

Abstract
We present a novel microfluidic solid-phase extraction (μSPE) device for the affinity enrichment of biotinylated membrane proteins from whole cell lysates. The device offers features that address challenges currently associated with the extraction and purification of membrane proteins from whole cell lysates, including the ability to release the enriched membrane protein fraction from the extraction surface so that they are available for downstream processing. The extraction bed was fabricated in PMMA using hot embossing and was comprised of 3,600 micropillars. Activation of the PMMA micropillars by UV/O3 treatment permitted generation of surface-confined carboxylic acid groups and the covalent attachment of NeutrAvidin onto the μSPE device surfaces, which was used to affinity select biotinylated MCF-7 membrane proteins directly from whole cell lysates. The inclusion of a disulfide linker within the biotin moiety permitted release of the isolated membrane proteins via DTT incubation. Very low levels (~20 fmol) of membrane proteins could be isolated and recovered with ~89% efficiency with a bed capacity of 1.7 pmol. Western blotting indicated no traces of cytosolic proteins in the membrane protein fraction as compared to significant contamination using a commercial detergent-based method. We highlight future avenues for enhanced extraction efficiency and increased dynamic range of the μSPE device using computational simulations of different micropillar geometries to guide future device designs.
Introduction
Membrane proteins play key roles in the pathology and physiology of biological cells, including regulating the trafficking of ions and solutes in/out of the cell, cell-to-cell interactions, and responses to stimuli through surface receptors.1 Specific modifications to membrane proteins have been linked to different pathologic states such as cancer, neurological disorders, and diabetes.2 Because of the interest in discovering and validating disease-specific protein signatures with diagnostic value or discovering new drug targets for personalized therapeutics, studies aimed at the identification, characterization, and quantification of membrane proteins has increased over the past few years. Most notably, several biopharmaceuticals that target membrane proteins are already being utilized for the treatment of tumors, lymphomas, and autoimmune diseases.3
Membrane proteins represent approximately one-third of all proteins encoded by the human genome.4,5 Yet, only a small fraction of the cell surface proteome has been characterized due to analytical challenges including: (i) Low abundance, especially compared to the cytosolic proteins;1,6 (ii) low frequency of tryptic cleavage sites in transmembrane domains;7 (iii) the heterogeneity of membrane proteins; and (iv) their hydrophobicity making them prone to precipitation and aggregation and thus, sensitive to solubilization.7,8 A number of analytical approaches have been developed to aid in the analysis of membrane proteins for example ultracentrifugation,9,10 affinity selection of modified or non-modified membrane proteins (antibody- or lectin-based approaches),11,12 two-phase partitioning13,14 and extraction.15,16 For example, detergent-based membrane protein recovery has been demonstrated to be as efficient as >90%; however, this efficiency was demonstrated for a mitochondrial membrane protein and recovery of a plasma membrane protein was only 50%.17 Two important issues are apparent: (i) It is imperative to specifically isolate plasma membrane proteins as signal pathways must be stimulated by external interaction;18 and (ii) the efficiency of detergent isolation intimately depends on the membrane protein’s complexity and hydrophobicity, thereby imparting variability in extraction efficiency.17 In general, the majority of detergent methods fail to produce highly pure isolates of membrane proteins due in large part to contamination from cytosolic proteins.18
Alternatively, affinity-based isolation of membrane proteins avoids such variability and has the potential to target plasma membrane proteins specifically. Approaches utilizing antibodies for affinity isolation are challenged by the fact that the appropriate antibodies must be available for the necessary targets; one runs the risk of neglecting portions of the membrane proteome.19 Recently, improved techniques for the enrichment of membrane proteins, both in vivo20 and in vitro,21 have been reported. These include the chemical capture of glycosylated membrane proteins,22 silica beads with the appropriate membrane protein-specific coatings,23,24 or cell surface biotinylation followed by solid-phase affinity extraction using surface immobilized avidin.20,25,26 Zhao et al. employed streptavidin-coated magnetic beads to enrich plasma membrane proteins that were obtained by lysing biotinylated cells from a human lung carcinoma cell line. The method resulted in a 400-fold enrichment of plasma membrane proteins relative to the endoplasmic reticulum, which was a major contaminant in the membrane fraction, dramatically reduced contamination from other cellular organelles, and as opposed to antibody-based methods, probed all portions of the membrane proteome accessible to surface labeling.27
A variety of microfluidic sold-phase extraction devices (μSPE) have been developed that employ modification of microchannel solid surfaces with molecular reagents that bear the desired affinity agent, the use of polymeric membranes as sorbents, or the incorporation of magnetic or silica beads.28,29 The first demonstration of μSPE was performed by introducing silica beads into a microchannel for the analysis of amino acids and peptides.29 Common to these μSPE devices, however, is the difficulty in handling whole cell lysates largely because impurities reduce the surface area available for specific isolation of the targets and cellular debris can cause clogging (i.e., device failure), especially when utilizing packed beads.30–33
We have previously demonstrated a simple and effective method for creating high surface area extraction beds that incorporate polymeric micropillars arrayed throughout a fluidic channel. The devices were made from thermoplastics and could be molded from metal masters in a single step.34–37 This dramatically simplified device production by eliminating the need for loading silica beads into small channels or the formation of monoliths. However, to the best of our knowledge, μSPE has yet to be applied for the analysis of membrane proteins from whole cell lysates. Our previous reports on using these μSPE devices were focused on analyzing nucleic acids.34,37
Herein, we present a μSPE device for the enrichment of membrane proteins by affinity selection from whole cell lysates. The μSPE device was fabricated by hot embossing into poly(methylmethacrylate), PMMA, and contained 3,600 micropillars within an extraction bed to provide high surface area. The extraction bed surfaces were covalently decorated with NeutrAvidin for selecting biotinylated membrane proteins from a cell lysate while minimizing background binding.38–43 Intact MCF-7 breast cancer cells were surface labeled with a membrane impermeable sulfo-NHS biotin reagent that ensured only membrane proteins were labelled and contained a disulfide linker that could later be cleaved by chemical reduction. The whole cell lysate was hydrodynamically passed through the μSPE device for extraction of the biotinylated membrane proteins, followed by release by cleaving the biotin moiety’s disulfide linker with 1,4-dithiothreitol (DTT). The isolated protein fraction was evaluated for membrane protein recovery and potential cytosolic protein contamination by a sandwich assay and Western blotting, respectively, both of which indicated highly efficient and pure membrane protein recoveries. We highlight the importance of membrane protein solubility for successful extraction, the ability to release extracted proteins for downstream profiling, and provide avenues for enhanced device performance through computational simulations of micropillar geometry and spacing to guide future device designs.
