures.
The actual insertion of the DBS electrode into the ventral intermediate nucleus of the thalamus, subthalamic nucleus (STN) or globus pallidus internus (GPi) during surgery has indeed been seen to sometimes cause a reduction or abolishment of tremor in ET or PD patients 99, 100 and personal observation] but also rigidity when surgery is performed under local anesthesia [personal observation]. This so‐called ‘microthalamotomy effect’ is believed to be mediated by the release of glutamate and adenosine 101, 102; such mechanical stimulation can induce activation of calcium signaling and ATP release in cultured astrocytes 103, 104, and thus it is possible that the adenosine and glutamate observed could have astrocyte origins.
The presence of mild reactive gliosis – defined by hypertrophy of astrocytes and increase in the production of the GFAP among other proteins 105 – around the implanted electrode as seen in several histological studies 106, 107, 108, 109, 110 has also implicated HFS‐induced astrocytic activation. After DBS electrode implantation, regional neuro‐inflammation in the rat was observed with concomitant memory impairment 111; in the macaque, reactive gliosis was seen after 3 months and 3 years 112; and in the human, reactive gliosis was observed in a postmortem study 12 years after implantation in an ET patient with good tremor control 106.
Induction of reactive astrocytosis may modulate local as well as widespread effects in neuronal circuits. It has been shown by Ortinski et al. 113 that reactive astrocytosis‐mediated deficits in inhibitory synaptic currents via a downregulation of glutamine synthetase and consequently GABA, triggering reversible hyperexcitability in hippocampal circuits. Implication of astrocyte activation by DBS was seen by Gradinaru et al. 114 in their optogenetic study on PD circuitry, where channelrhodopsin 2 (CHR2) was introduced specifically into astroglia of 6‐hydroxydopamine lesioned‐rodents through a lentiviral vector driven by a GFAP‐promoter. Optical stimulation of CHR2 activated astrocytes and triggered astrocytic Ca2+ waves, which resulted in an inhibition of neuronal firing in STN, though not sufficient enough to cause even trace responses in motor pathology 114. Although this group did not use DBS, it can be inferred that activated astrocytes alone are likely not responsible for mediating all the effects of DBS, but only together in a complex manner with neuronal modulation do they contribute to efficacy.
Glutamate plays a large role in astrocyte activation, and the evidence that it can be modulated by DBS is mounting. Dyskinesias in the naïve rat brought on by STN‐HFS were found to be correlated with increased phosphorylation of the NR2B subunit of the NMDA‐R in neurons of the STN, entopeduncular nucleus (correlate of the human GPi) and other locations 115; blockade of NR2B/NMDA‐Rs, found chiefly peri‐ or extrasynaptically 116, was found to suppress STN‐HFS‐induced dyskinesias, whereas blockade of noncompetitive type of NMDA‐R at the synapse was found to potentiate STN‐HFS‐induced dyskinesias 115, 117.
Such dyskinesias were also associated with altered expression of glutamate transporters in the substantia nigra and striatum, along with increased extracellular concentration of glutamate 117, 118, 119, 120, which has been found to persist after STN‐HFS ceases 121. The authors make the suggestion that extra‐synaptic NR2B/NMDA‐Rs play a key role in STN‐HFS‐induced dyskinesias, which corroborates with findings that stimulation frequencies of 100–200 Hz were found to maximally activate extrasynaptic NMDA‐Rs in hippocampal slices 122. This implies that not only are extra‐synaptic NMDA‐Rs preferentially activated by HFS and are critically linked to the production of results, but one can also contend that excessive extracellular glutamate plays a role in causing dyskinesias. Please refer to Figure 1 for an illustration of the HFS impact on astrocytic interaction with neurons.
Figure 1.
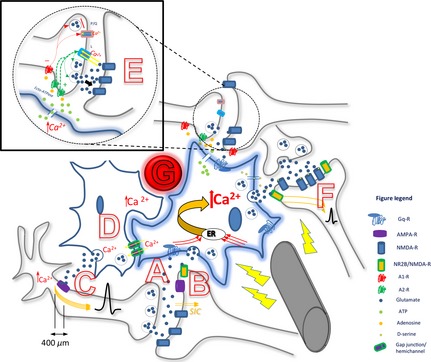
Summary diagram depicting the proposed simultaneity of deep brain stimulation (DBS) effects on axons, astrocyte–astrocyte and astrocyte–neuronal interactions. DBS electrode is here activating one local astrocyte (blue), leading to increased intracelluar Ca2+, causing hypermorphism and release of gliotransmitters, resulting in increased axonal flow and synaptic transmission. This astrocyte has multiple processes, each intimately participating at specific neuronal synapses; activation at one process/micro‐domain may or may not spread to others. Multiple processes are likely occurring simultaneously. (A) Secondary activation by DBS: Increased synaptic activity between axon and dendrite, with release of neurotransmitters (e.g., glutamate), is sensed by the astrocytic Gq protein‐coupled receptors on a process at the tripartite synapse (such as mGluR5), resulting in an increase in intracelluar Ca2+. This activation can be confined to one particular micro‐domain, or can be distributed throughout the astrocyte to all its processes. Each micro‐domain can express different gliotransmitters particular to the synapse. (B) Increased Ca2+ in astrocytes evokes postsynaptic slow inward currents (SICs) in nearby neurons through astrocytic glutamate release, which binds to the NR2B subunit of extrasynaptic NMDA‐R and increases neuronal excitability and increases probability of neurotransmitter release from neuronal presynaptic terminals. (C) Ca2+ activation in astrocyte causes release of extra‐synaptic glutamate; glutamate binds to peri‐synaptic AMPA‐R on presynaptic neuronal processes within 400 μm of axon hillock, causes influx of Ca2+ which broadens action potential (AP). (D) Astrocytes in clusters, coactivated and synchronized via gap junctions/hemichannels. (E) Astrocytic processes detect local synaptic activity – glutamate release – which causes a local Ca2+ evoked response mediated through mGluR5 activation; this leads to release of glial ATP which is is converted by ecto‐ATPase into adenosine. Adenosine then acts at presynaptic A1 coupled P/Q‐type calcium channels to incur synaptic depression while it also acts at presynaptic A2 coupled L‐type calcium channels to incur synaptic potentiation. Relative amounts of synaptic adenosine as well as interactions between each receptor determine the balance; here potentiation is depicted. Also, activation of presynaptic adenosine A2A receptors increases basal synaptic transmission. (F) Different gliotransmitters can be released at other processes due to Ca2+ activation in astrocyte. D‐serine and glutamate are released by the activated astrocyte, modulate NMDA‐R activity and synaptic plasticity. Glutamate released due to DBS astrocytic activation acts on neuronal extrasynaptic NR2B/NMDA‐R, causing increased neuronal excitability and probability of downstream synaptic transmission. (G) Blood vessel proximity to astrocytes – increased CBF linked to state changes of astrocyte activity; DBS induced changes in CBF and interaction with astrocytes not resolved.
