

Abstract
Clinical and animal studies implicate erythropoietin (EPO) and EPO receptor (EPOR) signaling in angiogenesis. In the eye, EPO is involved in both physiological and pathological angiogenesis in the retina. We hypothesized that EPOR signaling is important in pathological angiogenesis and tested this hypothesis using a rat model of oxygen-induced retinopathy that is representative of human retinopathy of prematurity. We first determined that EPOR expression and activation were increased and that activated EPOR was localized to retinal vascular endothelial cells (ECs) in retinas at postnatal day 18 (p18), when pathological angiogenesis in the form of intravitreal neovascularization occurred. In human retinal microvascular ECs, EPOR was up-regulated and activated by VEGF. Lentiviral-delivered shRNAs that knocked down Müller cell–expressed VEGF in the retinopathy of prematurity model also reduced phosphorylated EPOR (p-EPOR) and VEGFR2 (p-VEGFR2) in retinal ECs. In human retinal microvascular ECs, VEGFR2-activated EPOR caused an interaction between p-EPOR and p-VEGFR2; knockdown of EPOR by siRNA transfection reduced VEGF-induced EC proliferation in association with reduced p-VEGFR2 and p-STAT3; however, inhibition of VEGFR2 activation by siRNA transfection or semaxanib (SU5416) abolished VEGFA-induced proliferation of ECs and phosphorylation of VEGFR2, EPOR, and STAT3. Our results show that VEGFA-induced p-VEGFR2 activates EPOR and causes an interaction between p-EPOR and p-VEGFR2 to enhance VEGFA-induced EC proliferation by exacerbating STAT3 activation, leading to pathological angiogenesis.
Retinopathy of prematurity (ROP) is an important cause of vision loss and blindness in infants worldwide.1 Because of the limited ability to study human preterm infant eyes, models have been established in which newborn animals that normally vascularize their retinas after birth are exposed to oxygen stresses that lead to retinal features similar to human ROP.2 Based on such models of oxygen-induced retinopathy (OIR) and on observations in human infants, ROP has been described as having two phases.3,4 In phase I, infants experience delayed physiological retinal vascular development and sometimes vasoattenuation from high oxygen.5 Phase II is characterized by aberrant disordered developmental angiogenesis in the form of vasoproliferative intravitreal neovascularization (IVNV).2 Several angiogenic agonists and inhibitors have been recognized as potentially involved in human ROP. Of these, the most studied is vascular endothelial growth factor A (VEGFA).6–9
Besides being involved in human pathological angiogenic eye disease,10 VEGFA is also important in retinal vascular development.11,12 Inhibition of the bioactivity of VEGFA in preterm infants with severe ROP reduced the IVNV of phase II,13 but reports of persistent avascular retina and recurrent pathological angiogenesis raised concern.14 Furthermore, neutralizing VEGFA with an antibody similar to that used in human preterm infants with severe ROP initially reduced IVNV in the rat ROP model, but caused recurrent pathological angiogenesis in association with up-regulation of several angiogenic agonists, including erythropoietin (EPO).15
EPO is known mainly for hematopoiesis, and it has been used to treat anemia.16 However, a growing body of evidence indicates that EPO has other biological effects, including neuroprotective,17–19 antiapoptotic,19,20 antioxidative,20,21 and angiogenic22–24 properties. Evidence supporting the role of EPO in angiogenesis comes from clinical and animal studies. In clinical studies, proliferative diabetic retinopathy and severe ROP have been associated with increased EPO. In proliferative diabetic retinopathy, vitreous EPO was increased,25 and a promoter polymorphism in the EPO gene resulting in increased production of EPO was associated with severe diabetic retinopathy in a largely European-American population.26 In ROP, greater risk of severe ROP was associated with EPO treatment for anemia of prematurity.27,28 In a mouse OIR model, hyperoxia down-regulated EPO expression in the retina and decreased vascular stability in association with vaso-obliterated retina,29 and, after relative hypoxia, retinal EPO was increased and contributed to IVNV.30 EPO was also identified as a target in OIR in a study using a transgenic mouse in which hypoxia inducible factor 2α (HIF-2α; EPAS1, alias HLF) was knocked down,31 and EPO synergistically increased VEGFA-induced human retinal microvascular endothelial cell (hRMVEC) proliferation.15 However, EPO also promoted physiological retinal vascularization in a rat OIR model.32 Thus, the evidence is mixed, in that EPO has been associated with both physiological and pathological retinal angiogenesis. EPO is now being considered as a neuroprotective agent to promote cognitive development in preterm infants.33 Greater understanding is needed regarding EPO and EPO receptor (EPOR) signaling in ROP and developmental angiogenesis.
In the present study, we used a rat ROP model34 in which VEGFA is overexpressed by postnatal day 12 (p12) and causes IVNV at p18.35 Our research group previously found that the VEGFA signal is detected in Müller cells and developed a method using a lentiviral vector that targets Müller cells and knocks down VEGFA in vivo, thereby inhibiting IVNV without interfering with pup growth or serum VEGFA.36 The present investigation of the role of EPOR adapted lentiviral vector rat model. In phase I, EPOR activation was lower than in phase II, when VEGFA expression and VEGFR2 expression and activation were increased.37 In hRMVECs, VEGFA up-regulated and activated EPOR and (through crosstalk between activated VEGFR2 and EPOR) increased the activation of STAT3 to enhance angiogenesis. Our present findings support the hypothesis that VEGFA activates and causes an interaction between EPOR and VEGFR2 to contribute to pathological angiogenesis.
Materials and Methods
Rat ROP Model
The rat ROP model used in this study has been described previously.34 Within 6 hours of birth, newborn Sprague–Dawley pups and dam (Charles River Laboratories International, Wilmington, MA) were placed into a regulated oxygen environment (OxyCycler; BioSpherix, Lacona, NY) in which oxygen was cycled between 50% and 10% every 24 hours for 14 days, and then into ambient room air (RA) for 4 days. Phase I, delayed physiological retinal vascular development, occurred at p14 and phase II, IVNV, at p18. All animal studies were performed in compliance with the University of Utah (incorporating the current Guide for the Care and Use of Laboratory Animals) and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research. For each study, at least three different litters were analyzed for immunohistochemistry, quantitative real-time PCR (qPCR), or Western blotting; all litters had between 12 and 14 pups.