Experimental
Reagents and chemicals
Materials used in these studies included PMMA substrates for the fabrication of the μSPE devices and 250 μm thick cover plates (Plaskolite, Columbus, OH); 177 μm ID polyetheretherketone (PEEK) tubing (IDEX, Oak Harbor, WA); microcentrifuge tubes (Ambion, Foster City, CA); and 4–15% Western blotting gels with PVDF membranes (BioRad, Hercules, CA). Micro-90 and sodium dodecyl sulfate (SDS) were obtained from Fisher Scientific (Houston, TX). Nuclease-free H2O, reagent-grade isopropyl alcohol (IPA), 2-(4-morpholino)-ethane sulfonic acid (MES, pH = 5.0), and bovine serum albumin (BSA) were used as received and secured from Sigma-Aldrich (St. Louis, MO). 1× phosphate-buffered saline (PBS, pH = 8.2) was obtained from Life Technologies, Carlsbad, CA. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), sulfosuccinimidyl-2-(biotinamido)-ethyl-1,3′-dithiopropionate (sulfo-NHS-SS-biotin), NeutrAvidin, fluorescein-conjugated avidin (FITC-avidin), PageRuler Prestained Protein Ladder, the Mem-PER™ Plus Membrane Protein Extraction Reagent Kit, and the Biotin Quantification kit were all purchased from Pierce Biotechnology (Rockford, IL). Tris/Glycine/SDS buffer, β-mercaptoethanol, Tween-20, bromophenol blue, Tris-buffered saline, and the BioRad Mini-PROTEAN System were purchased from BioRad (Hercules, CA). 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate (CHAPS), thiourea, urea, magnesium acetate, Tris-HCl, glycerol, monoclonal anti-β-actin antibody and L-Lysine were also purchased from Sigma Aldrich. The ECL Western blotting detection kit and secondary antibody were obtained from GE Life Sciences (Pittsburgh, PA). Monoclonal anti-EpCAM antibodies were received from R&D Systems (Minneapolis, MN). MCF-7 cells were cultured according to ATCC protocols using MEM Alpha (1X)/insulin/10% FBS (fetal bovine serum) (Life Technologies, Carlsbad, CA). TrypLE express (Life Technologies) was used to detach cells from the flask surface.
Fabrication and design of the μSPE microfluidic device
A schematic of the fluidic chip is shown in Figure 1 along with a picture of the assembled device and SEM images. Fabrication of the microfluidic device involved the following major steps: (i) A brass master mold was fabricated by high precision micromilling (Kern MMP, Kern Micro- and Feinwerktechnik, Murnau-Westried, Germany). (ii) Hot embossing of the microfluidic structures was accomplished using the metal mold master, a HEX03 machine (JenOptik Mikrotechnik, Jena, Germany) and 3 mm thick PMMA substrates. For embossing, the substrate was heated to 180ºC with an applied pressure of 19 kN for 150 s. (iii) Post-processing of the microfluidic device included drilling 1 mm diameter sample reservoirs, device cleaning with 10% Micro-90, IPA, and DI water, and UV/O3 activation of the μSPE device and cover plate using a low pressure Hg lamp (22 mW/cm2 at 254 nm). (iv) Thermal fusion bonding of the cover plate to the substrate at 100ºC for 20 min. The embossed device consisted of three independent channels (100 μm height, 24 mm long and 1.4 mm wide) each containing 3,600 micropillars (100 μm height, 100 μm diameter and 50 μm pillar-to-pillar spacing) that served as the μSPE bed. Each bed had a total surface area of 1.10 cm2.
Figure 1.

(A) Illustration of the topographical layout of the PMMA μSPE device showing three separate beds with micropillars used for the affinity capture of biotinylated membrane proteins. (B) SEM image of the μSPE capture bed. (C) A photograph of the assembled PMMA μSPE device.
NeutrAvidin immobilization
NeutrAvidin was immobilized to the walls and pillars of the μSPE device by covalent coupling to pendant carboxylic acid groups generated by UV/O3 activation (as shown in Scheme 1 and outlined in Table S1). Briefly, NHS esters were formed by flooding the μSPE devices with EDC (6 mg mL−1) and NHS (60 mg mL−1) in 50 mM MES buffer (pH = 5.0) and incubating for 30 min at room temperature. The surface was rinsed with PBS then incubated with a 100 μL aliquot of NeutrAvidin (10 μM in PBS).
Scheme 1.

Overview of the on-chip extraction/purification of biotinylated membrane proteins from cell lysates using the μSPE device. (A) Micropillar activation and NeutrAvidin immobilization; (B) whole cell lysate infusion where the biotinylated membrane proteins are affinity captured while contaminating cytosolic proteins are eluted; and (C) FITC-avidin addition used to label unreacted biotin of selected membrane proteins followed by disulfide bond reduction releasing either FITC-avidin labeled membrane proteins or unlabeled membrane proteins from the μSPE bed. Symbols are defined in the legend.
Cell biotinylation and lysis
MCF-7 cells were washed with ice-cold PBS three times and incubated for 5–10 min in 3 mL TryplE express. Cells were centrifuged at 300× g for 10 min at 4ºC and resuspended at a concentration of 5 × 106 cells/mL in PBS. Eighty μL of sulfo-NHS-SS-biotin (10 mM, prepared immediately prior to use in nuclease-free H2O) was added to the cell suspension. Cells were incubated at room temperature for 30 min with constant mixing, centrifuged and resuspended in lysine (1 mg mL−1 in PBS) to quench the reaction, centrifuged and resuspended in ice-cold PBS and centrifuged to obtain a cell pellet. Cell lysis was performed by adding 50 μL of 4% CHAPS buffer (4% CHAPS, 7 M urea, 30 mM Tris-HCl, 2 M thiourea, and 5 mM magnesium acetate in 100 mL of nuclease-free H2O) to the pellet. Dialysis was performed using 7,00 MW cutoff cartridges (BioRad) and carried out overnight at 4ºC with two buffer (4% CHAPS) changes to further remove excess biotin.