How HFS impacts the role of astrocytes in regulating glutamate release and transport is indeed complex. We know that the excessive excitatory stimulation of NMDA, AMPA, and other glutamate receptors is neurotoxic. Astrocytic glutamate transporters play a critical role in cleaning up the released excitatory transmitters in extracellular space 123, 124, 125, 126. Our recent findings show that activation of delta‐opioid receptors (DOR) increases the expression and function of astrocytic glutamate transporters 39, suggesting that DOR may promote neuroprotection 45, 47, 127.
These findings relate directly to support recent theories that HFS may confer neuroprotection 128. In our studies involving electrical stimulation of the hypothalamus in the rabbit 129, 130, we observed an increased release of enkephalin, which is the only endogenous ligand for DOR. Such electrical stimulation likely caused DOR activation and increased astrocytic glutamate transport from the extracellular space. Very recent clinical observations that STN‐DBS improves survival in severe PD 131 may likely be due to astrocyte activation and an increased role of astrocytes in removing extracellular oxidative toxins.
Astrocytes as Players at the Tripartite Synapse with DBS
As their processes are ideally located in apposition with pre‐ and postsynaptic neuronal elements of synapses throughout the CNS 35, 132, 133, astrocytes have been implicated to be exquisitely involved in the regulation of basal synaptic transmission and plasticity in multiple brain locations 134, 135, 136, 137, 138. This was eloquently investigated by Panatier et al. 132 in the CA1 region of the hippocampus, where it was observed that astrocytic processes detect local synaptic activity – neurotransmitter release – which causes a local Ca2+‐ evoked response mediated through metabotropic glutamate subtype 5 receptor (mGluR5) activation. Such activated astrocytes were then found to increase the efficiency of basal synaptic transmission in CA1 pyramidal cells through activation of presynaptic adenosine A2A receptors 132.
These authors contend that such glial modulation may serve to adjust synaptic efficacy so that it can conform to various plasticity events that occur at specific regions in the brain, as adenosine release can either increase transmitter release, via presynaptic adenosine A2A receptor activation – or decrease it via adenosine A1 receptor activation 139, 140, 141.
Such differential modulation of glutamate and GABA release by astrocyte‐derived adenosine is hypothesized to contribute to the differential effects of DBS in the GPi and external segment of the globus pallidus (GPe). It is known that the GPi is inhibited by afferent GABAergic projections from the striatum and the GPe, and it is activated by a glutamatergic projection from the STN. A recent critique by Jantz and Watanabe 38 on the experimental findings of DBS‐induced inhibition of the GPi and excitation of the GPe in the normal monkey by Chiken and Nambu 142 highlights this point. Not only might the differences in the balance of GABAergic and glutamatergic inputs to these structures be a factor 142, but also the relative activation of A2A receptors, which are known to be expressed at higher levels in the GPe and striatum than in the GPi 143, 144, should certainly be considered. As A1 receptors are expressed widely in both the GPi and GPe, Jantz and Watanabe contend that the variation in adenosine receptor concentration can explain the different effects of DBS.
It is likely that both A1 and A2A receptors are also comodulated to differentially effect synaptic plasticity, as seen at the neuromuscular junction 141, and that such regulation may vary with long‐term potentiation (LTP) or depression (LTD) (more below). Group 1 mGluR (mGluR5) are the major subgroup of receptors implicated in the detection of glutamate by astrocytes in response to sustained neuronal activity 61, 145, and are most expressed in the striatum 146. Given their primarily peri‐ or extra‐synaptic location, the activation of these astrocytic mGluR5, resulting in astrocyte activation and release of gliotransmitters, is thought to be a key player in the expression of postsynaptic neuronal NMDA‐R and more permanent plastic effects.
It is yet unproven whether the local synaptic modulation brought upon by calcium signaling in an astrocytic process may extend to incur activation of the whole astrocyte, whereby multiple synapses in different spatial relations can be modulated, either uniformally or differentially, allowing an astrocyte to function as a ‘global network switchboard operator'. As astrocytes have such a particularly complex spatial relationship with adjacent neurons, maintaining several types of synapses 147, 148, it is likely that they interact with and differentially modulate many synapses according to the specific conditions at each, and thus use different transmitters to effect different forms of plasticity.
Astrocytes in Synaptic Plasticity after DBS
Neuroplasticity is believed to be fundamental to DBS efficacy. How else can the disparate temporal effects of efficacy in tremor vs. those in dystonia be explained?
In dystonia, it may take several weeks or months to achieve maximum clinical benefit in patients treated not only by GPi‐DBS 10, 12, 149, 150, 151, 152 but also following pallidotomy 153. Similarly, there is often not an observed quick loss of effect once DBS is turned off in dystonia, taking again weeks for presurgical symptoms to reemerge 150, 151. It is contended that such a slowly progressive clinical time course, as well as concomitant commensurate normalization of electrophysiological measures is due to a LTP synaptic plasticity 152.
The striatum is the source of control of inhibitory inputs within the basal ganglia (BG) circuit 154, 155, 156, and thus modulation via synaptic plasticity within the striatum can regulate striatal output [for reviews in normal and in disease states see refs 154, 157, 158. Activity‐dependent modifications in synaptic efficacy, such as LTP and LTD, represent key cellular substrates for adaptive motor control and procedural memory 158; each has been observed to be induced at medium spiny neuron (MSN) synapses by HFS 159, 160, 161.
In PD, the loss of dopamine afferents induces a compensatory over‐activity and structural remodeling of the corticostriatal system, as seen in rodent and nonhuman primate models of parkinsonism, to continue information relay despite a reduction in corticostriatal glutamatergic synapses 162, 163, 164, 165, 166. Commensurate with neuronal terminal reshaping, perisynaptic astrocytes have been found to be increased in number, and undergo morphological changes, where the appositions between the axo‐spinous complex and the astroglial processes were much tighter and continuous in histological studies of the MPTP primate striatum 166. Such structural changes in the tripartite synapse in disease states, whereby astrocytes contribute to the modulation of processing and integration of synaptic signaling through the formation of complex neuron–glia networks, allow them to be just as well‐poised to be modulated by deep brain stimulation as neurons.