For knockdown of Müller cell VEGFA, we used a lentivirus with a CD44 promoter that drives GFP and VEGFA shRNA when embedded within a microRNA 30 context. A subretinal injection of 1 μL of lentivirus (1 × 109 viral particles per milliliter) with either VEGFA shRNA or control luciferase shRNA was delivered at the beginning of the 50% oxygen cycle of the ROP model, on p8, as described previously.36 Each pup received the same type of lentivector in each eye, to reduce the potential for confounding from crossover effects. Eyes were processed for immunohistochemistry of phosphorylated VEGFR2 (p-VEGFR2), or EPOR (p-EPOR) and lectin.
Retinal Section Preparation and Staining
Anterior segments were removed from eyes after 10 minutes of fixation in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA); retinas were carefully dissected and placed into 4% paraformaldehyde for another 15 minutes, followed by overnight incubation in 30% sucrose. After immersion in optimal cutting temperature compound [Tissue-Tek OCT (Sakura Finetek, Torrance, CA); Electron Microscopy Sciences, Hatfield, PA], retinas were cut into 12-μm cryosections. Sections were incubated with rabbit anti–p-VEGFR2 (1:50; Abcam, Cambridge, MA) or anti–p-EPOR (1:50; Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C. After rinsing, sections were incubated for 1 hour with FITC-conjugated goat anti-rabbit secondary antibody (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA.) and Alexa Fluor 594–conjugated antibody for isolectin B4 (lectin) (1:500; Life Technologies, Carlsbad, CA), to stain vessels. Sections stained without primary antibodies were used as controls. Labeling for all sections was performed during the same experiment session. Images were captured using confocal microscopy (Olympus IX81; Olympus, Tokyo, Japan) at ×20 or ×40 magnification.
Cell Culture, Transfection, and Proliferation Assay
hRMVECs (Cell Systems, Kirkland, WA) were maintained in EGM-2 endothelial growth medium (Lonza, Walkersville, MD) supplemented with 5% fetal bovine serum. Cells of passage 3 to 5 were used for experiments. Confluent cells in six-well plates were starved for 16 hours and then treated with 20 ng/mL VEGFA or PBS for 24 hours for qPCR or were treated with VEGFA for 30 minutes for p-VEGFR2, p-EPOR, and p-STAT3.
For siRNA transfection, hRMVECs at 70% confluency were transfected in six-well plates with siRNAs targeting human EPOR, VEGFR2, or STAT3 (Life Technologies) using Lipofectamine 2000 reagent (Life Technologies). A silencer selective negative control siRNA was used as control.
For cell proliferation assays, hRMVECs were plated into 96-well plates at a density of 5000 cells per well. At 24 hours after transfection with siRNA, cells were starved in serum-free medium for 24 hours and then were incubated with 20 ng/mL VEGFA or control PBS for another 24 hours. To inhibit VEGFR2 or STAT3 activation, cells were pretreated with 5 μmol/L of the VEGFR2 tryosine-kinase inhibitor semaxanib (SU5416), 10 μmol/L of the JAK2/STAT3 inhibitor AG490 (both from Sigma-Aldrich, St. Louis, MO), or dimethyl sulfoxide (DMSO) vehicle control for 1 hour before addition of VEGFA. Cell number was measured with a Vybrant MTT cell proliferation assay kit (Life Technologies, Carlsbad, CA).
RNA Isolation and qPCR Analysis
Total RNA of cells was extracted by TRI Reagent (Sigma-Aldrich). For dissected retinas, total RNA was extracted using an RNeasy mini kit (Qiagen, Valencia, CA). RNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). cDNA was generated with the use of a high-capacity cDNA archive kit (Life Technologies). qPCR was performed on a Mastercycler ep realplex system (Eppendorf, Hauppauge, NY) with the use of SYBR Green master mix (Roche Diagnostics, Indianapolis, IN) and primers synthesized by the core research facility of the University of Utah. Expression levels for VEGFA were normalized to the mean value of internal control β-actin. The primers (forward and reverse, respectively) were rat EPOR 5′-CATTCTCGTCCTCATCTCACTG-3′ and 5′-AACTCATTCTCTGGGCTTGG-3′, and human EPOR 5′-CCCCAAGTTCGAGAGCAAAG-3′ and 5′-GGTAGGAGAAGCTGTAGTTGC-3′.
Protein Extraction and Western Blot Analysis
Dissected retinas and hRMVECs were lysed in radioimmunoprecipitation assay buffer with 1:100 protease inhibitors (Roche Diagnostics, Indianapolis, IN) and 2 mmol/L sodium orthovanadate and were homogenized and centrifuged at 15,700 × g for 10 minutes at 4°C. Total protein in the supernatant fluid was quantified by a bicinchoninic acid assay (Pierce BCA; Thermo Fisher Scientific, Waltham, MA). Membranes were incubated overnight at 4°C with primary antibodies to p-STAT3 and total STAT3 (1:1000; Cell Signaling Technology, Danvers, MA), p-EPOR and total EPOR (1:500; Santa Cruz Biotechnology), and p-VEGFR2 and total VEGFR2 (1:500; Santa Cruz Biotechnology). Blots were visualized, and the relative densities of bands were calculated as described previously,32 using gel analysis software (UN-SCAN-IT gel, version 6.1; Silk Scientific, Orem, UT). Relative activities were calculated as the ratio of phosphorylated to total protein or to β-actin and are expressed as fold difference compared with control. At least six samples from each group were measured and analyzed.
Statistical Analysis
Significant differences between treatment groups were determined by one-way analysis of variance using the Newman–Keuls multiple comparison post hoc test or a two-way analysis of variance for grouped comparison. A minimum value of P < 0.05 was considered statistically significant. For proliferation assays, experiments were performed three times with n = 3 per condition in each experiment. For qPCR and protein analyses, six individual samples were used (sometimes pooled from retinas of pups from the same group).
