

Highlights
-
•
l-Amino acid oxidase/monooxygenase (l-AAO/MOG) from Pseudomonas sp. AIU813 was studied.
-
•
l-Lysine oxidase activity of l-AAO/MOG increased with p-chloromercuribenzoate concentration.
-
•
Variant l-AAO/MOG C254I showed 5-times higher oxidase and decreased monooxygenase activities.
-
•
3D structure of l-AAO/MOG provided a structural basis for its biochemical characteristics.
Abbreviations: 4-AA, 4-aminoantipyrine; amid, amide hydrolase gene; CHCA, α-Cyano-4-hydroxycinnamic acid; FMOs, flavin monooxygenases; l-AAO, l-amino acid oxidase; l-AAO/MOG, l-amino acid oxidase/monooxygenase; laao/mog, l-amino acid oxidase/monooxygenase gene; LB, Luria–Bertani; LGOX, l-glutamate oxidase; MAO, flavin-containing monoamine oxidase; PAO, l-phenylalanine oxidase; p-CMB, p-chloromercuribenzoate; TFA, trifluoroacetic acid; TMO, l-tryptophan 2-monooxygenase; TOOS, N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline
Keywords: l-Amino acid oxidase/monooxygenase, Saturation mutagenesis, Crystallography, Flavin-containing monoamine oxidase family, Flavin monooxygenases
Abstract
In this study, it was shown for the first time that l-amino acid oxidase of Pseudomonas sp. AIU813, renamed as l-amino acid oxidase/monooxygenase (l-AAO/MOG), exhibits l-lysine 2-monooxygenase as well as oxidase activity. l-Lysine oxidase activity of l-AAO/MOG was increased in a p-chloromercuribenzoate (p-CMB) concentration-dependent manner to a final level that was five fold higher than that of the non-treated enzyme. In order to explain the effects of modification by the sulfhydryl reagent, saturation mutagenesis studies were carried out on five cysteine residues, and we succeeded in identifying l-AAO/MOG C254I mutant enzyme, which showed five-times higher specific activity of oxidase activity than that of wild type. The monooxygenase activity shown by the C254I variant was decreased significantly. Moreover, we also determined a high-resolution three-dimensional structure of l-AAO/MOG to provide a structural basis for its biochemical characteristics. The key residue for the activity conversion of l-AAO/MOG, Cys-254, is located near the aromatic cage (Trp-418, Phe-473, and Trp-516). Although the location of Cys-254 indicates that it is not directly involved in the substrate binding, the chemical modification by p-CMB or C254I mutation would have a significant impact on the substrate binding via the side chain of Trp-516. It is suggested that a slight difference of the binding position of a substrate can dictate the activity of this type of enzyme as oxidase or monooxygenase.
1. Introduction
l-Amino acid oxidase (EC 1.4.3.2, l-AAO) is a flavoprotein that catalyzes oxidative deamination of l-amino acid to produce α-keto acid, ammonia and hydrogen peroxide. l-AAOs were found in fungi [1], algae [2] and bacteria [3], and snake venoms [4]. l-AAOs from snake venoms have been extensively investigated, and the crystal structures of several of them, including l-AAO from Calloselasma rhodostoma, have been reported [5]. The crystal structure of some bacterial enzymes, l-AAO from Rhodococcus opacus [6], l-glutamate oxidase (EC 1.4.3.11, LGOX) from Streptomyces sp. X-119-6 [7], and l-phenylalanine oxidase (EC 1.13.12.9, PAO) from Pseudomonas sp. P-501 [8], have been also available. The crystal structure of tryptophan-2-monooxygenase (EC 1.13.12.3, TMO) from Pseudomonas savastanoi has recently been reported [9]. All of these l-AAOs are included in the flavin-containing monoamine oxidase (EC 1.4.3.4, MAO) family based on their structural similarity [10].
An l-AAO from Pseudomonas sp. AIU 813 was found to be specific to l-lysine, l-ornithine and l-arginine [11]. In the microbial l-AAOs, which are specific to these basic amino acids, the l-AAO from Trichoderma viride [12] and Trichoderma sp. [13] were investigated. The enzyme from Pseudomonas sp. AIU 813 catalyzes oxidative deamination of the α-group of only basic amino acids, and the reaction velocities toward each substrate were affected by the pH [11]. These results indicate that the enzyme is different from the above l-AAOs in the enzymatic properties.
Here, it was shown for the first time that l-AAO from Pseudomonas sp. AIU 813 exhibits l-lysine 2-monooxygenase as well as oxidase activity. Thus the enzyme was renamed as l-amino acid oxidase/monooxygenase (l-AAO/MOG) in this report. We focused on the enhancement of l-lysine oxidase activity of l-AAO/MOG in the presence of p-chloromercuribenzoate (p-CMB) [11]. The specific oxidase activity increased in a p-CMB concentration-dependent manner to a final level, and oxidase activity of the enzyme modified with p-CMB was five fold higher than that of the non-treated enzyme. Yamauchi et al. have demonstrated that when sulfhydryl group of l-lysine monooxygenase (EC 1.13.12.2) from Pseudomonas fluorescens Pf0-1 was modified with sulfhydryl reagents such as p-CMB, N-ethylmaleimide or HgCl2, the l-lysine monooxygenase activity was inhibited and the l-lysine oxidase activity was induced [14]. In this study, we investigated cloning of the gene, and the effect of changing cysteine residues, which are thought to be involved in the activity of this enzyme, to other amino acid residues in order to increase the oxidase activity of the enzyme. Moreover, we have determined the crystal structure of l-AAO/MOG and could locate the related cysteine residue in the active site, providing a structural basis for the conversion between oxidase and monooxygenase activities.