Modulation of the NMDA‐R is directly involved in these processes, and has been observed to occur on two time scales, and differentially involves particular gliotransmitters 167. Astrocytes incur short time scale modulation of NMDA‐R by releasing glutamate and by providing the NMDA‐R coagonist D‐serine, which like glycine, acts at separate and distinct NMDA‐R populations 167. Astrocyte‐derived D‐serine modulates the NMDA‐R current and is necessary for the induction of synaptic plasticity 138, 168, 169, 170, 171, 172, 173, as LTP was observed to be absent at NMDA‐R without astrocytic presence 173 but resumed upon exogenous D‐serine 138.
D‐serine is likely released by a Ca2+ trigger, possibly via vesicular routes 168, 174 and can incur dynamic changes in NMDA‐R mediated currents. Henneberger et al. 174 write that D‐serine can be delivered to distinct populations of NMDA‐Rs (e.g. synaptic, extrasynaptic) characterized by different subunit compositions and with variable affinity to coagonists; different micro‐domains on distinct astrocyte processes may differentially release D‐serine to specific NMDA‐Rs, which may in turn trigger different forms of plasticity 174, 175. Because D‐serine is known to cause internalization of NMDA‐R subunits 176, the authors infer that the release of D‐serine can be dynamic and provides the potential for second to second changes in NMDA‐R mediated currents.
Other longer time scale modulation of synaptic plasticity at the NMDA‐R by astrocytes has also recently been observed to involve the release of adenosine. Deng et al. 167 used astrocyte specific inducible transgenic mice (dnSNARE) brain slices to demonstrate that activation of adenosine A1R in cortex can affect neuronal excitability by regulating the surface expression of NMDA‐R through the activation of specific intracellular signaling pathways. Specifically, astrocytic activation of neuronal A1R led to downstream phosphorylation of the NR2A and NR2B subunits of the NMDA‐R, which, in turn, decreased the rate of its endocytosis 167. Due to such involvement in the trafficking of the NMDA‐R, adenosine is thus linked to a slower time scale of synaptic modulation, such as sleep–wake cycles and memory 167, 177; other global plastic changes can be implicated and might very well use this mechanism.
Combined with the differential activation of presynaptic receptors on causing potentiation or depression of neurotransmitter release, as influenced by its relative extra‐synaptic concentration (at least at the neuromuscular junction 141), it appears that there are many ways astrocyte‐derived adenosine can ultimately modulate synaptic transmission. Very likely, these mechanisms contribute to some of the temporal variability in effects seen after DBS. Together with the other gliotransmitters released upon activation, the astrocyte is well positioned to effect dynamic or delayed effects on neighboring neurons, which has implications on the overall function of the network 37, especially after an exogenous stimulus is applied.
Interestingly, our recent findings show that DOR activation promotes the expression of glutamate transporters [Liang J, Chao D, Chen S, Xia Y, 2010, unpublished data], neurotrophic factors [Liang J, Teng S, Chao D, Kim DH, Xia Y, 2011, unpublished data] and attenuated TNF‐α release/expression [Wang Q, Chao D, Chen T, Xia Y, unpublished data], suggesting that DOR activation protects the brain through an astrocytic mechanism via upregulating glutamate transporters and neurotrophic factors and downregulating TNF‐α activity, thus reducing brain injury. Several studies have demonstrated that activation of DOR reduces K+ leakage following hypoxic/ischemic insult 178, 179, 180. DOR activation has also been shown to confer neuroprotection and prevent cell death through protein kinase and mitogen‐activated protein kinase signaling pathways 181. As we have previously observed that electrical stimulation of the brain induces an increase in leu‐enkephalin, an endogenous DOR agonist, in the CSF, suggesting an increased release of enkephalin in the brain 129, we speculate that DBS may promote brain enkephalin‐DOR activity, thereby regulating astrocytic function. Indeed, we found that DOR activation downregulates astrocytic TNF‐α in hypoxic injury [Q. Wang et al. unpublished data]. This mechanism may, at least partially, contribute to the therapeutic efficacy of DBS, and ultimately underlie the observations of increased neuronal 128 and patient survival in PD 131 after DBS. Please refer to Figure 2 for an illustrative summary of these effects.
Figure 2.
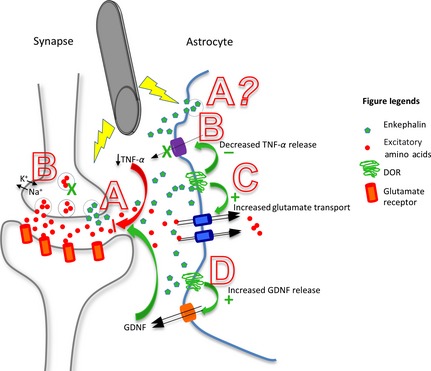
Summary diagram depicting the proposed role of astrocytic delta‐opioid receptors (DOR) in deep brain stimulation (DBS). Stimulation increases leu‐enkephalin release into the synapse, which then activates DOR located on the astrocyte at the tripartite synapse. DOR activation promotes neuroprotection here through three illustrated mechanisms. (A) Increased enkephalin release by neurons and possibly by astrocytes. (B) Decreased TNF‐α release, leading to decreased neuronal dysfunction and injury, possibly through stabilized ionic homeostasis and decreased excessive excitatory neurotransmission 182. (C) Increased expression and function of glutamate transporters to reduce excessive excitatory amino acids, thus attenuating neurotoxicity. (D) Increased release of astrocyte‐derived GDNF, which inhibits microglial activation and leads to decreased neuroinflammation.
Concluding Remarks
The currently available literature suggests that the efficacy of DBS depends upon the combined support of the complex neuronal–glial infrastructure. Whether or not this partnership is essential to DBS needs to be further proven, but mounting evidence supporting an astrocytic component cannot be excluded.
If we consider that not only may DBS induce axonal activation but also astrocytic activation, it becomes perhaps more plausible how synaptic plastic changes can occur over a wide network, which cannot be clearly rationalized by simple neuronal activation/inhibition theories alone.
It remains quite fascinating that such layers of complexity exist in these networks, can change in response to an electrical stimulus, and then produce such marked clinical amelioration of symptoms across a wide range of diseases. If one looks at the mechanisms of DBS in the context of a neurodegenerative/chronic inflammatory process such as PD, then it is plausible that the observed increased survival of DA neurons in the substantia nigra and increased clinical survival of PD patients is due to the conferred astrocytic neuroprotection via increased glutamate transport and decreased release of inflammatory cytokines, possibly through an upregulation of DOR. Conversely, if the mechanisms behind DBS efficacy are examined in tremor or other nondegenerative disease states, then astrocytic activation and release of gliotransmitters with resultant improved synaptic transmission and plasticity may be one of the dominant factors.