Results
EPOR Expression and Activation Are Increased in Retinal Vascular Endothelium in Association with IVNV in Rat ROP Model
We have previously reported that exogenous EPO restored developmental retinal vascularization by 40%.32 In addition, pathological angiogenesis recurred in the rat ROP model after treatment with anti-VEGFA antibody, in association with increased retinal EPO.15 These observations suggested a role for EPO in angiogenesis associated with the ROP model. To assess EPO signaling, we measured the expression of retinal EPO and EPOR at several time points, including phase I (p14) and phase II (p18) in the rat ROP model and in RA-raised control pups of the same developmental ages. Compared with RA, EPO protein was significantly decreased in the ROP model at p14 and p18 (Figure 1A); this is in contrast to retinal VEGFA, which is significantly increased at both p14 and p18 in the ROP model.38 Also, compared with RA, EPOR protein was significantly increased in the ROP model at p14 and p18 (Figure 1B). To determine whether EPOR was activated in the ROP model, Western blots were performed using anti–p-EPOR antibody. p-EPOR was increased in the retina in the ROP model at p18, but not at p14, compared with RA (Figure 1, C and D).
Figure 1.

EPOR expression and activation increased in vascular endothelium in rat ROP model. A and B: Protein analyses for EPO (A) and EPOR (B) in retinas from rat pups raised in room air (RA) and ROP model pups at time points from baseline to p18. C and D: Representative Western blots of p-EPOR in retina at p14 and p18 (C) and quantification of gels (D). E and F: Immunohistochemical staining of p-EPOR (green) (E) or p-VEGFR2 (green) (F) colabeling with lectin (red) in retinal cryosections at p18. The boxed region in each upper row corresponds to the adjacent image at higher magnification in the lower row. Negative controls are shown in Supplemental Figure S1A. Data are expressed as means ± SD (A and B) or as individual data points with means ± SD (D). ∗P < 0.05, ∗∗P < 0.01 versus RA at the same developmental age. †††P < 0.001, overall analysis of variance. Scale bars: 50 μm (E and F, upper rows); 10 μm (E and F, lower rows). GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
To determine the localization of EPOR activation, cryosections from RA and ROP retinas from p18 pups were colabeled with p-EPOR and lectin to stain vascular cells and, in adjacent sections, with p-VEGFR2 and lectin. p-EPOR colabeling with lectin was not strong at p18 in RA sections, but colabeled vessels were apparent at p18 in the ROP model (Figure 1E). By contrast, p-VEGFR2 colabeled with lectin-stained vessels in both RA and ROP sections, and appeared qualitatively greater in the ROP model (Figure 1F and Supplemental Figure S1A). Taken together, these results indicate that, like VEGFR2, EPOR is activated in ECs in the ROP model during phase II, when IVNV occurs.
EPOR in ECs Is Up-Regulated and Activated by VEGFA
The oxygen levels used in the ROP model result in retinal hypoxia, as indicated by increased conjugated pimonidazole compared with RA.39 Hypoxia induces HIF stabilization, which allows transcription of angiogenic factors,38 including VEGFA40 or EPO.41 In the ROP model, retinal VEGFA is increased at p14, compared with RA counterparts.37 By contrast, EPO protein was decreased during phase I (p14) (Figure 1A). Because both EPOR and VEGFR2 were activated in phase II, these findings raised the question of whether activation of EPOR in retinal ECs in the ROP model is affected by VEGFA. We therefore stimulated hRMVECs with VEGFA and measured EPOR expression and activation. Compared with PBS control, VEGFA increased EPOR mRNA expression (Figure 2A) and protein levels (Figure 2B) after overnight VEGFA treatment. VEGFA activated VEGFR2, beginning at 5 minutes of treatment (Supplemental Figure S2, A and B); EPOR activation peaked at 30 minutes of treatment (Figure 2C and Supplemental Figure S2, A and C), and STAT3 activation began at 30 minutes and peaked at 60 minutes of treatment (Supplemental Figure S2D). We therefore chose 30 minutes of stimulation with VEGFA for subsequent analyses of VEGFR2, EPOR, and STAT3 activation in the same cell lysate. Overnight treatment with 10 IU/mL EPO in hRMVECs also led to an increase in EPOR protein (Figure 2B), but the induction of EPOR by EPO was less than with VEGFA treatment; furthermore, 30 minutes of EPO treatment did not increase p-EPOR and p-VEGFR2 levels (Supplemental Figure S3).
Figure 2.

EPOR is up-regulated and activated by VEGFA in vitro and in vivo. A and B: qPCR of human EPOR (A) and Western blots of EPOR protein in hRMVECs stimulated by 20 ng/mL VEGFA or 10 IU/mL EPO for 18 hours (B). Human β-actin was used as an internal control. C: Western blots of p-EPOR and total EPOR in hRMVECs stimulated with 20 ng/mL VEGFA or PBS for 30 minutes. D and E: Quantification of Western blots of p-VEGFR2 (D) and p-EPOR (E) in retina. F: IHC of p-VEGFR2 or p-EPOR (blue) colabeled with lectin (red) in retinal sections from rat pups injected with lentivirus-delivered luciferase shRNA (Luc.shRNA) or VEGFA shRNA (VEGFA.shRNA) at p18 in rat ROP model. Negative controls are shown in Supplemental Figure S1B. Data expressed as means ± SD, representative of three or more independent experiments with n = 2-3 for each experiment. ∗P < 0.05 versus control. Scale bar = 50 μm.
Our research group has developed a lentivector gene therapy strategy to knock down overexpressed VEGFA in Müller cells in the rat ROP model.36 Retinal VEGFA was reduced to levels observed in the retina of RA-raised pups, and VEGFR2 activation was inhibited in vascular ECs, in association with reduced phase II IVNV.36 To assess whether knockdown of Müller cell–derived VEGFA reduces activated EPOR in the rat ROP model, we analyzed retinal lysates and retinal sections from p18 rat pup eyes in the ROP model that were knocked down for Müller cell VEGFA with a subretinal lentivector-driven shRNA. Compared with the control luciferase shRNA treatment, knockdown of Müller cell VEGFA reduced p-VEGFR2 (Figure 2D) in retinal lysates and reduced colabeling of p-VEGFR2 with lectin-stained ECs in retinal sections (Figure 2F), consistent with our previous findings.36 In the same retinas, p-EPOR in retinal lysates (Figure 2E) and colabeling with lectin in sections were also significantly reduced, compared with the respective luciferase shRNA controls (Figure 2F and Supplemental Figure S1B). These findings provide strong evidence that inhibiting VEGFA reduces p-VEGFR2 and p-EPOR in vascular endothelial cells in the ROP model.