2. Materials and methods
2.1. Materials
l-Lysine, l-arginine, l-ornithine, and other amino acids were purchased from Wako Pure Chemical Industries (Osaka, Japan). Restriction endonucleases and the ligation reaction mixture were obtained from Toyobo (Osaka, Japan) and Takara Bio (Shiga, Japan). All other chemicals were purchased from Kanto Kagaku Co. (Tokyo, Japan), Nacalai Tesque Inc. (Kyoto, Japan), Sigma–Aldrich Co. (MO, USA), or Tokyo Kasei Kogyo (Tokyo, Japan), unless otherwise stated, and were of the highest commercially available grade.
2.2. Bacterial strains, plasmids, and culture conditions
The Pseudomonas sp. AIU 813 strain was used for preparation of genomic DNA using Wizard Genomic DNA Purification Kit (Promega, Madison, WI). The strain was grown aerobically at 30 °C in tryptone/glucose/yeast (TGY) medium (0.5% peptone, 0.5% yeast extract, 0.1% KH2PO4, 0.1% d-glucose) for 12 h. Escherichia coli BL21(DE3) was obtained from Merck KGaA (Darmstadt, Germany) and was grown aerobically at 37 °C in Luria–Bertani (LB) medium; ampicillin (100 μg/mL) was added if the strain harboured a plasmid carrying the ampicillin-resistance gene.
2.3. Gene isolation and cloning of l-AAO/MOG
Chromosomal DNA was isolated using a cell culture preparation of genomic DNA from Pseudomonas sp. AIU 813 and was used to amplify a DNA fragment containing the full-length coding region of the l-AAO/MOG gene (laao/mog) as template, using primers P1 and P2; the sequences of these oligonucleotides are listed in Table S1. The product was purified, cloned into pT7 Blue T, and sequenced using an ABI PRISM 310 genetic analyser (PE Applied Biosystems, CA, USA). Based on the partial sequence obtained, primers P3 and P4 were designed to amplify the full-length laao/mog from the genomic DNA of Pseudomonas sp. AIU 813. The product obtained was digested with NdeI and XhoI and then ligated into NdeI/XhoI-digested pET15b, generating pET15b-laao/mog.
2.4. Isolation of recombinant l-AAO/MOG
Recombinant l-AAO/MOG was prepared from E. coli BL21(DE3) harbouring pET15b-laao/mog, which was disrupted by sonication. The cell-free extract was dialyzed against 20 mM potassium phosphate buffer (pH 8.0) containing 300 mM KCl and 5 mM imidazole, and the protein solution was purified using the Profinia™ Protein Purification System (Bio-Rad, CA, USA) according to the manufacturer’s instructions using a Profinia™ native IMAC cartridge for the His-tagged proteins. The purified enzyme was finally dialyzed and stored in 20 mM potassium phosphate buffer (pH 7.0).
2.5. Identification of reaction products from l-amino acids
A reaction mixture (2.0 mL) containing 1.04 U l-AAO/MOG, 0.2 M each l-amino acid in 40 mM potassium phosphate buffer (pH 7.0) was incubated at 30 °C for 24 h, and the reaction was stopped by boiling for 5 min. The reaction mixture was then filtrated and applied to a TSKgel SP-2SW column (4.6 × 250 nm, Tosoh Corp., Tokyo, Japan) to separate the reaction products from each l-amino acid. The eluate containing each product was concentrated by a SpeedVac concentrator and applied to an HCT Ultra mass spectrometric instrument (Bruker Daltonics GmbH, Bremen, Germany). Electrospray ionization (ESI-) mass spectrometry (MS) was performed according to the method of Isobe et al. [15].
2.6. Detection of cysteine residues modified with p-CMB in l-AAO/MOG
The purified l-AAO/MOG (10 ng) treated with the following two independent conditions: (i) no treatment; and (ii) modification with p-CMB, and then denatured with an 8 M urea solution (10 μL). Each reaction mixture was diluted with a 50 mM ammonium bicarbonate solution (80 μL), reacted with trypsin (350 ng, Promega, Madison, WI, USA), and incubated at 37 °C overnight. The tryptic digested samples were desalted and concentrated to approximately 8 μL by a ZipTip C18 (Millipore, Billerica, MA, USA). The concentrated sample (0.5 μL) was mixed with 0.5 μL of an α-Cyano-4-hydroxycinnamic acid (CHCA) solution (5 mg/mL in 50% acetonitrile containing 0.1% trifluoroacetic acid (TFA)) on a MALDI target plate and analyzed after air-drying. The mass spectra were collected by both reflectron positive ion mode and linear positive ion mode with an AXIMA-CFR™-plus MALDI-TOF MS instrument (Shimadzu/Kratos, Manchester, UK).