Together, the plastic changes in these glial–neuronal interactions network‐wide likely underlie the range of effects seen. Much more needs to be explored in vivo in both electrophysiological and imaging domains, but the evidence is mounting.
Aknowlegments
YX was supported by NIH (AT‐004422 and HD‐034852) and Vivian L Smith Neurologic Foundation.
Conflict of Interest
The authors declare no conflict of interests.
References
- 1. Benabid AL, Pollak P, Louveau A, Henry S, de Rougemont J. Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl Neurophysiol 1987;50:344–346. [DOI] [PubMed] [Google Scholar]
- 2. Deep‐Brain Stimulation For Parkinson's Disease Study Group . Deep‐brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson's disease. N Engl J Med 2001;345:956–963. [DOI] [PubMed] [Google Scholar]
- 3. Krack P, Batir A, Van Blercom N, et al. Five‐year follow‐up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson's disease. N Engl J Med 2003;349:1925–1934. [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez‐Oroz MC, Obeso JA, Lang AE, et al. Bilateral deep brain stimulation in Parkinson's disease: a multicentre study with 4 years follow‐up. Brain 2005;128:2240–2249. [DOI] [PubMed] [Google Scholar]
- 5. Benabid AL, Pollak P, Gervason C, et al. Long‐term suppression of tremor by chronic stimulationof the ventral intermediate thalamic nucleus. Lancet 1991;337:403–406. [DOI] [PubMed] [Google Scholar]
- 6. Hubble JP, Busenbark KL, Wilkinson S, Penn RD, Lyons K, Koller WC. Deep brain stimulation for essential tremor. Neurology 1996;46:1150–1153. [DOI] [PubMed] [Google Scholar]
- 7. Ondo W, Jankovic J, Schwartz K, Almaguer M, Simpson RK. Unilateral thalamic deep brain stimulation for refractory essentialtremor and Parkinson's disease tremor. Neurology 1998;51:1063–1069. [DOI] [PubMed] [Google Scholar]
- 8. Koller WC, Lyons KE, Wilkinson SB, Troster AI, Pahwa R. Long‐term safety and efficacy of unilateral deep brain stimulationof the thalamus in essential tremor. Mov Disord 2001;16:464–468. [DOI] [PubMed] [Google Scholar]
- 9. Cif L, El Fertit H, Vayssiere N, et al. Treatment of dystonic syndromes by chronic electrical stimulation of the internal globus pallidus. J Neurosurg Sci 2003;47:52–55. [PubMed] [Google Scholar]
- 10. Yianni J, Bain P, Giladi N, et al. Globus pallidus internus deep brain stimulation for dystonic conditions:a prospective audit. Mov Disord 2003;18:436–442. [DOI] [PubMed] [Google Scholar]
- 11. Krauss JK, Yianni J, Loher TJ, Aziz TZ. Deep brain stimulationfor dystonia. J Clin Neurophysiol 2004;21:18–30. [DOI] [PubMed] [Google Scholar]
- 12. Vidailhet M, Vercueil L, Houeto JL, et al. Bilateral deep‐brain stimulation of the globuspallidus in primary generalized dystonia. N Engl J Med 2005;352:459–467. [DOI] [PubMed] [Google Scholar]
- 13. Schulder M, Sernas T, Mahalick D, Adler R, Cook S. Thalamic stimulation in patients with multiple sclerosis. Stereotact Funct Neurosurg 1999;72:196–201. [DOI] [PubMed] [Google Scholar]
- 14. Berk C, Carr J, Sinden M, Martzke J, Honey CR. Thalamic deep brain stimulation for the treatment of tremor due to multiple sclerosis: a prospective study of tremor and quality of life. J Neurosurg 2002;97:815–820. [DOI] [PubMed] [Google Scholar]
- 15. Hooper J, Taylor R, Pentland B, Whittle IR. A prospective studyof thalamic deep brain stimulation for the treatment of movementdisorders in multiple sclerosis. Br J Neurosurg 2002;16:102–109. [DOI] [PubMed] [Google Scholar]
- 16. Bittar RG, Hyam J, Nandi D, et al. Thalamotomy versus thalamic stimulation for multiple sclerosis tremor. J Clin Neurosci 2005;12:638–642. [DOI] [PubMed] [Google Scholar]
- 17. Temel Y, Visser‐Vandewalle V. Surgery in Tourette syndrome. Mov Disord 2004;19:3–14. [DOI] [PubMed] [Google Scholar]
- 18. Diederich NJ, Kalteis K, Stamenkovic M, Pieri V, Alesch F. Efficient internal pallidal stimulation in Gilles de la Tourette syndrome:a case report. Mov Disord 2005;20:1496–1499. [DOI] [PubMed] [Google Scholar]
- 19. Houeto JL, Karachi C, Mallet L., et al. Tourette's syndrome and deep brain stimulation. J Neurol Neurosurg Psychiatry 2005;76:992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenberg BD, Malone DA, Friehs GM, et al. Three‐year outcomes in deep brain stimulation for highly resistant obsessive‐compulsive disorder. Neuropsychopharm 2006;31:2384–2393. [DOI] [PubMed] [Google Scholar]
- 21. Mallet L, Polosan M, Jaafari N, et al. Subthalamic nucleus stimulation in severe obsessive‐compulsive disorder. N Engl J Med 2008;359:2121–2134. [DOI] [PubMed] [Google Scholar]
- 22. Denys D, Mantione M, Figee M, van den Munckhof P, Koerselman F, Westenberg H. Deep brain stimulation of the nucleus accumbens for treatment‐refractory obsessive‐compulsive disorder. Arch Gen Psych 2010;67:1061–1068. [DOI] [PubMed] [Google Scholar]
- 23. Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment‐resistant depression. Neuron 2005;45:651–660. [DOI] [PubMed] [Google Scholar]
- 24. Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH. Subcallosal cingulate gyrus deep brain stimulation for treatment‐resistant depression. Biol Psych 2008;64:461–467. [DOI] [PubMed] [Google Scholar]
- 25. Malone DA Jr, Dougherty DD, Rezai AR, et al. Deep brain stimulation of the ventral capsule/ventral striatum for treatment‐resistant depression. Biol Psych 2009;65:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sillay KA, Sani S, Starr PA. Deep brain stimulation for medically intractable cluster headache. Neurobiol Dis 2010;38:361–368. [DOI] [PubMed] [Google Scholar]
- 27. Fontaine D, Lanteri‐Minet M, Ouchchane L, et al. Anatomical location of effective deep brain stimulation electrodes in chronic cluster headache. Brain 2010;133:1214–1223. [DOI] [PubMed] [Google Scholar]
- 28. Chabardès S, Kahane P, Minotti L, Koudsie A, Hirsch E, Benabid AL. Deep brain stimulation in epilepsy with particular reference to the subthalamic nucleus. Epileptic Disord 2002;4(Suppl 3):S83–S93. [PubMed] [Google Scholar]
- 29. Pereira EA, Green AL, Stacey RJ, Aziz TZ. Refractory epilepsy and deep brain stimulation. J Clin Neurosci 2012;19:27–33. [DOI] [PubMed] [Google Scholar]
- 30. Machado A, Haber SN, Sears N, Greenberg B, Malone D, Rezai A. Functional topography of the ventral striatum and anterior limb of the internal capsule determined by electrical stimulation of awake patients. Clin Neurophysiol 2004;120:1941–1948. [DOI] [PubMed] [Google Scholar]