Activation of EPOR by VEGFA Enhances VEGFA-Mediated EC Proliferation
EpoR−/− rescued mice were reported to have a significant reduction in ischemia-induced VEGFA and VEGFR2 expression.23 Our results also suggest that crosstalk exists, but that VEGFA–VEGFR2 signaling can regulate EPOR expression and activation. To assess crosstalk between EPOR and VEGFR2, we transfected hRMVECs with EPOR siRNA or control siRNA and then stimulated them with VEGFA or PBS (control). EPOR siRNA significantly reduced total EPOR in a dose-dependent pattern (Figure 3A), achieving maximum reduction at 50 pmol EPOR siRNA; this dose was used for the later experiments. We then determined whether knockdown of EPOR affects VEGFA-induced EC proliferation. Compared with hRMVECs transfected with control siRNA, VEGFA-induced hRMVEC proliferation was partially reduced after EPOR siRNA transfection (Figure 3B). By contrast, inhibition of VEGFR2 by the VEGFR2 inhibitor SU5416 (Figure 3C) or by siRNA transfection (Figure 3D) abolished VEGFA-induced proliferation. These results indicate that VEGFA-activated EPOR augments EC proliferation caused by stimulation with VEGFA, but that knockdown of EPOR does not fully abolish VEGFA-induced EC proliferation.
Figure 3.
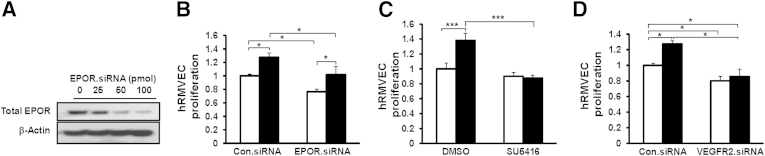
Knockdown of EPOR reduces VEGFA-induced EC proliferation, and inhibition of VEGFR2 activation inhibits it, in hRMVECs. A: Western blots of total EPOR in hRMVECs transfected with 0 to 100 pmol EPOR siRNA. B–D: VEGFA-induced proliferation assay in hRMVECs transfected with control siRNA (Con.siRNA) or EPOR siRNA (B), in hRMVECs treated with SU5416 (C), and in hRMVECs transfected with Con.siRNA or VEGFR2 siRNA (D). Data are expressed as means ± SD, representative of three or more independent experiments. White bars, PBS; black bars, VEGFA (B–D). ∗P < 0.05, ∗∗∗P < 0.001.
Activation of EPOR Interacts with VEGFR2 to Regulate STAT3 Activation
One means whereby EPOR can affect VEGFA-induced angiogenesis is through an interaction between the receptors. To determine whether there was an interaction between activated EPOR and VEGFR2, coimmunoprecipitation of EPOR and VEGFR2 was determined after stimulation with VEGFA or PBS control. After stimulation with VEGFA, both p-VEGFR2 and p-EPOR levels increased (Figure 4A), compared with control, and in the same cell lysate coimmunoprecipitation of VEGFR2 and p-EPOR likewise increased (Figure 4B). Activation of VEGFR2 led to reduced total VEGFR2 in lysates, which is believed to represent increased ubiquitinization of VEGFR2 on activation of the receptor.42 To determine whether VEGFR2 activation is required for the interaction of p-EPOR and VEGFR2, hRMVECs were pretreated with the VEGFR2 inhibitor SU5416 for 1 hour before stimulation with VEGFA. SU5416 effectively blocked VEGFA-induced p-VEGFR2 and also p-EPOR (Figure 4A). In the same cell lysate, the VEGFA-induced coimmunoprecipitation of p-EPOR and VEGFR2 was abolished by pretreatment with the SU5416 (Figure 4B). hRMVECs transfected with EPOR siRNA failed to exhibit VEGFA-induced phosphorylation of EPOR or an interaction between p-EPOR and p-VEGFR2 (Figure 4C). These findings support the notion that an interaction between the receptors occurs in response to VEGFA and that activation of either receptor is needed for the interaction.
Figure 4.

VEGFA-activated EPOR interacts with p-VEGFR2 in hRMVECs. A: Western blots of phosphorylated and total VEGFR2 and EPOR in hRMVECs treated with PBS or VEGFA in the presence of DMSO control or SU5416. B and C: Coimmunoprecipitation of p-EPOR and VEGFR2 in hRMVECs treated with PBS or VEGFA in the presence of DMSO control or SU5416 (B) or in hRMVECs transfected with Con.siRNA or EPOR.siRNA (C). Gels are representative of three or more independent experiments with n = 2 for each experiment. IB, immunoblot (Western blot); IP, immunoprecipitation.
Based on the findings that VEGFA-mediated EC proliferation was augmented through EPOR, we explored signaling pathways affected by EPOR or VEGFR2 activation. Previously, we used an ROP model with supplemental oxygen (ROP + SO model),43 originally developed by Berkowitz and Zhang,44 in which rat pups are placed into 28% instead of 21% oxygen after repeated fluctuations in oxygen concentration as a way to study the role of supplemental oxygen in ROP. We had found that the 28% oxygen reduces total retinal VEGFA, compared with the standard 21% oxygen model.39 By using the 28% oxygen model, we could unmask signaling effects dominated by VEGFA-induced signaling and found that activation of STAT3 was important in phase II IVNV in the ROP + SO model.43 In the present study, therefore, we focused on EC-activated STAT3.
Compared with control siRNA, EPOR siRNA inhibited VEGFA-induced p-EPOR (Figure 5, A and B) and p-STAT3 (Figure 5, E and F), and reduced p-VEGFR2 (Figure 5, C and D). Similarly, VEGFA-induced p-VEGFR2 (Figure 4A), p-EPOR (Figure 4A), and p-STAT3 (Figure 6, A and B) were reduced to baseline in hRMVECs pretreated with the VEGFR2 inhibitor SU5416. We further found that knockdown of VEGFR2 by siRNA transfection inhibited VEGFA-induced p-EPOR (Figure 6, C and E), but knockdown of STAT3 by siRNA did not affect VEGFA-induced p-VEGFR2 and p-EPOR (Figure 6, C–E). These findings provide evidence that p-VEGFR2 is required for VEGFA-induced p-EPOR. Furthermore, activation of p-STAT3 occurred downstream of p-VEGFR2 and p-EPOR. Inhibition of STAT3 with AG490 reduced VEGFA-induced hRMVEC proliferation to baseline (Figure 6F). Taken together, these findings provide evidence that VEGFA activates EPOR and induces an interaction between activated EPOR and VEGFR2, which augments angiogenesis through activation of STAT3.