2.7. Creation of saturation site-directed mutants
Each round of saturation site-directed mutagenesis was carried out using oligonucleotide primers P5, P6, P7, P8 and P9 (Table S1). The target amino acid positions (Cys-254, 280, 331, 342, and 413) were coded by NNS. The reactions were performed as described in the Stratagene Quik Change™ site-directed mutagenesis protocol [16]. After the reaction, 10 U of DpnI was added to the products and incubated at 37 °C for 2 h to digest the template DNA. Approximately 10 ng of mutant library plasmid DNA was used to transform competent E. coli BL21(DE3) cells following a standard protocol [17]. A 200 μL aliquot from each E. coli BL21(DE3) transformation was plated on agar plates and incubated overnight at 37 °C.
2.8. Library screening
The colonies mutated in each cysteine residue were picked from the agar plates and placed into 200 μL LB broth supplemented with ampicillin (100 μg/mL) for selection, in 96-well plates, by using a Colony Picker PM-1 robot (Microtec Co. Ltd., Chiba, Japan). Cells were grown at 37 °C and 200 rpm, and 0.5 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) was used for induction when the cells reached an A600 of 0.5–0.7. After incubation at 30 °C and 200 rpm for 12 h, the cells were collected by centrifugation at 3220g for 30 min, and disrupted by sonication. The crude cell extracts were obtained by centrifugation at 3220g for 30 min, and the supernatant was used for the activity assay during the primary screening. Supernatant aliquots (20 μL) were transferred into new 96-well plates for screening. Each of these lysate aliquots was incubated with 40 μmol l-lysine in a color reagent consisting of 0.6 μmol 4-aminoantipyrine (4-AA), 1.94 μmol N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3-methylaniline (TOOS) sodium salt dehydrate, 6.7 units horseradish peroxidase, and 0.1 mmol potassium phosphate, pH 7.0; the mutants possessing high activity were selected. In the next round of screening, the same reaction mixture was added to the native and p-CMB-treated lysate, respectively. Formation of hydrogen peroxide was spectrophotometrically analyzed at 30 °C for 10 min by measuring the absorbance at 555 nm. Spectrophotometric measurements were performed with a UV/visual spectrophotometer U-3210 system (Hitachi High-Technologies Co., Tokyo, Japan).
2.9. Saturation mutagenesis of Cys-254
Site-saturation mutagenesis experiments of Cys-254 were carried out with the same PCR procedure as described in “Creation of saturation site-directed mutants”. Four of the 50 μL of purified PCR product was directly transformed into 200 μL of E. coli BL21(DE3) competent cells. The transformant was incubated overnight in 5 mL LB medium containing 100 μg/mL ampicillin. The randomized plasmid library was isolated by mini-prep and used for DNA sequencing. The mutations were confirmed using an ABI PRISM 310 genetic analyser.
2.10. Screening, purification, and expression of 5-aminopentaneamide amide hydrolase
Strains exhibiting 5-aminopentaneamide amide hydrolase activity were screened from our collection that had been originally isolated from soil. An isolated strain was first incubated in a test tube containing 3 mL of a valeramide medium (pH 7.0) consisting of 0.2% K2HPO4, 0.1% NaCl, 0.02% MgSO4·H2O, 0.001% CaCl2, 1.0% glycerol, 0.1% trace element solution, 0.1% vitamin solution, and 0.5% valeramide, at 30 °C for 48 h with shaking [18].
The purification was carried out in 20 mM phosphate buffer (pH 7.0) at 4 °C, unless otherwise stated. Bacterial strains were grown in 20 L medium. After 24 h incubation, the culture was centrifuged at 10,000g for 15 min. Pellets were suspended in 20 mM phosphate buffer (pH 7.0), and the cell suspension was sonicated using a Kubota Insonator 201M sonicator (Kubota Corp., Tokyo, Japan) for 20 min, on an ice bath. The lysed suspension was then ultracentrifuged for 60 min at 105,000g. A clear supernatant was directly applied onto a Q Sepharose column (GE Healthcare, CT, USA). The Q Sepharose fractions with amide hydrolase activity were then further purified through an Octyl Sepharose column (GE Healthcare). The Octyl Sepharose fractions were applied onto a RESOURCE PHE column (GE Healthcare). After dialysis with 20 mM phosphate buffer, pH 7.0 at 4 °C, the RESOURCE PHE fractions were run through a MonoQ5/5 anion exchange column (GE Healthcare). The amide hydrolase activity of these purified fractions was assayed at room temperature by measuring the ammonia liberated from valeramide degradation. The ammonia concentrations were determined by the indophenol reaction using an Ammonia-Test-Wako kit (Wako Pure Chemical Ind.).