- 31. Greenberg BD, Gabriëls LA, Malone DA Jr, et al. Deep brain stimulation of the ventral internal capsule/ventral striatum for obsessive‐compulsive disorder: worldwide experience. Mol Psychiatry 2010;15:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Limousin P, Greene J, Pollak P, Rothwell J, Benabid AL, Frackowiak R. Changes in cerebral activity pattern due to subthalamic nucleus or internal pallidum stimulation in Parkinson's disease. Ann Neurol 1997;42:283–291. [DOI] [PubMed] [Google Scholar]
- 33. Haegelen C, García‐Lorenzo D, Le Jeune F, et al. SPECT and PET analysis of subthalamic stimulation in Parkinson's disease: analysis using a manual segmentation. J Neurol 2010;257:375–382. [DOI] [PubMed] [Google Scholar]
- 34. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 1999;22:208–215. [DOI] [PubMed] [Google Scholar]
- 35. Ventura R, Harris KM. Three‐dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 1999;19:6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takano T, Tian GF, Peng W, et al. Astrocyte‐mediated control of cerebral blood flow. Nat Neurosci 2006;9:260–267. [DOI] [PubMed] [Google Scholar]
- 37. Vedam‐Mai V, van Battum EY, Kamphuis W, et al. Deep brain stimulation and the role of astrocytes. Mol Psychiatry 2012;17:124–131. [DOI] [PubMed] [Google Scholar]
- 38. Jantz JJ, Watanabe M. Pallidal deep brain stimulation modulates afferent fibers, efferent fibers, and glia. J Neurosci 2013;33:9873–9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mena MA, Garcia de Yebenes J. Glial cells as players in parkinsonism: the “good”, the “bad”, and the “mysterious” glia. Neuroscientist 2008;14:544–560. [DOI] [PubMed] [Google Scholar]
- 40. Niranjan R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's Disease: focus on astrocytes. Mol Neurobiol 2013; PMID:23783559. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 41. Solano RM, Casarejos MJ, Menendez‐Cuervo J, Rodriguez‐Navarro JA, Garcia de Yebenes J, Mena MA. Glial dysfunction in parkin null mice: effects of aging. J Neurosci 2008;28:598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 2009;85:352–370. [DOI] [PubMed] [Google Scholar]
- 43. Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson's disease. Neurotherapeutics 2010;7:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niranjan R, Nath C, Shukla R. Melatonin attenuated mediators of neuroinflammation and alpha‐7 nicotinic acetylcholine receptor mRNA expression in lipopolysaccharide (LPS) stimulated rat astrocytoma cells, C6. Free Radic Res 2012;46:1167–1177. [DOI] [PubMed] [Google Scholar]
- 45. Tian X, Hua F, Sandhu HK, et al. Effect of δ‐opioid receptor activation on BDNF‐TrkB vs. TNF‐α in the mouse cortex exposed to prolonged hypoxia. Int J Mol Sci 2013;14:15959–15976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tian X, Guo J, Zhu M, Li M, Wu G, Xia Y. δ‐Opioid receptor activation rescues the functional TrkB receptor and protects the brain from ischemia‐reperfusion injury in the rat. PLoS ONE 2013;8:e69252. doi: 10.1371/journal.pone.0069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He X, Sandhu HK, Yang Y, et al. Neuroprotection against hypoxia/ischemia: δ‐opioid receptor‐mediated cellular/molecular events. Cell Mol Life Sci 2013;70:2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pribiag H, Stellwagen D. TNF‐α downregulates inhibitory neurotransmission through protein phosphatase 1‐dependent trafficking of GABAA receptor. J Neurosci 2013;33:15879–15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Husain S, Abdul Y, Potter DE. Non‐analgesic effects of opioids: neuroprotection in the retina. Curr Pharm Des 2012;18:6101–6108. [DOI] [PubMed] [Google Scholar]
- 50. Bennett DA, Beckett LA, Murray AM, et al. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med 1996;334:71–76. [DOI] [PubMed] [Google Scholar]
- 51. Kasten M, Chade A, Tanner CM. Epidemiology of Parkinson's disease. Handb Clin Neurol 2007;83:129–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA. Cerebrovascular disease pathology and parkinsoniam signs in old age. Stroke 2011;42:3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karunasinghe RN, Lipski J. Oxygen and glucose deprivation (OGD)‐induced spreading depression in the Substantia Nigra. Brain Res 2013;1527:209–221. [DOI] [PubMed] [Google Scholar]
- 54. Rodriguez‐Grande B, Blackabey V, Gittens B, Pinteaux E, Denes A. Loss of substance P and inflammation precede delayed neurodegeneration in the substantia nigra after cerebral ischemia. Brain Behav Immun 2013;29:51–61. [DOI] [PubMed] [Google Scholar]
- 55. Lin B, Levy S, Raval AP, Perez‐Pinzon MA, Defazio RA. Forebrain ischemia triggers GABAergic system degeneration in substantia nigra at chronic stages in rats. Cardiovasc Psychiatry Neurol 2010;2010:506952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rocha SM, Cristovao AC, Campos FL, Fonseca CP, Baltazar G. Astrocyte‐derived GDNF is a potent inhibitor of microglial activation. Neurobiol Dis 2012;47:407–415. [DOI] [PubMed] [Google Scholar]
- 57. Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 2010;11:227–238. [DOI] [PubMed] [Google Scholar]
- 58. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci 1998;10:2129–2142. [DOI] [PubMed] [Google Scholar]
- 59. Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 2004;24:6920–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte‐mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1998;1:683–692. [DOI] [PubMed] [Google Scholar]
- 61. Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004;443:729–743. [DOI] [PubMed] [Google Scholar]
- 62. Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci 2004;24:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jourdain P, Bergersen LH, Bhaukaurally K, et al. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 2007;10:331–339. [DOI] [PubMed] [Google Scholar]
- 64. Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007;317:1083–1086. [DOI] [PubMed] [Google Scholar]
- 65. Sasaki T, Kuga N, Namiki S, Matsuki N, Ikegaya Y. Locally synchronized astrocytes. Cereb Cortex 2011;21:1889–1900. [DOI] [PubMed] [Google Scholar]
- 66. Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med 2005;11:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor‐dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci 1998;18:6822–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cotrina ML, Lin JHC, Lopez‐Garcıa JC, Naus CCG, Nedergaard M. ATP‐mediated glia signaling. J Neurosci 2000;20:2835–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ye ZC, Wyeth MS, Baltan‐Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci 2003;23:3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Retamal MA, Froger N, Palacios‐Prado N, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 2007;27:13781–13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K(+) channels and gap junction coupling. Glia 2012;60:1192–1202. [DOI] [PubMed] [Google Scholar]