Figure 5.

Knockdown of EPOR reduces VEGFA-induced p-VEGFR2 and inhibits VEGFA-induced p-EPOR and p-STAT3 in hRMVECs. Representative Western blots, with quantification of densitometry, for p-EPOR (A and B), p-VEGFR2 (C and D), and p-STAT3 (E and F) in hRMVECs transfected with Con.siRNA or EPOR.siRNA and treated with PBS or VEGFA. Data are expressed as means ± SD, representative of three or more independent experiments with n = 2 for each experiment. White bars, PBS; black bars, VEGFA (in panels B, D, and F). ∗P < 0.05, ∗∗P < 0.01.
Figure 6.

STAT3 is the downstream target of activated VEGFR2 and EPOR, and activation of STAT3 mediates VEGFA-induced EC proliferation. A and B: Representative Western blots (A) and quantification (B) of p-STAT3 and total STAT3 in hRMVECs treated with PBS or VEGFA in the presence of control DMSO or SU5416. Quantification of densitometry of p-STAT3 is normalized to total STAT3. C–E: Representative Western blots (C) with quantification (D and E) of p-VEGFR2, p-EPOR, total VEGFR2, and total STAT3 in hRMVECs transfected with Con.siRNA, VEGFR2.siRNA, or STAT3.siRNA and treated with PBS or with 20 ng/mL VEGFA for 30 minutes. Quantification of densitometry of p-VEGFR2 is normalized to total VEGFR2 (D) and that of p-EPOR is normalized to β-actin (E). F: Cell proliferation assay in hRMVECs treated with PBS or VEGFA in the presence of DMSO or AG490 Data are expressed as means ± SD, representative of three or more independent experiments with n = 2 for each experiment. White bars, PBS; black bars, VEGFA (in panels B, D, E and F). ∗P < 0.05, ∗∗∗P < 0.001.
Discussion
With the present study, we provide evidence that VEGFA activates VEGFR2, which then phosphorylates EPOR and forms an interaction with p-EPOR to exacerbate STAT3 activation and mediate the pathological angiogenesis seen in phase II ROP. We provide data using the relevant ROP model in rat showing that p-EPOR–labeled ECs were increased during phase II IVNV, but not in phase I. Furthermore, in the rat model with knockdown for Müller cell VEGFA, p-EPOR labeling in ECs was significantly reduced, compared with luciferase shRNA control. Knockdown of EPOR only partially reduced VEGFA-induced EC proliferation, whereas inhibition of VEGFR2 by either of two methods (siRNA transfection or tyrosine kinase inhibition) appeared to reduce VEGFA-induced EC proliferation to control levels. We further show that VEGFA induced an interaction between EPOR and VEGFR2, which triggered activation of STAT3 in ECs to induce EC proliferation. These findings suggest that EPOR may enhance VEGFR2 signaling and thus potentially lead to pathological angiogenesis in the setting of increased VEGFA, as in ROP, whereas in the physiological angiogenesis of normal development, VEGFA concentration is not as elevated and does not activate EPOR or cause its interaction with VEGFR2.
Recombinant EPO has been used to treat anemia of prematurity,16 but several studies have indicated an association between severe ROP and EPO use.28,45 EPO has since been shown to have other properties besides hematopoiesis,46,47 including neuroprotection through interactions between EPOR and the β common receptor and angiogenesis in ischemic limb models through interactions between VEGFR2 and EPOR.19,48 In terms of ROP, inhibition of phase II IVNV with a neutralizing antibody to VEGFA caused recurrent pathological angiogenesis in association with increased retinal EPO expression.15 Studies in the mouse OIR model demonstrated that EPO is a target for phase II IVNV,30,31 but can be protective if delivered as an intraperitoneal injection before hyperoxia-induced vaso-obliteration in phase I.29 Furthermore, prolyl hydroxylase inhibitors can increase EPO and reduce phase I vaso-obliteration and phase II IVNV in the mouse OIR49 and rat ROP50 models. Increased size of the avascular retina has been associated with increased severity of ROP in several clinical studies,51,52 suggesting that restoring physiological retinal vascularization and reducing avascular retina could reduce IVNV in severe ROP. Based on such studies, it was proposed that the timing of EPO administration is important and that EPO delivered early might protect against severe ROP. However, even though exogenous administration of EPO restored physiological retinal vascularization in phase I of the ROP model,32 phase II IVNV was not reduced (unpublished data). Also, in a multicenter clinical trial of darbepoietin (a derivative of erythropoietin) delivered systemically by weekly subcutaneous injections beginning immediately after birth to improve cognitive development in preterm infants (n = 102), darbepoietin neither increased nor reduced the occurrence of severe ROP.33 The study population was small, but these findings show that timing of EPO administration is not the only consideration involved in ROP.
Previous studies have indicated a role for EPOR in cancer cell growth53 and in EPO–EPOR signaling in angiogenesis.54 In EPOR knockout mice rescued by permitting EPO signaling of hematopoiesis (Epor−/− rescued mice), EPOR–EPOR signaling regulated angiogenesis by up-regulating VEGFA and its receptors in a hindlimb ischemia model, suggesting crosstalk between the signaling pathways.23,55 In the present study, we found evidence that VEGFA can also up-regulate EPOR and activate EPOR through p-VEGFR2, leading to an interaction between the receptors and pathological angiogenesis. We previously reported that activation of STAT3 caused phase II IVNV in a model of ROP + SO.43 Our present findings provide evidence that STAT3 is activated in ECs by VEGFA, leading to IVNV through an interaction between activated VEGFR2 and EPOR (which accounts for the phase II IVNV observed previously43). There are several potential mechanisms whereby STAT3 activation can increase IVNV. Activated STAT3 dimers can translocate to the nucleus and regulate transcription of genes involved in angiogenesis by binding to the gene promoters. In a previous study in the rat ROP model,32 after repeated oxygen fluctuations, VEGFA activated STAT3 in Müller cells; after translocation to the nucleus, p-STAT3 dimers bound the EPO promoter and down-regulated expression of EPO, in association with delayed physiological retinal vascular development in phase I. Thus, VEGFA-activated STAT3 may also be involved in up-regulating EPOR in ECs by directly binding to the EPOR promoter.