The amide hydrolase gene (amid) of Pseudomonas sp. was amplified using the primers P10 and P11, with NdeI and BamHI sites added to the forward and reverse primers, respectively (Table S1). The amide hydrolase expression plasmid was constructed by cloning of this NdeI/BamHI-digested fragment into the corresponding site of pET23a. pET23a-amid was then transformed into the host E. coli BL21(DE3), and the expressed protein was purified as described above.
2.11. Enzyme assays of oxidase and monooxygenase activities
The l-AAO/MOG activities in the presence of l-lysine, l-arginine, or l-ornithine as substrates were measured using color development method with 4-AA, TOOS and horseradish peroxidase [11]. One unit of enzyme activity was defined as the amount of enzyme catalysing the formation of 1 μmol of hydrogen peroxide per minute. Moreover, l-lysine oxidase and monooxygenase activities were measured by coupling with the purified recombinant amide hydrolase. The assay mixtures contained 20 mM potassium phosphate buffer, pH 7.0, 40 mM l-amino acid, 10 U catalase, 10 U amide hydrolase and an appropriate amount of enzyme. Reactions were carried out at 30 °C for different time intervals, stopped by heat treatment at 100 °C for 2 min, and then assessed using an Ammonia-Test-Wako kit. The amount of ammonia produced by l-lysine monooxygenase reaction was calculated from that of the reaction without amide hydrolase (l-lysine oxidase alone) and with amide hydrolase (both l-lysine oxidase and monooxygenase). Protein concentration was determined using BSA as the standard [19], with 1 unit being defined as the amount of enzyme producing 1 μmol of ammonia per minute.
2.12. Crystallography
Wild-type l-AAO/MOG protein for crystallization was expressed using the E. coli BL21(DE3) harbouring pET15b-laao/mog, as described in the “Gene isolation and cloning of l-AAO/MOG”. The native protein was purified by column chromatography using Ni-NTA superflow (QIAGEN, Hilden, Germany) with 10–500 mM imidazole. After desalting, the protein was further purified on Mono Q 10/100 GL and HiLoad 16/600 Superdex 200 pg columns (GE Healthcare, Buckinghamshire, England). Selenomethionine (Se-Met) substituted l-AAO/MOG protein was expressed in E. coli BL21 CodonPlus (DE3)-RIL-X (Novagen, Madison, WI) in Se-Met core medium (Wako Pure Chemical Ind.) supplemented with 10 g/L d-glucose, 250 mg/L MgSO4·7H2O, 4.2 mg/L FeSO4·7H2O, 10 mL/L KAO & Michaylak vitamin solution (Sigma–Aldrich, St Louis, MO), 100 μg/mL kanamycin, and 50 mL/L seleno-l-methionine at 20 °C. The Se-Met substituted l-AAO/MOG was purified with the same procedure for the native protein. Crystals of native l-AAO/MOG were obtained by sitting-drop vapor diffusion at 20 °C by mixing 1 μL of protein solution, consisting of 20 mg/mL l-AAO/MOG in 20 mM HEPES–NaOH (pH 7.6), and 1 μL reservoir solution, consisting of 8% polyethylene glycol (PEG) 4000 and 0.1 M Na-acetate (pH 4.6). The Se-Met substituted crystals were obtained using reservoir solution consisting of 16% PEG 3350, 0.02 M citric acid, and 0.08 M Bis-Tris propane–HCl (pH 8.8). Crystals were cryoprotected in the reservoir solutions supplemented with 20% (w/v) glycerol and were flash-cooled at 100 K in a stream of nitrogen gas. X-ray diffraction data were collected at the BL1A and NW12A beamline at the Photon Factory and Photon Factory-AR, High Energy Accelerator Research Organization, Tsukuba, Japan. Diffraction data were processed using HKL2000 [20]. Initial phases were calculated using Autosol program of PHENIX [21]. An initial model was built using BUCCANEER [22] and ARP/wARP [23]. Manual model rebuilding and refinement were achieved using Coot [24] and Refmac5 [25]. The quality of the final structure was analyzed using MolProbity [26]. Four outliers in the Ramachandran plot are all in the B chain (Table 1). All of the outlier residues (Asp335, Asp406, Asp466, and Pro467) are located near the protein surface, and the electron density was somewhat ambiguous in these regions. Molecular graphic figures were prepared using PyMol (DeLano Scientific, Palo Alto, CA). Phylogenetic tree was drawn using the multiple alignment results of the ClustalW2 server and FigTree version 1.4.0.
Table 1.