The extent of biotinylation was quantified using a commercial kit. Briefly, biotinylated membrane proteins were added to a solution of avidin and 2-(4′-hydroxyazobenzene)-2-carboxylic acid (HABA). Displacement of HABA molecules reduced colorimetric absorption at 500 nm as measured with an Ultrospec 4000 UV/Vis spectrophotometer (Pharmacia Biotech). To aid in the determination of the extent of biotinylation of MCF-7 membrane proteins, we took a stock solution of biotinylated cells (5 × 106 MCF-7 biotinylated cells per mL) and labeled the cells with 20 μL FITC-avidin (50 μg mL−1 in PBS). The cells were then washed with PBS five times. The cells were lysed, and the lysate was evaluated using a fluorometric assay (as detailed below) to determine the concentration of FITC-avidin in the cell lysate, which was taken as the concentration of biotinylated membrane proteins (2.7:1 avidin:membrane protein stoichiometric ratio). We performed the same experiment with a stock solution of cells that were not biotinylated to determine if non-specific binding of FITC-avidin occurred. The fluorescence signal for the non-biotinylated proteins was undetectable, as the FITC-avidin could not bind to the cells because they were absent of any biotin moiety.
Membrane protein extraction using the μSPE device
The steps employed in our μSPE device and assay of membrane proteins from whole cell lysates are shown in Scheme 1 and outline in Table S1. The cell lysate (5 × 106 MCF-7 biotinylated cells per mL) was infused into the affinity bed at a volumetric flow rate of 5.0 μL/min so that biotinylated membrane proteins could be affinity selected by the surface-confined NeutrAvidin. The surface was then rinsed with a high salt (1 M KCl) and high pH (0.1 M Na2CO3, pH = 11.5) wash to remove any loosely-bound cytosolic proteins. In some cases, we checked for the affinity selection of biotinylated membrane proteins by counter staining with a 100 μL solution consisting of FITC-avidin (50 μg/mL in PBS). Figure S1 shows fluorescence images of biotinylated MCF-7 cells incubated with fluorescein-labeled avidin. The device was rinsed with 100 μL PBS prior to imaging at 20× magnification using a fluorescence microscope with excitation at 488 nm and a 300 ms exposure time. The microscope was a 200M inverted microscope (Zeiss) that contained a single band filter set (Omega Optical), an XBO 75 Xe arc lamp, and a Cascade 1K EMCCD camera (Photometrics). When noted, a proprietary solubilization buffer included with the Mem-PER™ Plus Membrane Protein Extraction Reagent Kit that was added to the cell lysate (initially in 4% CHAPS) prior to infusion.
Membrane protein extraction
Membrane proteins were extracted using a Mem-PER™ Plus Membrane Protein Extraction Reagent kit following the manufacturer’s protocol. See the SI for details on this procedure.
Release of captured biotinylated membrane proteins from capture surface
After affinity selection of the biotinylated membrane proteins by the μSPE device, a 300 mM solution of DTT (in 4% CHAPS) was continuously infused into the SPE bed at a flow rate of 5.0 μL/min for 2 h to release the selected membrane proteins by reducing the disulfide bond carried in the sulfo-NHS-biotin reagent. Infusion was done in the dark to prevent photobleaching of FITC-avidin that was used to determine the efficiency of the release process. A total of 100 μL of PBS was then infused into the μSPE device and the chip was then imaged as outlined above. We further verified that the extracted proteins were indeed released from the affinity bed by measuring the fluorescence of the resulting effluent that was collected during the DTT infusion/rinse. A Horiba Jobin Yvon Fluorolog-3 spectrofluorometer was utilized to form a calibration curve (R2 = 0.9972) of FITC-avidin molecules to evaluate the concentration of the eluted biotinylated membrane proteins. The entrance and exit slits were set at 5 mm with a photomultiplier tube voltage of 950 V. Excitation/emission wavelengths of 491/520 nm were employed.
Protein analysis by Western blotting
Gel runs for the blotting assay employed the BioRad Mini-PROTEAN System. The procedure is summarized here. Five mL of 3× Laemmli sample buffer (6% SDS, 30% glycerol, 187.5 mM Tris-HCl, 15% β-mercaptoethanol, 0.006% bromophenol blue) was added to each protein fraction to prepare them for gel electrophoresis. The fractions were heated at 95°C for 5 min, cooled on ice and briefly vortexed before being placed on the gel. A 4–15% BioRad precast gel was used along with a PageRuler Prestained Protein Ladder that had a molecular weight range of 10–250 kDa. The running buffer (Tris/Glycine/SDS) was used to rinse the wells of the gel and the gel was placed in a gel box along with the running buffer. Five μL of the PageRuler was added to the well and 50 μL of each protein sample was added to the remaining wells. The gel was run for ~35 min at 200 V until the dye front could no longer be seen.
A PVDF membrane was prepared by incubating in methanol for 30 s, rinsed briefly in ddH2O, and then incubated in ice-cold transfer buffer (20% methanol, 10× Tris/Glycine/SDS buffer, ddH2O) for 5 min. The gel was removed from the cassette case and placed on the PVDF membrane and both were sandwiched together with a transfer cassette. The PVDF/gel was placed back into the gel box along with the transfer buffer and run for 70 min at 250 mA. The membrane was removed from the cassette and rinsed briefly with TBS and Tween-20 buffer (0.1% TBST, TBS, Tween-20, ddH2O). The membrane was blocked in 5% milk (dry milk, 0.1% TBST) for 1 h and then incubated overnight at 4°C with the primary antibody (anti-beta-actin or anti-EpCAM antibodies) suspended in 5% dry milk and 0.1% TBST. After incubation, the antibody solution was decanted from the membrane. The membrane was washed five times for 5 min with the 0.1% TBST buffer and blocked for 5 min in 5% milk. The membrane was incubated for 1 h at room temperature with the secondary antibody (1 μL secondary antibody + 5 mL 5% milk). The membrane was washed five times with 0.1% TBST for 5 min and lastly with TBS for 5 min. The membrane was placed on a piece of plastic wrap and 2.5 mL of an ECL solution was pipetted over the membrane and incubated for 5 min making sure that no part of the membrane dried out. The membrane was removed from the ECL solution and excess solution was carefully blotted away. The membrane was placed in a plastic sleeve and was exposed to film in a darkroom for 30 s and visualized.