Our findings also support the notion that VEGFA activates VEGFR2 (which in turn activates EPOR) in phase II ROP, potentially through another downstream signaling pathway or through an adaptor protein that facilitates an interaction between the receptors (Figure 7). Although current clinical trials show preliminary evidence that ROP is neither increased nor decreased when EPO derivatives are administered early,33 the present study does not directly address whether exogenous EPO can safely be used in preterm infants. Our findings provide evidence of a potential role for EPOR and VEGFR2 interactions, induced by VEGFA in phase II IVNV in ROP and in other conditions associated with pathological angiogenesis.
Figure 7.

The hypothetical signaling pathway in pathological angiogenesis regulated by interactions of activated VEGFR2 and EPOR. VEGFA activates VEGFR2, which then phosphorylates EPOR and forms an interaction with p-EPOR to exacerbate STAT3 activation and mediate pathological angiogenesis as seen in phase II ROP.
Acknowledgments
We thank Dr. John Flannery for providing the lentivector construct pFmCD44.1GW and Dr. Scott Hammond for helping us design miR30-embedded VEGFA shRNA.
Footnotes
Supported by NIH grants R01-EY015130 and R01-EY017011 (M.E.H.), March of Dimes grant 6-FY13-75 (M.E.H.), and an unrestricted grant from Research to Prevent Blindness, Inc., to the Department of Ophthalmology & Visual Sciences, University of Utah.
Z.Y. and H.W. contributed equally to this work.
Disclosures: None declared.
Supplemental Data
Negative controls for immunolabeling for p-VEGFR2 and p-EPOR shown in Figure 1, E and F in retinal sections from p18 pups in the ROP model (A) and for p-VEGFR2 and p-EPOR shown in Figure 2F in sections of retinas treated with lentivector-delivered luciferase shRNA or VEGFA shRNA (B).
Representative Western blots (A) and quantification of p-VEGFR2 normalized to total VEGFR2 (B) and of p-EPOR normalized to β-actin (C) in hRMVECs incubated with 20 ng/mL VEGFA for 0, 5, 10, 15, 30, and 60 minutes, respectively. D: Representative Western blots of p-STAT3 and total STAT3 in hRMVECs under the same conditions. Data are expressed as means ± SEM. n = 4. ∗P < 0.05, ∗∗P < 0.001 versus baseline, analysis of variance.
Western blotting was performed for p-VEGFR2, total VEGFR2, p-EPOR, and β-actin in hRMVECs treated with PBS, 20 ng/mL VEGFA, or 10 U/mL EPO for 30 minutes. Quantification of p-VEGFR2 normalized to total VEGFR2 (A) and p-EPOR normalized to β-actin (B). Data are expressed as means ± SD. n = 4. ∗∗P < 0.01, ∗∗∗P < 0.001 versus control, analysis of variance.
Supplemental Data
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2013.12.023.
References
- 1.Gilbert C. Retinopathy of prematurity: a global perspective of the epidemics, population of babies at risk and implications for control. Early Hum Dev. 2008;84:77–82. doi: 10.1016/j.earlhumdev.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Hartnett M.E., Penn J.S. Mechanisms and management of retinopathy of prematurity. N Engl J Med. 2012;367:2515–2526. doi: 10.1056/NEJMra1208129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith L.E.H., Wesolowski E., McLellan A., Kostyk S.K., D’Amato R., Sullivan R., D’Amore P.A. Oxygen induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. [PubMed] [Google Scholar]
- 4.Ashton N. Retrolental fibroplasia now retinopathy of prematurity. Br J Ophthalmol. 1984;68:689. [Google Scholar]
- 5.Shah P.K., Narendran V., Kalpana N. Aggressive posterior retinopathy of prematurity in large preterm babies in South India. Arch Dis Child Fetal Neonatal Ed. 2012;97:F371–F375. doi: 10.1136/fetalneonatal-2011-301121. [DOI] [PubMed] [Google Scholar]
- 6.Pierce E.A., Avery R.L., Foley E.D., Aiello L.P., Smith L.E.H. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA. 1995;92:905–909. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pierce E., Foley E., Lois E.H. Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol. 1996;114:1219–1228. doi: 10.1001/archopht.1996.01100140419009. [Erratum appeared in Arch Ophthalmol 1997, 115:427] [DOI] [PubMed] [Google Scholar]
- 8.Penn J.S., Madan A., Caldwell R.B., Bartoli M., Caldwell R.W., Hartnett M.E. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–371. doi: 10.1016/j.preteyeres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng E.W.M., Adamis A.P. Anti-VEGF aptamer (pegaptanib) therapy for ocular vascular diseases. Ann N Y Acad Sci. 2006;1082:151–171. doi: 10.1196/annals.1348.062. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N., Damico L., Shams N., Lowman H., Kim R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina. 2006;26:859–870. doi: 10.1097/01.iae.0000242842.14624.e7. [DOI] [PubMed] [Google Scholar]
- 11.Chan-Ling T., Gock B., Stone J. The effect of oxygen on vasoformative cell division: evidence that ‘physiological hypoxia’ is the stimulus for normal retinal vasculogenesis. Invest Ophthalmol Vis Sci. 1995;36:1201–1214. [PubMed] [Google Scholar]
- 12.Stone J., Itin A., Alon T., Pe’er J., Gnessin H., Chan-Ling T., Keshet E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15:4738–4747. doi: 10.1523/JNEUROSCI.15-07-04738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mintz-Hittner H.A., Kennedy K.A., Chuang A.Z., BEAT-ROP Cooperative Group Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364:603–615. doi: 10.1056/NEJMoa1007374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J., Blair M.P., Shapiro M.J., Lichtenstein S.J., Galasso J.M., Kapur R. Reactivation of retinopathy of prematurity after bevacizumab injection. Arch Ophthalmol. 2012;130:1000–1006. doi: 10.1001/archophthalmol.2012.592. [Erratum appeared in Arch Ophthalmol 2013, 131:212] [DOI] [PubMed] [Google Scholar]