Data collection and refinement statistics.
| A. data collection | Native l-AAO/MOG | Se-met l-AAO/MOG |
|---|---|---|
| Beamline | BL1A | NW12A |
| Wavelength (Å) | 1.000 | 0.97921 |
| Space group | P21212 | I222 |
| Unit cell (Å) | a = 118.9 | a = 74.2 |
| b = 141.4 | b = 101.1 | |
| c = 76.0 | c = 151.7 | |
| Resolution (Å)a | 50.00–1.90 | 50.00–2.20 |
| (1.93–1.90) | (2.24–2.20) | |
| Total reflections | 619,854 | 420,419 |
| Unique reflections | 98,486 (4930) | 55,946 (2810) |
| Completeness (%)a | 97.5 (99.9) | 99.9 (100.0) |
| Redundancya | 6.3 (6.2) | 7.5 (7.4) |
| Mean I/σ (I)a | 24.7 (3.2) | 27.0 (3.0) |
| Rsym (%)a | 8.2 (50.7) | 9.7 (59.2) |
| B. Refinement | ||
| Resolution (Å) | 30.4–1.9 | |
| No. of reflections | 93,180 | |
| R-factor/Rfree (%) | 20.9/25.3 | |
| No. of atoms | 8987 | |
| No. of solvents | 405 (Water), 2 (FAD) | |
| RMSD from ideal values | ||
| Bond lengths (Å) | 0.021 | |
| Bond angles (°) | 2.09 | |
| Average B-factor (Å2) | ||
| Protein (chain A/B) | 28.7/29.3 | |
| FAD (chain A/B) | 18.1/19.2 | |
| Water | 30.3 | |
| Ramachandran Plot (%) | ||
| Favored (chain A/B) | 98.3/96.8 | |
| Allowed (chain A/B) | 1.7/2.4 | |
| Outlier (chain A/B) | 0.0/0.7 | |
Values for highest resolution shell are given in parentheses.
2.13. Accession numbers
The nucleotide sequence for Pseudomonas sp. AIC 813 l-AAO/MOG and Pseudomonas sp. 5-aminopentaneamide amide hydrolase genes have been deposited in DDBJ/EMBL/GenBank under accession numbers Genbank: AB830473 and Genbank: AB830474. The atomic coordinates and structure factors (PDB: 3WE0) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
3. Results
3.1. Nucleotide sequence and deduced primary structure
We purified an l-AAO/MOG from Pseudomonas sp. AIU 813; the N-terminal sequence of the intact protein was found to be MNNNRHPADGKKPI [11]. On the basis of this information, we cloned the gene encoding the entire Pseudomonas sp. AIU 813 l-AAO/MOG. The open reading frame encodes a polypeptide of 560 amino acid residues, as expected, with a predicted molecular mass of 62 kDa. The amino acid sequence of l-AAO/MOG from Pseudomonas sp. AIU813 had high identity to putative l-lysine 2-monooxygenase of Pseudomonas putida ATCC 12633 (BAG54787.1, 94%), and exhibits 30.1% identity to TMO from P. savastanoi (EFI00478.1). All these sequences showed a highly conserved dinucleotide-binding motif GxGxxG.
3.2. Identification of products from l-amino acids
Each l-amino acid was incubated with 1.04 U of l-AAO/MOG, and the reaction products were separated by HPLC with a TSKgel SP-2SW column. Then, MS of each reaction product was analyzed to demonstrate the generation of corresponding keto-acid and amide from l-amino acids. In the case of l-lysine oxidation, it was confirmed that l-lysine was eluted at 8.7 min from a TSKgel SP-2SW column. The other peaks eluted at 3.8 min and 11.5 min were analyzed by MS. The molecular ion peak of the products eluted at 3.8 and 11.5 min was observed with m/z(+) values identical to that of 6-amino-2-oxohexanoic acid and 5-aminopentanamide, respectively. In HPLC analysis of oxidation products of l-ornithine, l-ornithine was eluted at 8.5 min and one product was eluted at 10.8 min, which was same as 4-aminobutanamide (amide from l-ornithine). The molecular mass of the other oxidation product, which was eluted at 3.1 min, was coincident with the theoretical value of 5-amino 2-oxopentanoic acid (α-keto acid from l-ornithine). Two types of oxidation products of l-arginine were also confirmed by MS analyses (Table 2). These results indicate that l-AAO from Pseudomonas sp. AIU 812 exhibits l-amino acid monooxygenase activity as well as oxidase activity (Fig. 1). Therefore, we changed the name of this enzyme from l-AAO to l-AAO/MOG in this report.
Table 2.
Identification of oxidation products from l-amino acids.
| l-Amino acid | Elution time (min) |
Reaction product | |
|---|---|---|---|
| l-Amino acid | Products (oxidation or monooxygenation) | Molecular mass (product name) | |
| l-Lysine | 8.7 | 3.8 (Oxidation) | 145.1 (6-Amino-2-oxoheanoic acid) |
| 11.5 (Monooxygenation) | 116.1 (5-Aminopentanamide) | ||
| l-Ornithine | 8.5 | 3.1 (Oxidation) | 131.1 (5-Amino-2-oxopentanoic acid) |
| 10.8 (Monooxygenation) | NDa (4-aminobutanamide) | ||
| l-Arginine | 10.8 | 3.7 (Oxidation) | 173.1 (5-Guanidino-2-oxopentanoic acid) |
| 18.7 (Monooxygenation) | 144.1 (4-Guanidinobutanamide) | ||
ND, not determine. MS analysis of l-ornithine monooxygenation product was not carried out, because the product was eluted at the same time as 4-aminobutanamide standard by HPLC with a TSKgel SP-2SW column.
Fig. 1.