Computational fluid dynamics (CFD) simulations and diffusion analysis
Different micropillar geometries were assessed for the isolation of membrane proteins by CFD simulations using COMSOL Multiphysics 4.3a. Briefly, three numerically tractable model geometries (with only a few rows of micropillars) were tested: (I) Circular pillars with radii of 100 μm and pillar-to-pillar spacing of 50.0 μm, (II) diamond pillars with side lengths of 20.0 μm and pillar-to-pillar spacing of 20.0 μm, which is similar to a previously published device;44 and (III) circular pillars with radii of 10.0 μm and pillar-to-pillar spacing of 20.0 μm, which was also tested to determine the effects of pillar shape (circular vs. diamond). For all geometries, steady-state laminar velocity fields were solved (see Figure S2). Due to computational limits, entire μSPE beds could not be simulated via COMSOL.
The effects of protein diffusion throughout an entire bed’s length were evaluated using an analytical solution to Fick’s 2nd law. The time-dependent position probability packet of a protein, initially centered between two pillars, was evaluated over a bed’s length, L, according to its velocity (extracted from the CFD simulations), and the probability of immobilization was taken as the area of the Gaussian packet outside the fluidic channel’s walls. We took into account pillar shape by applying a path correction factor, C, to the effective length traveled, where Leff = C × L. For a circular pillar, protein travels about a half perimeter, yielding C = π/2 ≈ 1.57, and for a diamond, the protein travels about a triangle, where there is a smaller effective length given by . These path correction factors can be shown to be independent of pillar size or L. Details on this model’s derivation and implementation are given in the SI.
Results and Discussion
The μSPE device utilized affinity selection for the specific isolation of membrane proteins from whole cell lysates. The affinity selection utilized NeutrAvidin molecules that were immobilized within the fabricated μSPE bed. Prior to cell lysis, the intact biological cells (MCF-7) were biotinylated. A disulfide moiety was incorporated into the biotinylation reagent so that membrane proteins could be released following affinity selection for downstream analysis (Scheme 1, Table S1). The reducing agent cleaves the disulfide bond and as a result, releases the proteins with an attached residue of 104 g/mole per protein for each biotinylated site. In addition, it will reduce disulfides directly within proteins that contain such linkages. We will demonstrate both the efficiency of membrane protein extraction from whole cell lysates and the purity of the isolated fractions using this μSPE device compared to a detergent-based method. We will also present numerical simulations to guide future device designs for improved extraction efficiency and expanded dynamic range.
Solubilization, isolation, and release of biotinylated membrane proteins using the μSPE device
We first biotinylated membrane proteins found on MCF-7 cells using a membrane impermeable sulfo-NHS biotin reagent containing a disulfide linker. The success of biotinylation was confirmed by imaging whole cells labeled with fluorescent FITC-avidin (see Figure S1). Cells were then lysed with the whole cell lysate containing both cytosolic and biotinylated membrane proteins, which were subsequently passed through the μSPE bed that was decorated with NeutrAvidin molecules (Scheme 1, Table S1). NeutrAvidin molecules were covalently anchored to the μSPE bed walls through the surface-confined carboxylic acids and accessible primary amine groups found on NeutrAvidin. Our group has shown that after UV/O3 activation of PMMA, carboxylic acid functional groups are generated.45
After removing potential cytosolic contaminants via a high salt and high pH wash, FITC-avidin was introduced into the μSPE device, which bound to free biotin molecules found on the affinity selected proteins (2.7 biotin molecules per membrane protein) in a sandwich-type assay (Scheme 1, Table S1, and Figure 2A), which permitted direct observation of membrane proteins isolated in the μSPE bed. Note that control images (FITC-avidin incubated with the μSPE bed without first passing through the cell lysate) indicated minimal nonspecific adsorption of the dye-labeled avidin (Figure 2B).
Figure 2.

(A) Fluorescence image of poorly solubilized membrane proteins isolated on the μSPE device. (B) Control image of the μSPE bed incubated with FITC-avidin without first infusing the cell lysate showing minimal nonspecific adsorption of the dye-labeled avidin complex. (C) Fluorescence image of well-solubilized membrane proteins isolated on the μSPE bed. (D) μSPE bed after release of membrane proteins with DTT.
The cell lysate/FITC-avidin sandwich indicated that the membrane proteins isolated in the μSPE device were aggregated (Figure 2A) when introduced into the lysis buffer without CHAPS, likely due to poor solubilization of the membrane proteins. Solubilization of the membrane protein fraction is critical to the μSPE device’s performance. If poorly solubilized, membrane proteins may appear as globular deposits on the surface of the μSPE bed as shown in Figure 2A. Consequently, cytosolic contaminants could become trapped within the deposits as well as lipid contaminants. In addition, extraction may be enabled by mixed mechanisms including the specific biotin/avidin interaction and non-specific interactions (i.e., hydrophobic/hydrophobic). Under the operation of these non-specific interactions, the ability to release isolated membrane proteins by reduction of the disulfide moiety may be compromised. To ensure proper solubilization of the membrane proteins, we added a solubilization buffer to the 4% CHAPS lysis solution. Processing the cell lysate with this solubilization buffer showed much more uniform membrane protein coverage on the micropillars with fluorescence visible along all sides of the micropillars as well as the floor of the bed (Figure 2C).
The specificity of the membrane protein’s extraction to the NeutrAvidin moieties permitted us to reduce the disulfide bond in the biotin linker and release extracted membrane proteins (and FITC-avidin molecules from the sandwich complex) from the μSPE bed. After release, the FITC-avidin’s fluorescence signal in the μSPE bed returned to the micropillar’s innate autofluorescence level (Figure 2D). This loss in fluorescence signal corresponded to an increase in the fluorescence signal of the chip effluent following DTT mediated release (Figure 3). The amount of FITC-avidin released into the effluent was used to determine the biotinylated protein recovery. After biotinylation, cells were labeled with FITC-avidin, washed, lysed, and analyzed with a fluorometer. From ~500,000 cells mL−1, 24.1 pmol mL−1 of biotinylated membrane proteins were obtained, which corresponded to ~3 × 107 biotinylated membrane protein molecules per cell.