- 72. Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience 2004;129:1045–1056 Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008;322:1551–1555. [DOI] [PubMed] [Google Scholar]
- 74. Gomez‐Gonzalo M, Losi G, Chiavegato A, et al. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol 2010;8:e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Agulhon C, Petravicz J, McMullen AB, et al. What is the role of astrocyte calcium in neurophysiology? Neuron 2008;59:932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Charles AC, Merrill JE, Dirksen ER, Sanderson MJ. Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 1991;6:983–992. [DOI] [PubMed] [Google Scholar]
- 77. Cornell‐Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long‐range glial signaling. Science 1990;247:470–473. [DOI] [PubMed] [Google Scholar]
- 78. Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol 2004;2:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009;323:1211–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang X, Lou N, Xu Q, et al. Astrocytic Ca2 + signaling evoked by sensory stimulation in vivo. Nat Neurosci 2006;9:816–823. [DOI] [PubMed] [Google Scholar]
- 81. Sasaki T, Matsuki N, Ikegaya Y. Action‐potential modulation during axonal conduction. Science 2011;331:599–601. [DOI] [PubMed] [Google Scholar]
- 82. Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y. Large‐scale calcium waves traveling through astrocytic networks in vivo. J Neurosci 2011;31:2607–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hinterberger T, Veit R, Strehl U, et al. Brain areas activated in fMRI during selfregulation of slow cortical potentials (SCPs). Exp Brain Res 2003;152:113–122. [DOI] [PubMed] [Google Scholar]
- 84. He BJ, Raichle ME. The fMRI signal, slow cortical potential and consciousness. Trends Cogn Sci 2009;13:302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Khader P, Schicke T, Roder B, Rosler F. On the relationship between slow cortical potentials and BOLD signal changes in humans. Int J Psychophysiol 2008;67:252–261. [DOI] [PubMed] [Google Scholar]
- 86. Nagai Y, Critchley HD, Featherstone E, Fenwick PB, Trimble MR, Dolan RJ. Brain activity relating to the contingent negative variation: an fMRI investigation. Neuroimage 2004;10:1232–1241. [DOI] [PubMed] [Google Scholar]
- 87. Filosa JA, Bonev AD, Straub SV, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 2006;9:1397–1403. [DOI] [PubMed] [Google Scholar]
- 88. Zonta M, Angulo MC, Gobbo S, et al. Neuron‐to‐astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 2003;6:43–50. [DOI] [PubMed] [Google Scholar]
- 89. Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol 2010;72:335–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci 2006;26:2862–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004;431:195–199. [DOI] [PubMed] [Google Scholar]
- 92. Sasaki T, Matsuki N, Ikegaya Y. Metastability of active CA3 networks. J Neurosci 2007;27:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Perlmutter JS, Mink JW, Bastian AJ, et al. Blood flow responses to deep brain stimulation of thalamus. Neurology 2002;58:1388–1394. [DOI] [PubMed] [Google Scholar]
- 94. Kefalopoulou Z, Paschali A, Markaki E, et al. Regional cerebral blood flow changes induced by deep brain stimulation in secondary dystonia. Acta Neurochir 2010;152:1007–1014. [DOI] [PubMed] [Google Scholar]
- 95. Wyckhuys T, Staelens S, Van NB, et al. Hippocampal deep brain stimulation induces decreased rCBF in the hippocampal formation of the rat. Neuroimage 2010;52:55–61. [DOI] [PubMed] [Google Scholar]
- 96. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature 2010;468:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bekar L, Libionka W, Tian GF, et al. Adenosine is crucial for deep brain stimulation‐mediated attenuation of tremor. Nat Med 2008;14:75–80. [DOI] [PubMed] [Google Scholar]
- 98. Tawfik VL, Chang SY, Hitti FL, et al. Deep brain stimulation results in local glutamate and adenosine release: investigation into the role of astrocytes. Neurosurgery 2010;67:367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mann JM, Foote KD, Garvan CW, et al. Brain penetration effects of microelectrodes and DBS leads in STN or GPi. J Neurol Neurosurg Psych 2009;80:794–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Morishita T, Foote KD, Wu SS, et al. Brain penetration effects of microelectrodes and deep brain stimulation leads in ventral intermediate nucleus stimulation for essential tremor. J Neurosurg 2010;112:491–496. [DOI] [PubMed] [Google Scholar]
- 101. Shon YM, Lee KH, Goerss SJ, et al. High frequency stimulation of the subthalamic nucleus evokes striatal dopamine release in a large animal model of human DBS neurosurgery. Neurosci Lett 2010;475:136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chang SY, Shon YM, Agnesi F, Lee KH. Microthalamotomy effect during deep brain stimulation: potential involvement of adenosine and glutamate efflux. Conf Proc IEEE Eng Med Biol Soc 2009;2009:3294–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell‐specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci USA 2006;103:10058–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci 1998;18:4022–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia 2005;50:427–434. [DOI] [PubMed] [Google Scholar]
- 106. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Long‐term deep brain stimulation for essential tremor: 12‐year clinicopathologic follow‐up. Mov Disord 2010;25:232–238. [DOI] [PubMed] [Google Scholar]
- 107. Haberler C, Alesch F, Mazal PR, et al. No tissue damage by chronic deep brain stimulation in Parkinson's disease. Ann Neurol 2000;48:372–376. [PubMed] [Google Scholar]
- 108. Pilitsis JG, Metman LV, Toleikis JR, Hughes LE, Sani SB, Bakay RA. Factors involved in long‐term efficacy of deep brain stimulation of the thalamus for essential tremor. J Neurosurg 2008;109:640–646. [DOI] [PubMed] [Google Scholar]
- 109. Stock G, Sturm V, Schmitt HP, Schlor KH. The influence of chronic deep brain stimulation on excitability and morphology of the stimulated tissue. Acta Neurochir 1979;47:123–129. [DOI] [PubMed] [Google Scholar]
- 110. Sun DA, Yu H, Spooner J, et al. Postmortem analysis following 71 months of deep brain stimulation of the subthalamic nucleus for Parkinson disease. J Neurosurg 2008;109:325–329. [DOI] [PubMed] [Google Scholar]