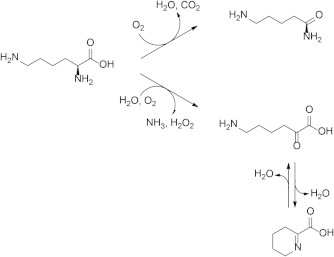
Reaction scheme of l-lysine oxidative deamination (oxidase) and oxidative decarboxylation (monooxygenase).
3.3. Detection of cysteine residues modified with p-CMB
The tryptic peptides derived from l-AAO/MOG were analyzed by MALDI-TOF MS. The specific peptide peaks were observed at m/z 1250.01, 1963.11, 2917.37, 2978.37 and 3216.06 in untreated l-AAO/MOG (Table S2(i)). These values fit well with the theoretical m/z values (m/z 1249.56, 1962.92, 2916.42, 2979.39 and 3,215.54) calculated from the amino acid sequence of l-AAO/MOG. In contrast, the peak of m/z 1570.61, 2283.91, 3238.42, 3300.78, and 3858.87 were observed in l-AAO/MOG modified p-CMB (Table S2(ii)), and mass shift of approximately m/z 320 is appeared in peptide including each cysteine residue. These results show that the six cysteine residues (Cys-254, 280, 321, 331, 342, and 413) in the enzyme were modified with p-CMB.
3.4. Isolation of l-amino acid oxidase unaffected by p-CMB
Five cysteine residues (Cys-254, 280, 331, 342, and 413) in l-AAO/MOG are conserved in the amino acid sequence alignment with other putative l-lysine 2-monooxygenases (Fig. S1). In order to confirm the effects of modification of the sulfhydryl group of cysteine residues, saturation mutagenesis studies were carried out on the 5 cysteine residues. Libraries of 4500 clones were obtained from each saturation mutagenesis cycle, using laao/mog from Pseudomonas sp. AIU 813 as a template. The screening strategy was adapted for high throughput by using a color development method. The assay was performed in 96-well microplates, containing the substrate (l-lysine), mutant enzyme extracts, and the color reagent assay mixture. To validate the assay, a test plate containing the parental extract in different wells was prepared. The activities of clones were evaluated and 225 mutants from the first screening library were selected due to their increased activity as compared to that of the recombinant wild type.
During the second screening, strains that showed no effect of p-CMB on their l-lysine oxidase activities were picked from the 225 mutants; of these, only 1 mutant showed significant improvement in activity. The sequencing of this mutant plasmid revealed the presence of 1 mutation in the nucleotide sequence; the mutation changed a Cys-254 to an Ile. The mutated plasmid was again transferred into E. coli BL21(DE3) and selected cells were subjected to further studies.
3.5. Effects of sulfhydryl reagents on l-lysine oxidation
The oxidase activities of recombinant wild-type enzyme (l-AAO/MOG) and the C254I mutant enzyme (l-AAO/MOG C254I) were purified from E. coli BL21(DE3) harbouring pET15b-laao/mog and pET15b-laao/mog C254I (l-AAO/MOG C254I gene) as a carboxy-terminal His6-tagged protein, and the protein was purified to near homogeneity in SDS–PAGE.
The substrate specificity of recombinant l-AAO/MOG and l-AAO/MOG C254I were determined spectrophotometrically by using 40 mM l-amino acids and d-amino acids as substrates. l-AAO/MOG and l-AAO/MOG C254I also oxidized l-lysine, l-arginine and l-ornithine but other l-amino acids and d-amino acids were not, being consistent with the substrate specificity of the native l-AAO/MOG purified from Pseudomonas sp. AIU 813 [11]. The specific activities of the recombinant wild type enzyme for l-lysine, l-ornithine and l-arginine were 0.532, 0.146, and 0.108 U/mg, respectively. These enzymes were examined as a function of p-CMB concentration (Fig. 2). As the amount of added p-CMB increased, the oxidase activity of the recombinant l-AAO/MOG increased. In contrast, l-lysine oxidase activity (2.5 U/mg) of the l-AAO/MOG C254I was 5 times higher than that of the recombinant wild type in the absence of p-CMB and was not affected by addition of p-CMB.
Fig. 2.
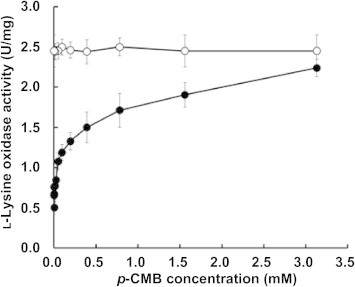
Effects of sulfhydryl reagents on l-lysine oxidation. The oxidase activity followed the hydrogen peroxide was determined as described under experimental procedures. The enzyme, preincubated with varying amounts of p-CMB, was added to the assay mixture containing 40 mM l-lysine and 20 mM potassium phosphate buffer, pH 7.0 (n = 3). Black circles, wild-type l-AAO/MOG; white circles, l-AAO/MOG C254I.