Figure 3.
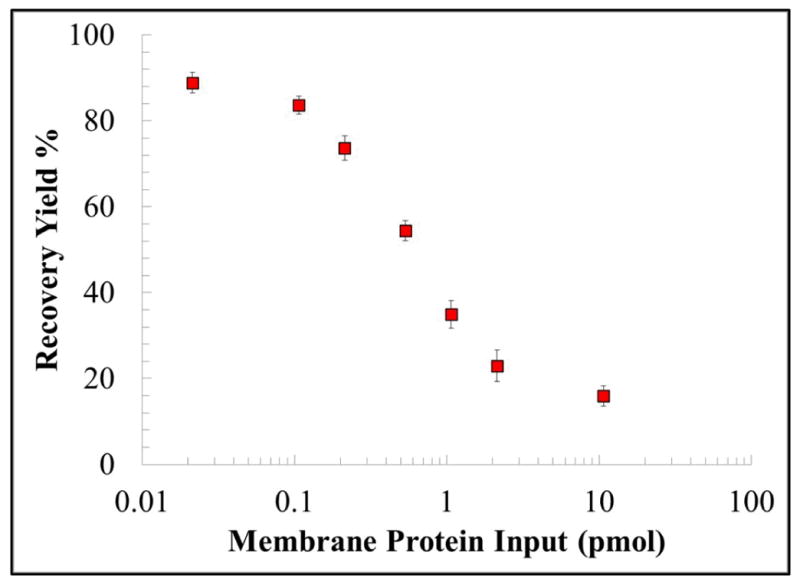
The recovery of biotinylated MCF-7 membrane proteins loaded onto the μSPE device. The total amount of protein (pmol) before and after μSPE purification was estimated from fluorescence data, which measured proteins that were biotinylated. Error bars in the graph represent standard deviations from three replicate runs.
With increasing amounts of biotinylated membrane proteins infused through the μSPE bed, we observed decreased recovery. The recovery was found to be 88.9 ±2.4% when 0.02 pmol of biotinylated membrane proteins were processed. The μSPE data compared favorably to recoveries using the detergent-based technique, which recovered ~50% of the membrane proteins. Also, recovery via the detergent-based technique is highly variable depending on the complexity and hydrophobicity of the membrane protein,17 whereas the efficiency of the μSPE bed is dependent on the efficiency of biotinylation rather than hydrophobicity, permitting efficient sampling of nearly all membrane proteins.27 When 10.7 pmol was processed, only 16.0 ±2.3% of protein was recovered, indicating that the μSPE bed was saturated with biotinylated membrane proteins (Figure 3). From Figure 3, the data suggested that the maximum amount of protein that could be loaded onto the μSPE bed was approximately 1.7 pmol. The theoretical load of immobilized NeutrAvidin, where NeutrAvidin is assumed to be a hard sphere with radii of 2.6 nm and is immobilized in a close packed hexagonal arrangement,46 the maximum load of NeutrAvidin was calculated to be 6.8 pmol. Assuming a 1:1 ratio between NeutrAvidin molecules and biotinylated membrane protein, the activated PMMA μSPE bed’s maximum recovery when saturated by biotinylated membrane proteins was approximately 25% relative to theoretical calculations. This observed difference may be attributed to inefficient UV/O3 activation of the PMMA μSPE bed, which we have demonstrated previously.47 Utilization of cyclic olefin copolymer (COC) as the fluidic substrate instead of PMMA should improve UV/O3 activation efficiency, generating a higher and more uniform carboxylic acid surface density leading to higher loads of NeutrAvidin for more efficient recovery of biotinylated material and a larger dynamic range.47 Furthermore, the device’s dynamic range can be extended by fabricating μSPE beds with smaller and more densely packed pillars, which should increase the available surface area and also decrease diffusional distances.37
Purity of membrane protein fractions obtained from the μSPE device and a bench-top detergent extraction method
We assessed the purity of the membrane proteins recovered from the μSPE device by Western blotting and staining for actin, a highly abundant cytosolic protein (~1 × 108 per cell)48 and the epithelial cell adhesion molecule (EpCAM), which is a highly expressed membrane protein found in MCF-7 cells (>400,000 per cell49). The presence of an actin band in the membrane protein fraction would indicate the presence of cytosolic impurities in the membrane protein fraction, while an EpCAM band in this same fraction would indicate successful isolation of membrane proteins. These results secured using the μSPE device were directly compared to a commercial, detergent-based extraction protocol.
Membrane and cytosolic protein fractions obtained by the detergent-based technique are shown in Figure 4. The Western blot clearly showed the presence of actin with intense bands in the total cell lysate (T) and the cytosolic fraction (C). But, there was also the presence of actin in the membrane protein fraction (M), suggesting relatively high cytosolic contamination when attempting to isolate membrane proteins using the detergent-based technique. The same Western blot analysis was also performed after processing an MCF-7 whole cell lysate using the μSPE bed. In this case, no actin band was observed in the Western blots for the membrane protein fraction. We subsequently stained for EpCAM and confirmed the presence of this membrane protein in the fraction isolated via μSPE. Considering the abundance of actin relative to EpCAM, the absence of an actin band clearly indicated highly pure membrane protein fractions isolated using μSPE.
Figure 4.
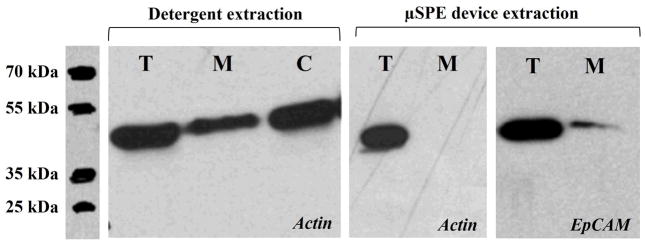
Actin Western blots demonstrating for detergent-based extraction and the μSPE extraction using actin as the model cytosolic protein. Also shown is the EpCAM Western blot of the membrane protein fraction eluted from the μSPE device to show that there were membrane proteins from the MCF-7 cell lysate in the fraction. For these Western blots, approximately 5 × 106 MCF-7 cells were lysed and taken to a total volume of 1.0 mL. This lysate was either directly loaded onto the gel (30 μL) for Western analysis or diluted ~1000-fold with 100 μL processed using the μSPE device. Due to the limited bed capacity of the μSPE device, the EpCAM band intensity was much weaker for the μSPE device compared to direct processing of the lysate.