- 111. Hirshler YK, Polat U, Biegon A. Intracranial electrode implantation produces regional neuroinflammation and memory deficits in rats. Exp Neurol 2010;222:254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Griffith RW, Humphrey DR. Long‐term gliosis around chronically implanted platinum electrodes in the Rhesus macaque motor cortex. Neurosci Lett 2006;406:81–86. [DOI] [PubMed] [Google Scholar]
- 113. Ortinski PI, Dong J, Mungenast A, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 2010;13:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science 2009;324:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Quintana A, Melon C, Kerkerian‐Le Goff L, Salin P, Savasta M, Sgambato‐Faure V. Forelimb dyskinesia mediated by high‐frequency stimulation of the subthalamic nucleus is linked to rapid activation of the NR2B subunit of N‐methyl‐D‐aspartate receptors. Eur J Neurosci 2010;32:423–434. [DOI] [PubMed] [Google Scholar]
- 116. Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharm 2007;53:362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sgambato‐Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacologicaldopamine replacement and deep brain stimulation for the treatment of Parkinson's disease. Prog Neurobiol 2012;96:69–86. [DOI] [PubMed] [Google Scholar]
- 118. Dupre KB, Ostock CY, Eskow Jaunarajs KL, et al. Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp Neurol 2011;229:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mela F, Marti M, Bido S, Cenci MA, Morari M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to l‐DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol Dis 2012;45:573–582. [DOI] [PubMed] [Google Scholar]
- 120. Seifert G, Steinhauser C. Neuron‐astrocyte signaling and epilepsy. Exp Neurol 2013;244:4–10. [DOI] [PubMed] [Google Scholar]
- 121. Lee KH, Kristic K, van HR, et al. High‐frequency stimulation of the subthalamic nucleus increases glutamate in the subthalamic nucleus of rats as demonstrated by in vivo enzyme‐linked glutamate sensor. Brain Res 2007;1162:121–129. [DOI] [PubMed] [Google Scholar]
- 122. Harris AZ, Pettit DL. Recruiting extrasynaptic NMDA receptors augments synaptic signaling. J Neurophysiol 2008;99:524–533. [DOI] [PubMed] [Google Scholar]
- 123. Murugan M, Ling EA, Kaur C. Dysregulated glutamate uptake by astrocytes causes oligodendroglia death in hypoxic perventricular white matter damage. Mol Cell Neurosci 2013;56C:342–354. [DOI] [PubMed] [Google Scholar]
- 124. Stobart JL, Anderson CM. Multifunctional role of astrocytes as gatekeeps of neuronal energy supply. Front Cell Neurosci 2013;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. López‐Bayghen E, Ortega A. Glial glutamate transporters: new actors in brain signaling. UBMB Life 2011;63:816–823. [DOI] [PubMed] [Google Scholar]
- 126. Kim K, Lee SG, Kegelman TP, et al. Role of excitatory amino acid transporter‐2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol 2011;226:2484–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yang Y, Zhi F, He X, et al. δ‐opioid receptor activation and microRNA expression of the rat cortex in hypoxia. PLoS ONE 2012;7:e51524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Spieles‐Engemann AL, Behbehani MM, Collier TJ, et al. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis 2010;39:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Xia Y, Zhang AZ, Cao XD. Increased leu‐enkephalin immunoreactivity in cerebrospinal fluid during stimulation of hypothalamic defence area in rabbits. Sheng Li Xue Bao 1988;40:379–381. [PubMed] [Google Scholar]
- 130. Xia Y, Zhang AZ, Cao XD, Tang QM, Xu XR. Inhibitory effect of analogous electroacupuncture on sympathetic cardiovascular response to stimulation of hypothalamic defence area in rabbits. J Tradit Chin Med 1987;7:211–214. [PubMed] [Google Scholar]
- 131. Ngoga D, Mitchell R, Kausar J, Hodson J, Harries A, Pall H. Deep brain stimulation improves survival in severe Parkinson's disease. J Neurol Neurosurg Psych 2014;85:17–22. [DOI] [PubMed] [Google Scholar]
- 132. Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basasl transmission at central synapses. Cell 2011;146:785–798. [DOI] [PubMed] [Google Scholar]
- 133. Witcher MR, Kirov SA, Harris KM. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 2007;55:13–23. [DOI] [PubMed] [Google Scholar]
- 134. Gordon GRJ, Iremonger KJ, Kantevari S, Ellis‐Davies GCR, MacVicar BA, Bains JS. Astrocyte‐mediated distributed plasticity at hypothalamic glutamate synapses. Neuron 2009;64:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gourine AV, Kasymov V, Marina N, et al. Astrocytes control breathing through pH‐dependent release of ATP. Science 2010;329:571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Matsui K, Jahr CE. Differential control of synaptic and ectopic vesicular release of glutamate. J Neurosci 2004;24:8932–8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Matsui K, Jahr CE, Rubio ME. High‐concentration rapid transients of glutamate mediate neural‐glial communication via ectopic release. J Neurosci 2005;25:7538–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Panatier A, Theodosis DT, Mothet J‐P, et al. Glia‐derived D‐serine controls NMDA receptor activity and synaptic memory. Cell 2006;125:775–784. [DOI] [PubMed] [Google Scholar]
- 139. Pascual O, Casper KB, Kubera C, et al. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005;310:113–116. [DOI] [PubMed] [Google Scholar]
- 140. Serrano A, Haddjeri N, Lacaille J‐C, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci 2006;26:5370–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Todd KJ, Darabid H, Robitaille R. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 2010;30:11870–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chiken S, Nambu A. High‐frequency pallidal stimulation disrupts information flow through the pallidum by GABAergic inhibition. J Neurosci 2013;33:2268–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Luquin N, Sierra S, Rico AJ, et al. Unmasking adenosine 2A receptors (A2ARs) in monkey basal ganglia ouput neurons using cholera toxin subunit B (CTB). Neurobiol Dis 2012;47:347–357. [DOI] [PubMed] [Google Scholar]
- 144. Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol 2009;193:589–615. [DOI] [PubMed] [Google Scholar]
- 145. D'Ascenzo M, Fellin T, Terunuma M, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA 2007;104:1995–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Testa CM, Standaert DG, Young AB, Penney JB Jr. Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci 1994;14:3005–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 2002;22:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Halassa MM, Fellin T, Takano H, Dong J‐H, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci 2007;27:6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long‐term results. J Neurosurg 2004;101:189–194. [DOI] [PubMed] [Google Scholar]