3.6. Saturation mutagenesis of residue Cys-254
The results of saturation mutagenesis are summarized graphically in Fig. 3. Each variant was expressed in E. coli BL21(DE3), and the enzymatic activities of the purified enzymes were investigated by measuring oxidase and monooxygenase activities, at pH 7.0. In order to measure monooxygenase activity, 5-aminopentaneamide amide hydrolase was purified (Table S3), and the gene was cloned from Pseudomonas sp. Because the recombinant enzyme used in the coupling reaction results in the production of 5-aminopentanoate and ammonia, l-lysine oxidase and monooxygenase activities were measured by the detection of ammonia. Fifteen variants showed higher l-lysine oxidase activity than the recombinant wild-type enzyme. The highest l-lysine oxidase activity was observed with the C254I variant (Fig. 3A). Many variants showed lower l-lysine monooxygenase activity than the recombinant wild-type enzyme, and the monooxygenase activity of l-AAO/MOG C254I mutant decreased remarkably (Fig. 3B).
Fig. 3.
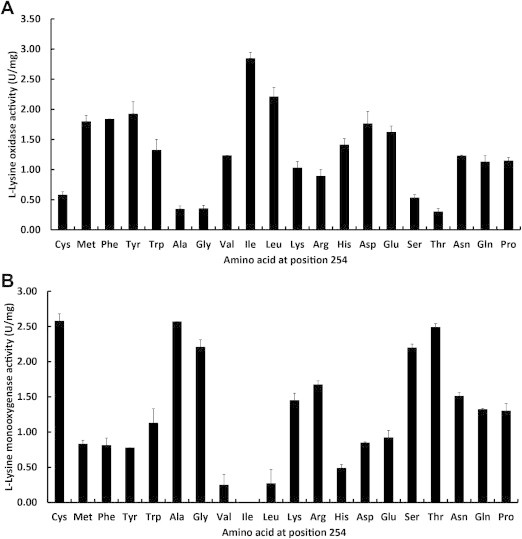
Saturation mutagenesis of residue 254. The oxidase and monooxygenase activities for l-lysine were determined as described under experimental procedures (n = 3). (A) l-Lysine oxidase activities; (B) l-Lysine monooxygenase activities.
3.7. Crystal structure
The crystal structure of l-AAO/MOG was determined at 1.9 Å resolution and refined to an R factor of 20.9% (Rfree = 25.3%) (Table 1). The native crystal of l-AAO/MOG belongs to the space group P21212 and contains two molecules in the asymmetric unit (Fig. 4A). l-AAO/MOG consists of three domains (Fig. 4B), and a topology diagram is shown in Fig. S2. FAD binding domain (blue; residues 38–83, 264–358, 475–538) is made up of 8 α helices and 7 β strands. FAD was strongly bound to the protein during the purification steps, and the electron density for the two FAD molecules in the asymmetric units was clearly observed without additional soaking into the crystals. Average B-factors of the FAD molecules were lower than the protein and the water molecules in the structure (Table 1). Substrate binding domain (red; residues 12–37, 84–147, 258–263, 359–474, 539–560) consists of 12 β strands and 4 α helices. Within this domain, a region connecting β16 and α15 (residues 419–428) was disordered and not included in the final model. Helical domain (green; residues 148–257) is located between the other two domains. All of the three domains are involved in the dimer interface. From a Dali structural similarity search, TMO from P. savastanoi [9] showed the highest similarity (Z score = 41.0 and RMSD = 2.1 Å for 506 Cα atoms). The 2nd hit was l-AAO from C. rhodostoma [5] (Z score = 33.6 and RMSD = 2.4 Å for 437 Cα atoms) although it shows a quite low sequence identity with l-AAO/MOG (20.9%). PAO from Pseudomonas sp. P-501 [8] shows a relatively low structural similarity (Z score = 33.1 and RMSD = 2.8 Å for 497 Cα atoms), and it was the 5th hit of the structural similarity search. l-AAO/MOG is also structurally similar to human monoamine oxidase A (7th hit, Z score = 29.8 and RMSD = 2.6 Å for 404 Cα atoms) as well as snake venom l-AAOs (Fig. S3A), indicating that it is a member of MAO family.
Fig. 4.