Computational modeling of micropillar geometry and membrane protein extraction
To further increase the device’s dynamic range and the efficiency for recovering membrane proteins, we conducted computation modeling to guide future designs of the μSPE device. Specifically, we were interested in investigating how micropillar geometry and spacing may affect the efficiency of membrane protein extraction. The simulations were carried out using computational fluid dynamics (CFD) with COMSOL Multiphysics and a numerical analysis using Fick’s 2nd law governing diffusion. For CFD simulations, numerically tractable geometries composed of only a few staggered rows of micropillars (as opposed to the thousands occupying the μSPE bed), were tested. Three different geometries were evaluated: (I) Circular micropillars with the same dimensions as the μSPE device shown in Figure 1; (II) small, diamond micropillars (20 μm side length) spaced by 20 μm, which is similar to a device we have used previously;37,50 and (III) circular micropillars with analogous dimensions as geometry II (20 μm pillar radii, 20 μm pillar-to-pillar spacing). The steady-state velocity fields (shown in Figure S2) were comparable in all geometries, which is not surprising given the low Reynolds number for these devices. Additionally, the velocities between the pillars were nearly uniform regardless of pillar position, indicating uniform protein distribution throughout all μSPE beds. Average linear velocities through the beds were extracted from the CFD simulations to assess diffusion occurring on the length scale of the entire μSPE bed, which would be numerically intractable to model using CFD simulations alone.
For cases with diffusion, a protein with its initial position centered between two pillars and described by a Gaussian probability packet that spreads over time according to its diffusion constant was propagated over a time scale proportional to its velocity and effective path length through the μSPE bed. The normalized area of the Gaussian packet outside the bounds of the fluidic pathways (overlapping with a micropillar itself) was taken as probable extraction onto a micropillar’s surface. The results for several flow rates through geometries I–III are shown in Figure 5. Two sets of results are shown; the first simulation less accurately assumes that the protein travels in a straight path through the bed (ignoring micropillars altogether), while the second uses an effective bed length corrected by a factor (C), which included the distance required to circumnavigate a micropillar that was dependent on the micropillar’s shape. Further details are provided in the SI.
Figure 5.

Illustration of the path correction factor (C) for both circular and diamond shaped micropillars. The probability of protein-post interaction (Pi) for Geometries I–III, both with (solid black or white, where ) and without (solid grey, where C = 0) the path correction factor applied to the μSPE bed’s length.
As the flow rate increased, the probability of protein interaction with the pillar surface (Pi) decreased for all geometries. However, this dependency was less pronounced for Geometries II and III, which only had 20 μm pillar-to-pillar spacing and required less diffusion to occur for protein-pillar interaction. Comparison between Figures 3 and 5 indicated good agreement (an experimental recovery of 88.9 ±2.1% vs. 68.0% theoretically), especially because this diffusion model only considered a protein centered exactly between two pillars, a worst-case scenario requiring the largest transverse diffusion to occur for protein-pillar interaction, whereas well-solubilized proteins are homogeneously distributed throughout the interstitial space between the pillars. The model indicated that smaller pillar-to-pillar spacing, regardless of the micropillar shape, should increase membrane protein recovery.
Lastly, inclusion of the path correction factor increased the time for diffusion and improved the probability of extraction and more so for circular pillars compared to diamond-shaped pillars, which have a larger perimeter (C = π/2 ≈ 1.57) than diamond pillars ( ). However, this effect was minor when comparing Geometries II and III (an improvement of only 0.6% at 10 μL min−1 infusion) as the small pillar-to-pillar spacing induced higher recovery even when the path correction factor was ignored. Thus, future designs, especially those integrated with downstream protein separation and analysis, should employ μSPE beds with smaller, more densely packed pillars with smaller pillar-to-pillar spacing than that employed herein. This would also result in a higher surface area to improve recovery and the dynamic range.
Conclusion
A polymer microfluidic chip was designed, fabricated, and evaluated for the solid-phase extraction and purification of membrane proteins from whole cell lysates. The device contained 3,600 micropillars that provided a higher surface area for protein extraction compared to an open channel of the same dimensions, could be replicated from a mold master in a single step, and did not require complex post-processing steps for its operation, such as the addition of functionalized beads or the chemical formation of monolithic supports.
MCF-7 cells were biotinylated with a membrane impermeable reagent and then lysed. Whole cell lysates were processed through the μSPE device, where biotinylated membrane proteins were specifically selected using immobilized NeutrAvidin. Inclusion of a disulfide moiety within the biotinylation reagent framework permitted release of the extracted membrane proteins following reduction of the disulfide linkage. The μSPE assay produced significantly lower levels of cytosolic protein contamination compared to a commercially-available detergent method. Furthermore, we were able to recover ~89% of biotinylated membrane proteins from a whole cell lysate. Thus, we demonstrated efficient recovery of highly pure fractions of membrane proteins that can be released for downstream analysis. The μSPE device comprised a simplified workflow to allow for the generation of information regarding a small but important portion of the proteome that is typically difficult to analyze.18,51,52 We also provided several avenues to increase both the recovery and dynamic range of the device including polymer choice and computational simulations indicating the benefits of small circular pillars with reduced pillar-to-pillar spacing.
The results secured using this μSPE device for the extraction and purification of membrane proteins will provide an attractive approach that can be integrated to other devices for future studies directed toward determining potential therapeutic targets or selection agents for various cell types due to the higher purity membrane protein fractions isolated and the ability to process small numbers of cells. For example, we have previously demonstrated microfluidic cell isolation units for isolating extremely rare, circulating tumor cells from whole blood patient samples with high purity.53,54 These microfluidic devices can be coupled to the μSPE device detailed in this manuscript to isolate plasma membrane proteins from these rare cells for downstream multi-dimensional electrophoresis for protein separation,55,56 solid-phase proteolytic digestion57 and mass spectrometry for protein identification.58–60 We are currently developing an integrated system incorporating these previously described devices with the μSPE device for top-down proteomic analysis of membrane proteins from rare cells, such as circulating tumor cells.