- 150. Ruge D, Tisch S, Hariz MI, et al. Deep brain stimulation effects in dystonia: time course of electrophysiological changes in early treatment. Mov Disord 2011;26:1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ruge D, Cif L, Limousin P, et al. Shaping reversibility? Long‐term deep brain stimulation in dystonia: the relationship between effects on electrophysiology and clinical symptoms. Brain 2011;134:2106–2115. [DOI] [PubMed] [Google Scholar]
- 152. Tisch S, Limousin P, Rothwell JC, et al. Changes in forearm reciprocal inhibition following pallidal stimulation for dystonia. Neurology 2006;66:1091–1093. [DOI] [PubMed] [Google Scholar]
- 153. Lozano AM, Kumar R, Gross RE, et al. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord 1997;12:865–870. [DOI] [PubMed] [Google Scholar]
- 154. Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron 2008;60:543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gustafson N, Gireesh‐Dharmaraj E, Czubayko U, Blackwell KT, Plenz D. A comparative voltage and current‐clamp analysis of feedback and feedforward synaptic transmission in the striatal microcircuit in vitro. J Neurophysiol 2006;95:737–752. [DOI] [PubMed] [Google Scholar]
- 156. Tepper JM, Koos T, Wilson CJ. GABAergic microcircuits in the neostriatum. Trends Neurosci 2004;27:662–669. [DOI] [PubMed] [Google Scholar]
- 157. Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci 2008;28:6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Picconi B, Piccoli G, Calabresi P. Synaptic dysfunction in Parkinson's disease. Adv Exp Med Biol 2012;970:553–572. [DOI] [PubMed] [Google Scholar]
- 159. Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Longterm synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci 1992;12:4224–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Charpier S, Deniau JM. In vivo activity‐dependent plasticity at cortico‐striatal connections: evidence for physiological long‐term potentiation. Proc Natl Acad Sci USA 1997;94:7036–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lovinger DM, Tyler EC, Merritt A. Short‐ and long‐term synaptic depression in rat neostriatum. J Neurophysiol 1993;70:1937–1949. [DOI] [PubMed] [Google Scholar]
- 162. Galarraga E, Bargas J, Martinez‐Fong D, Aceves J. Spontaneous synaptic potentials in dopamine‐denervated neostriatal neurons. Neurosci Lett 1987;81:351–355. [DOI] [PubMed] [Google Scholar]
- 163. Gubellini P, Picconi B, Bari M, et al. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci 2002;22:6900–6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Liang L, DeLong MR, Papa SM. Inversion of dopamine responses in striatal medium spiny neurons and involuntary movements. J Neurosci 2009;28:7537–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Marti M, Sbrenna S, Fuxe K, Bianchi C, Beani L, Morari M. In vitro evidence for increased facilitation of striatal acetylcholine release via pre‐ and postsynaptic NMDA receptors in hemiparkinsonian rats. J Neurochem 1999;72:875–878. [DOI] [PubMed] [Google Scholar]
- 166. Villalba RM, Smith Y. Neuroglial plasticity at the striatal glutamatergic synapses in Parkinson's disease. Front Syst Neurosci 2011;5:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Deng Q, Terunuma M, Fellin T, Moss SJ, Haydon PG. Astrocytic activation of A1 receptors regulates the surface expression of NMDA receptors through a Src kinase dependent pathway. Glia 2011;59:1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long‐term potentiation depends on release of D‐serine from astrocytes. Nature 2010;463:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mothet JP, Parent AT, Wolosker H, et al. D‐serine is an endogenous ligand for the glycine site of the N‐methyl‐D‐aspartate receptor. Proc Natl Acad Sci USA 2000;97:4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Schell MJ, Molliver ME, Snyder SH. D‐serine, an endogenous synaptic modulator—Localization to astrocytes and glutamate‐stimulated release. Proc Natl Acad Sci USA 1995;92:3948–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Schell MJ, Brady RO, Molliver ME, Snyder SH. D‐Serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci 1997;17:1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Stevens ER, Esguerra M, Kim PM, et al. D‐serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci USA 2003;100:6789–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Yang Y, Ge W, Chen Y, et al. Contribution of astrocytes to hippocampal long‐term potentiation through release of D‐serine. Proc Natl Acad Sci USA 2003;100:15194–15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Henneberger C, Bard L, Rusakov DA. D‐Serine: a key to synaptic plasticity? Int J Biochem Cell Biol 2012;44:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Liu L, Wong TP, Pozza MF, et al. Role of NMDA Receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004;304:1021–1024. [DOI] [PubMed] [Google Scholar]
- 176. Nong Y, Huang YQ, Ju W, et al. Glycine binding primes NMDA receptor internalization. Nature 2003;422:302–307. [DOI] [PubMed] [Google Scholar]
- 177. Florian C, Vecsey CG, Halassa MM, Haydon PG, Abel T. Astrocyte‐derived adenosine and A1 receptor activity contribute to sleep loss‐induced deficits in hippocampal synaptic plasticity and memory in mice. J Neurosci 2011;31:6956–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Chao D, Donnelly DF, Feng Y, Bazzy‐Asaad A, Xia Y. Cortical delta‐opioid receptors potentiate K+ homeostasis during anoxia and oxygen‐glucose deprivation. J Cereb Blood Flow Metab 2007;27:356–368. [DOI] [PubMed] [Google Scholar]
- 179. Chao D, Bazzy‐Asaad A, Balboni G, Xia Y. delta‐, but not mu‐, opioid receptor stabilizes K(+) homeostasis by reducing Ca(2 + ) influx in the cortex during acute hypoxia. J Cell Physiol 2007;212:60–67. [DOI] [PubMed] [Google Scholar]
- 180. Chao D, Bazzy‐Asaad A, Balboni G, Salvadori S, Xia Y. Activation of DOR attenuates anoxic K+ derangement via inhibition of Na+ entry in mouse cortex. Cereb Cortex 2008;18:2217–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ma MC, Qian H, Ghassemi F, Zhao P, Xia Y. Oxygen‐sensitive {delta}‐opioid receptor‐regulated survival and death signals: novel insights into neuronal preconditioning and protection. J Biol Chem 2005;280:16208–16218. [DOI] [PubMed] [Google Scholar]
- 182. Rossi S, Motta C, Studer V, et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult Scler 2013; PMID: 23886826. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]