- 15.McCloskey M., Wang H., Jiang Y., Smith G.W., Strange J., Hartnett M.E. Anti-VEGF antibody leads to later atypical intravitreous neovascularization and activation of angiogenic pathways in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2013;54:2020–2026. doi: 10.1167/iovs.13-11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strauss R.G. Anaemia of prematurity: pathophysiology and treatment. Blood Rev. 2010;24:221–225. doi: 10.1016/j.blre.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Digicaylioglu M., Lipton S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 18.Erbayraktar S., Grasso G., Sfacteria A., Xie Q.W., Coleman T., Kreilgaard M., Torup L., Sager T., Erbayraktar Z., Gokmen N., Yilmaz O., Ghezzi P., Villa P., Fratelli M., Casagrande S., Leist M., Helboe L., Gerwien J., Christensen S., Geist M.A., Pedersen L.Ø., Cerami-Hand C., Wuerth J.P., Cerami A., Brines M. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci USA. 2003;100:6741–6746. doi: 10.1073/pnas.1031753100. [Erratum appeared in Proc Natl Acad Sci USA 2003, 100:9102] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leist M., Ghezzi P., Grasso G., Bianchi R., Villa P., Fratelli M., Savino C., Bianchi M., Nielsen J., Gerwien J., Kallunki P., Larsen A.K., Helboe L., Christensen S., Pedersen L.Ø., Nielsen M., Torup L., Sager T., Sfacteria A., Erbayraktar S., Erbayraktar Z., Gokmen N., Yilmaz O., Cerami-Hand C., Xie Q., Coleman T., Cerami A., Brines M. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305:239–242. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q., Pfister F., Dorn-Beineke A., vom Hagen F., Lin J., Feng Y., Hammes H.P. Low-dose erythropoietin inhibits oxidative stress and early vascular changes in the experimental diabetic retina. Diabetologia. 2010;53:1227–1238. doi: 10.1007/s00125-010-1727-7. [DOI] [PubMed] [Google Scholar]
- 21.Guneli E., Cavdar Z., Islekel H., Sarioglu S., Erbayraktar S., Kiray M., Sokmen S., Yilmaz O., Gokmen N. Erythropoietin protects the intestine against ischemia/reperfusion injury in rats. Mol Med. 2007;13:509–517. doi: 10.2119/2007-00032.Guneli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kertesz N., Wu J., Chen T.H.P., Sucov H.M., Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. 2004;276:101–110. doi: 10.1016/j.ydbio.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Nakano M., Satoh K., Fukumoto Y., Ito Y., Kagaya Y., Ishii N., Sugamura K., Shimokawa H. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res. 2007;100:662–669. doi: 10.1161/01.RES.0000260179.43672.fe. [DOI] [PubMed] [Google Scholar]
- 24.Bennis Y., Sarlon-Bartoli G., Guillet B., Lucas L., Pellegrini L., Velly L., Blot-Chabaud M., Dignat-Georges F., Sabatier F., Pisano P. Priming of late endothelial progenitor cells with erythropoietin before transplantation requires the CD131 receptor subunit and enhances their angiogenic potential. J Thromb Haemost. 2012;10:1914–1928. doi: 10.1111/j.1538-7836.2012.04835.x. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe D., Suzuma K., Matsui S., Kurimoto M., Kiryu J., Kita M., Suzuma I., Ohashi H., Ojima T., Murakami T., Kobayashi T., Masuda S., Nagao M., Yoshimura N., Takagi H. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med. 2005;353:782–792. doi: 10.1056/NEJMoa041773. [DOI] [PubMed] [Google Scholar]
- 26.Tong Z., Yang Z., Patel S., Chen H., Gibbs D., Yang X. Promoter polymorphism of the erythropoietin gene in severe diabetic eye and kidney complications. Proc Natl Acad Sci USA. 2008;105:6998–7003. doi: 10.1073/pnas.0800454105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suk K.K., Dunbar J.A., Liu A., Daher N.S., Leng C.K., Leng J.K., Lim P., Weller S., Fayard E. Human recombinant erythropoietin and the incidence of retinopathy of prematurity: a multiple regression model. J AAPOS. 2008;12:233–238. doi: 10.1016/j.jaapos.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Brown M.S., Barón A.E., France E.K., Hamman R.F. Association between higher cumulative doses of recombinant erythropoietin and risk for retinopathy of prematurity. J AAPOS. 2006;10:143–149. doi: 10.1016/j.jaapos.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Chen J., Connor K.M., Aderman C.M., Smith L.E. Erythropoietin deficiency decreases vascular stability in mice. J Clin Invest. 2008;118:526–533. doi: 10.1172/JCI33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J., Connor K.M., Aderman C.M., Willett K.L., Aspegren O.P., Smith L.E.H. Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest Ophthalmol Vis Sci. 2009;50:1329–1335. doi: 10.1167/iovs.08-2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morita M., Ohneda O., Yamashita T., Takahashi S., Suzuki N., Nakajima O., Kawauchi S., Ema M., Shibahara S., Udono T., Tomita K., Tamai M., Sogawa K., Yamamoto M., Fujii-Kuriyama Y. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003;22:1134–1146. doi: 10.1093/emboj/cdg117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H., Byfield G., Jiang Y., Smith G.W., McCloskey M., Hartnett M.E. VEGF-mediated STAT3 activation inhibits retinal vascularization by down-regulating local erythropoietin expression. Am J Pathol. 2012;180:1243–1253. doi: 10.1016/j.ajpath.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohls R.K., Christensen R.D., Kamath-Rayne B.D., Rosenberg A., Wiedmeier S.E., Roohi M., Lacy C.B., Lambert D.K., Burnett J.J., Pruckler B., Schrader R., Lowe J.R. A randomized, masked, placebo-controlled study of darbepoetin alfa in preterm infants. Pediatrics. 2013;132:e119–e127. doi: 10.1542/peds.2013-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penn J.S., Tolman B.L., Lowery L.A. Variable oxygen exposure causes preretinal neovascularisation in the newborn rat. Invest Ophthalmol Vis Sci. 1993;34:576–585. [PubMed] [Google Scholar]