Supplementary Material
Acknowledgments
The authors would like to thank the University Cancer Research Fund (UCRF) of the University of North Carolina and the National Institutes of Health (R21CA173279) for partial financial support of this work. KNB would like to thank the National Science Foundation for support through a pre-doctoral fellowship.
References
- 1.Wu CC, Yates JR., 3rd Nat Biotechnol. 2003;21:262–267. doi: 10.1038/nbt0303-262. [DOI] [PubMed] [Google Scholar]
- 2.Cooper EC, Jan LY. Proc Natl Acad Sci U S A. 1999;96:4759–4766. doi: 10.1073/pnas.96.9.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brekke OH, Sandlie I. Nat Rev Drug Discov. 2003;2:52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- 4.Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. BMC Biol. 2009;7:50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallin E, von Heijne G. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macher BA, Yen TY. Mol Biosyst. 2007;3:705–713. doi: 10.1039/b708581h. [DOI] [PubMed] [Google Scholar]
- 7.Zheng YZ, Foster LJ. J Proteomics. 2009;72:12–22. doi: 10.1016/j.jprot.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Tan S, Tan HT, Chung MC. Proteomics. 2008;8:3924–3932. doi: 10.1002/pmic.200800597. [DOI] [PubMed] [Google Scholar]
- 9.Castle JD. Curr Protoc Immunol. 2003;Chapter 8(Unit 8):1B. doi: 10.1002/0471142735.im0801bs56. [DOI] [PubMed] [Google Scholar]
- 10.Huber Lukas A, Pfaller K, Vietor I. Circul Res. 2003;92:962–968. doi: 10.1161/01.RES.0000071748.48338.25. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh D, Krokhin O, Antonovici M, Ens W, Standing KG, Beavis RC, Wilkins JA. J Proteome Res. 2004;3:841–850. doi: 10.1021/pr049937f. [DOI] [PubMed] [Google Scholar]
- 12.Lawson EL, Clifton JG, Huang F, Li X, Hixson DC, Josic D. Electrophoresis. 2006;27:2747–2758. doi: 10.1002/elps.200600059. [DOI] [PubMed] [Google Scholar]
- 13.Schindler J, Lewandrowski U, Sickmann A, Friauf E, Nothwang HG. Mol Cell Proteomics. 2006;5:390–400. doi: 10.1074/mcp.T500017-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Schindler J, Nothwang HG. Proteomics. 2006;6:5409–5417. doi: 10.1002/pmic.200600243. [DOI] [PubMed] [Google Scholar]
- 15.McCarthy FM, Burgess SC, van den Berg BH, Koter MD, Pharr GT. J Proteome Res. 2005;4:316–324. doi: 10.1021/pr049842d. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy FM, Cooksey AM, Burgess SC. Methods Mol Biol. 2009;528:110–118. doi: 10.1007/978-1-60327-310-7_7. [DOI] [PubMed] [Google Scholar]
- 17.Qoronfleh MW, Benton B, Ignacio R, Kaboord B. J Biomed Biotechnol. 2003;2003:249–255. doi: 10.1155/S1110724303209244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordwell SJ, Thingholm TE. Proteomics. 2010;10:611–627. doi: 10.1002/pmic.200900521. [DOI] [PubMed] [Google Scholar]
- 19.Vuong GL, Weiss SM, Kammer W, Priemer M, Vingron M, Nordheim A, Cahill MA. Electrophoresis. 2000;21:2594–2605. doi: 10.1002/1522-2683(20000701)21:13<2594::AID-ELPS2594>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 20.Rybak JN, Ettorre A, Kaissling B, Giavazzi R, Neri D, Elia G. Nat Methods. 2005;2:291–298. doi: 10.1038/nmeth745. [DOI] [PubMed] [Google Scholar]
- 21.Elia G. Proteomics. 2008;8:4012–4024. doi: 10.1002/pmic.200800097. [DOI] [PubMed] [Google Scholar]
- 22.Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R, Bibel M, Schiess R, Aebersold R, Watts JD. Nat Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson JM, Ackerman WEt, Tewari AK, Kniss DA, Vandre DD. Anal Biochem. 2009;387:87–94. doi: 10.1016/j.ab.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonson AB, Schnitzer JE. J Thromb Haemost. 2007;5:183–187. doi: 10.1111/j.1538-7836.2007.02551.x. [DOI] [PubMed] [Google Scholar]
- 25.Scheurer SB, Rybak JN, Roesli C, Brunisholz RA, Potthast F, Schlapbach R, Neri D, Elia G. Proteomics. 2005;5:2718–2728. doi: 10.1002/pmic.200401163. [DOI] [PubMed] [Google Scholar]
- 26.Tang X, Yi W, Munske GR, Adhikari DP, Zakharova NL, Bruce JE. J Proteome Res. 2007;6:724–734. doi: 10.1021/pr060480e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y, Zhang W, Kho Y, Zhao Y. Anal Chem. 2004;76:1817–1823. doi: 10.1021/ac0354037. [DOI] [PubMed] [Google Scholar]
- 28.Figeys DDA, Aebersold R. J Chromatogr A. 1997;763:295–306. doi: 10.1016/s0021-9673(96)00847-3. [DOI] [PubMed] [Google Scholar]
- 29.Jemere AB, Oleschuk RD, Ouchen F, Fajuyigbe F, Harrison DJ. Electrophoresis. 2002;23:3537–3544. doi: 10.1002/1522-2683(200210)23:20<3537::AID-ELPS3537>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 30.McClain MA, Culbertson CT, Jacobson SC, Allbritton NL, Sims CE, Ramsey JM. Analytical Chemistry. 2003;75:5646–5655. doi: 10.1021/ac0346510. [DOI] [PubMed] [Google Scholar]
- 31.Ramsey JD, Collins GE. Analytical Chemistry. 2005;77:6664–6670. doi: 10.1021/ac0507789. [DOI] [PubMed] [Google Scholar]
- 32.Hagan KA, Reedy CR, Bienvenue JM, Dewald AH, Landers JP. Analyst. 2011;136:1928–1937. doi: 10.1039/c0an00922a. [DOI] [PubMed] [Google Scholar]
- 33.Kutter JP, Jacobson SC, Ramsey JM. Journal of Microcolumn Separations. 2000;12:93–97. [Google Scholar]
- 34.Witek MA, Llopis SD, Wheatley A, McCarley RL, Soper SA. Nucleic Acids Res. 2006:34. doi: 10.1093/nar/gkl146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park DS, Hupert ML, Witek M, Guy J, Datta P, You BH, Soper SA, Murphy MC. Ieee. Proceedings of the Ieee Twentieth Annual International Conference on Micro Electro Mechanical Systems; 2007. pp. 95–98. [Google Scholar]
- 36.Park DS, Hupert ML, Witek MA, You BH, Datta P, Guy J, Lee JB, Soper SA, Nikitopoulos DE, Murphy MC. Biomed Microdev. 2008;10:21–33. doi: 10.1007/s10544-007-9106-y. [DOI] [PubMed] [Google Scholar]
- 37.Witek MA, Hupert ML, Park DSW, Fears K, Murphy MC, Soper SA. Anal Chem. 2008;80:3483–3491. doi: 10.1021/ac8002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brett PJ, Tiwana H, Feavers IM, Charalambous BM. J Biol Chem. 2002;277:20468–20476. doi: 10.1074/jbc.M200387200. [DOI] [PubMed] [Google Scholar]
- 39.Glover BP, McHenry CS. Cell. 2001;105:925–934. doi: 10.1016/s0092-8674(01)00400-7. [DOI] [PubMed] [Google Scholar]
- 40.Guo Y, Guettouche T, Fenna M, Boellmann F, Pratt WB, Toft DO, Smith DF, Voellmy R. J Biol Chem. 2001;276:45791–45799. doi: 10.1074/jbc.M105931200. [DOI] [PubMed] [Google Scholar]
- 41.Hiller Y, Gershoni JM, Bayer EA, Wilchek M. Biochem J. 1987;248:167–171. doi: 10.1042/bj2480167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unson MD, Newton GL, Arnold KF, Davis CE, Fahey RC. J Clin Microbiol. 1999;37:2153–2157. doi: 10.1128/jcm.37.7.2153-2157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wojciechowski M, Sundseth R, Moreno M, Henkens R. Clin Chem. 1999;45:1690–1693. [PubMed] [Google Scholar]
- 44.Witek MA, Hupert ML, Park DS, Fears K, Murphy MC, Soper SA. Anal Chem. 2008;80:3483–3491. doi: 10.1021/ac8002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarley RL, Vaidya B, Wei SY, Smith AF, Patel AB, Feng J, Murphy MC, Soper SA. Journal of the American Chemical Society. 2005;127:842–843. doi: 10.1021/ja0454135. [DOI] [PubMed] [Google Scholar]
- 46.Lahiri J, Isaacs L, Tien J, Whitesides GM. Anal Chem. 1999;71:777–790. doi: 10.1021/ac980959t. [DOI] [PubMed] [Google Scholar]
- 47.Jackson JM, Witek MA, Hupert ML, Brady C, Pullagurla S, Kamande J, Aufforth RD, Tignanelli CJ, Torphy RJ, Yeh JJ, Soper SA. Lab Chip. 2014 [Google Scholar]
- 48.Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. 4. Section 1.2 W. H. Freeman; New York: 2000. [Google Scholar]
- 49.Rao CG, Chianese D, Doyle GV, Miller MC, Russell T, Sanders RA, Jr, Terstappen LW. Int J Oncol. 2005;27:49–57. [PubMed] [Google Scholar]
- 50.Witek MA, Llopis SD, Wheatley A, McCarley RL, Soper SA. Nucleic Acids Res. 2006;34:e74. doi: 10.1093/nar/gkl146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gauthier DJ, Lazure C. Expert Rev Proteomics. 2008;5:603–617. doi: 10.1586/14789450.5.4.603. [DOI] [PubMed] [Google Scholar]
- 52.Josic D, Clifton JG. Proteomics. 2007;7:3010–1029. doi: 10.1002/pmic.200700139. [DOI] [PubMed] [Google Scholar]
- 53.Jackson JM, Witek MA, Hupert ML, Brady C, Pullagurla S, Kamande J, Aufforth RD, Tignanelli CJ, Torphy RJ, Yeh JJ, Soper SA. Lab on a chip. 2014;14:106–117. doi: 10.1039/c3lc50618e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamande JW, Hupert ML, Witek MA, Wang H, Torphy RJ, Dharmasiri U, Njoroge SK, Jackson JM, Aufforth RD, Snavely A, Yeh JJ, Soper SA. Anal Chem. 2013;85:9092–9100. doi: 10.1021/ac401720k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shadpour H, Soper SA. Anal Chem. 2006;78:3519–3527. doi: 10.1021/ac0600398. [DOI] [PubMed] [Google Scholar]
- 56.Osiri JK, Shadpour H, Park S, Snowden BC, Chen ZY, Soper SA. Electrophoresis. 2008;29:4984–4992. doi: 10.1002/elps.200800496. [DOI] [PubMed] [Google Scholar]
- 57.Lee J, Soper SA, Murray KK. Analyst. 2009;134:2426–2433. doi: 10.1039/b916556h. [DOI] [PubMed] [Google Scholar]
- 58.Musyimi HK, Narcisse DA, Zhang X, Stryjewski W, Soper SA, Murray KK. Anal Chem. 2004;76:5968–5973. doi: 10.1021/ac0489723. [DOI] [PubMed] [Google Scholar]
- 59.Musyimi HK, Guy J, Narcisse DA, Soper SA, Murray KK. Electrophoresis. 2005;26:4703–4710. doi: 10.1002/elps.200500317. [DOI] [PubMed] [Google Scholar]
- 60.Musyimi HK, Soper SA, Murray KK. On-line and Off-line MALDI from a Microfluidic Device. 2008 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.