- 35.Budd S., Byfield G., Martiniuk D., Geisen P., Hartnett M.E. Reduction in endothelial tip cell filopodia corresponds to reduced intravitreous but not intraretinal vascularization in a model of ROP. Exp Eye Res. 2009;89:718–727. doi: 10.1016/j.exer.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H., Smith G.W., Yang Z., Jiang Y., McCloskey M., Greenberg K., Geisen P., Culp W.D., Flannery J., Kafri T., Hammond S., Hartnett M.E. Short hairpin RNA-mediated knockdown of VEGFA in Müller cells reduces intravitreal neovascularization in a rat model of retinopathy of prematurity. Am J Pathol. 2013;183:964–974. doi: 10.1016/j.ajpath.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Budd S.J., Thompson H., Hartnett M.E. Association of retinal vascular endothelial growth factor with avascular retina in a rat model of retinopathy of prematurity. Arch Ophthalmol. 2010;128:1014–1021. doi: 10.1001/archophthalmol.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budd S.J., Hartnett M.E. Increased angiogenic factors associated with peripheral avascular retina and intravitreous neovascularization: a model of retinopathy of prematurity. Arch Ophthalmol. 2010;128:589–595. doi: 10.1001/archophthalmol.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito Y., Uppal A., Byfield G., Budd S., Hartnett M.E. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Invest Ophthalmol Vis Sci. 2008;49:1591–1598. doi: 10.1167/iovs.07-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll V.A., Ashcroft M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006;66:6264–6270. doi: 10.1158/0008-5472.CAN-05-2519. [DOI] [PubMed] [Google Scholar]
- 41.Frede S., Freitag P., Geuting L., Konietzny R., Fandrey J. Oxygen-regulated expression of the erythropoietin gene in the human renal cell line REPC. Blood. 2011;117:4905–4914. doi: 10.1182/blood-2010-07-298083. [DOI] [PubMed] [Google Scholar]
- 42.Ewan L.C., Jopling H.M., Jia H., Mittar S., Bagherzadeh A., Howell G.J., Walker J.H., Zachary I.C., Ponnambalam S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 43.Byfield G., Budd S., Hartnett M.E. The role of supplemental oxygen and JAK/STAT signaling in intravitreous neovascularization in a ROP rat model. Invest Ophthalmol Vis Sci. 2009;50:3360–3365. doi: 10.1167/iovs.08-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berkowitz B.A., Zhang W. Significant reduction of the panretinal oxygenation response after 28% supplemental oxygen recovery in experimental ROP. Invest Ophthalmol Vis Sci. 2000;41:1925–1931. [PubMed] [Google Scholar]
- 45.Ohlsson A., Aher S.M. Early erythropoietin for preventing red blood cell transfusion in preterm and/or low birth weight infants. Cochrane Database Syst Rev. 2012;9:CD004863. doi: 10.1002/14651858.CD004863.pub3. [DOI] [PubMed] [Google Scholar]
- 46.Aapro M., Jelkmann W., Constantinescu S.N., Leyland-Jones B. Effects of erythropoietin receptors and erythropoiesis-stimulating agents on disease progression in cancer. Br J Cancer. 2012;106:1249–1258. doi: 10.1038/bjc.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Junk A.K., Mammis A., Savitz S.I., Singh M., Roth S., Malhotra S., Rosenbaum P.S., Cerami A., Brines M., Rosenbaum D.M. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brines M., Cerami A. Discovering erythropoietin’s extra-hematopoietic functions: biology and clinical promise. Kidney Int. 2006;70:246–250. doi: 10.1038/sj.ki.5001546. [DOI] [PubMed] [Google Scholar]
- 49.Sears J.E., Hoppe G., Ebrahem Q., Anand-Apte B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc Natl Acad Sci USA. 2008;105:19898–19903. doi: 10.1073/pnas.0805817105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trichonas G., Lee T.J., Hoppe G., Au J., Sears J.E. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy in the rat 50/10 model. Invest Ophthalmol Vis Sci. 2013;54:4919–4926. doi: 10.1167/iovs.13-12171. [DOI] [PubMed] [Google Scholar]
- 51.Cryotherapy for Retinopathy of Prematurity Cooperative Group Multicenter trial of cryotherapy for retinopathy of prematurity: natural history ROP: ocular outcome at 5(1/2) years in premature infants with birth weights less than 1251 g. Arch Ophthalmol. 2002;120:595–599. doi: 10.1001/archopht.120.5.595. [DOI] [PubMed] [Google Scholar]
- 52.Flynn J.T. Retinopathy of prematurity. Pediatr Clin North Am. 1987;34:1487–1516. doi: 10.1016/s0031-3955(16)36370-2. [DOI] [PubMed] [Google Scholar]
- 53.Paragh G., Kumar S.M., Rakosy Z., Choi S.C., Xu X., Acs G. RNA interference-mediated inhibition of erythropoietin receptor expression suppresses tumor growth and invasiveness in A2780 human ovarian carcinoma cells. Am J Pathol. 2009;174:1504–1514. doi: 10.2353/ajpath.2009.080592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morita T., Kourembanas S. Endothelial cell expression of vasconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J Clin Invest. 1999;96:2676–2682. doi: 10.1172/JCI118334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satoh K., Kagaya Y., Nakano M., Ito Y., Ohta J., Tada H., Karibe A., Minegishi N., Suzuki N., Yamamoto M., Ono M., Watanabe J., Shirato K., Ishii N., Sugamura K., Shimokawa H. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation. 2006;113:1442–1450. doi: 10.1161/CIRCULATIONAHA.105.583732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Negative controls for immunolabeling for p-VEGFR2 and p-EPOR shown in Figure 1, E and F in retinal sections from p18 pups in the ROP model (A) and for p-VEGFR2 and p-EPOR shown in Figure 2F in sections of retinas treated with lentivector-delivered luciferase shRNA or VEGFA shRNA (B).
Representative Western blots (A) and quantification of p-VEGFR2 normalized to total VEGFR2 (B) and of p-EPOR normalized to β-actin (C) in hRMVECs incubated with 20 ng/mL VEGFA for 0, 5, 10, 15, 30, and 60 minutes, respectively. D: Representative Western blots of p-STAT3 and total STAT3 in hRMVECs under the same conditions. Data are expressed as means ± SEM. n = 4. ∗P < 0.05, ∗∗P < 0.001 versus baseline, analysis of variance.
Western blotting was performed for p-VEGFR2, total VEGFR2, p-EPOR, and β-actin in hRMVECs treated with PBS, 20 ng/mL VEGFA, or 10 U/mL EPO for 30 minutes. Quantification of p-VEGFR2 normalized to total VEGFR2 (A) and p-EPOR normalized to β-actin (B). Data are expressed as means ± SD. n = 4. ∗∗P < 0.01, ∗∗∗P < 0.001 versus control, analysis of variance.