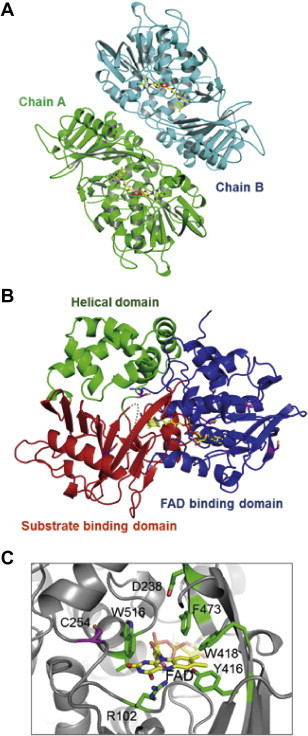
Crystal structure of l-AAO/MOG. (A) Dimer structure contained in the asymmetric unit. A and B chains are colored green and cyan, respectively. FAD molecules are shown as yellow sticks. (B) Monomer structure. FAD binding domain, substrate binding domain, and helical domain are colored blue, red, and green, respectively. Six Cys residues are shown as magenta. A disordered region (residues 419–428) is indicated by a grey dash line. (C) The active site. An “aromatic cage” is formed by Phe-418, Phe-473, Trp-516, and re-face of the isoalloxazine ring of FAD. Cys-254, the critical residue for conversion of l-AAO/MOG activities, is positioned behind Trp-516. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.8. Active site
Three aromatic residues (Trp-418, Phe-473, and Trp-516) are present at the active site, forming a hydrophobic pocket with the re face of the isoalloxazine ring of FAD (Fig. 4C). This structural feature is called ‘aromatic cage’ and is generally found at the catalytic center of MAO family enzymes [10]. The distance between Trp516 and Phe473 (7.9 Å) is similar to that of monoamine oxidase B (7.8 Å between Tyr398 and Tyr435), indicating that they play a steric role in substrate binding [27]. The isoalloxazine ring of FAD in l-AAO/MOG adopts an almost a planar conformation, suggesting that the cofactor is in an oxidized state [28]. Cys-254, which is the critical residue for the conversion of oxidase and monooxygenase activities, is located at the ‘back’ side of Trp-516. The distance between Cys-254 and Trp-516 is 3.6 Å.
3.9. Evolutionary relationship
l-AAO/MOG exhibits two activities, oxidative deamination (oxidase) and oxidative decarboxylation (monooxygenase), to the canonical substrate (l-Lys) while it shows structural similarity to MAO family enzymes. Therefore, we investigated a possible evolutionary relationship of this enzyme with flavin monooxygenases (FMOs) as well as MAO family enzymes. FMOs have been classified into six classes (A–F) [29]. The FAD binding domain of three classes (class A, B, and F) has the same fold with those of MAO family enzymes, being included in an ‘FAD/NAD(P)-binding domain’ superfamily in SCOP database [30] (Fig. S3B). Therefore, we constructed a phylogenetic tree containing both MAO family enzymes and class A, B, and F FMOs (Fig. S4). In the phylogenetic tree, a branch including l-AAO/MOG, TMO, and PAO is clearly separated from other MAO family enzymes and FMOs.
4. Discussion
In previous report, characterization of the new l-amino acid oxidase, which is specific to basic amino acids, from Pseudomonas sp. AIU813 was studied [11]. The amino acid sequence of this enzyme was similar to that of l-lysine 2-monooxygenase, and it was confirmed that the enzyme showed 2-monooxygenase activity as well as oxidase activity by the HPLC and MS analysis. l-Lysine oxidase activity of the enzyme increased in a p-CMB concentration-dependent manner like l-lysine monooxygenase from P. fluorescens Pf0-1 [14]. This report describes screening of an l-AAO/MOG mutant that is unaffected by p-CMB, and enzymatic comparison of wild type and the variant. Moreover, we determined the crystal structure and located the key residue for the activity conversion.
The role of sulfhydryl groups in flavoprotein enzymes has been extensively investigated; these groups have been implicated in the binding of substrate [31] and the flavin coenzyme [32,33], as well as in the electron transfer in oxidation-reduction reactions [34–37]. Many flavoprotein monooxygenases [33,38–43], including l-lysine monooxygenase [44], are known to be inhibited by some sulfhydryl-blocking reagents, suggesting that sulfhydryl groups play a significant role in these monooxygenases. In the l-AAO/MOG from Pseudomonas sp. AIU 813, cysteine residue modified by p-CMB plays a different role with sulfhydryl groups of above flavoproteins. As described in this paper, modification of the sulfhydryl groups of the l-AAO/MOG of Pseudomonas sp. AIU 813 altered the activities of this enzyme quantitatively as well as qualitatively; more specifically, it resulted in a decrease in the monooxygenase activity and an increase in the oxidase activity (Fig. 3). The oxidase activity of the l-AAO/MOG C254I mutant was 5 times higher than that of the wild type in their specific activities, and treatment of the mutant enzyme with p-CMB did not induce oxidase activity. Therefore, we analyzed the modification of the sulfhydryl group of the C254 residue in this enzyme; this residue is related to conversion of oxidase and monooxygenase activities.
l-Lysine oxidase activity of l-AAO/MOG from Pseudomonas sp. AIU 813 incubated with p-CMB was enhanced, and in contrast the monooxygenase activity was declined. In the saturation mutagenesis analysis at C254 in l-AAO/MOG, the similar interconversions between oxidase and monooxygenase were shown by the substitutions of Cys254 to aromatic amino acids or branched chain amino acids (Fig. 3). It seems that the size of the side-chains of the residue 254 greatly affect the oxidase and monooxygenase activities of l-AAO/MOG. However, the mutations to Trp, Tyr, Phe, and Phe showed less effect than that of C254I (Fig. 3), suggesting that the too large side chain is not optimal for the conversion to oxidase.
We also determined a high-resolution three-dimensional structure of l-AAO/MOG to provide a structural basis for its biochemical characteristics, such as substrate specificity and activity conversion. Some l-AAOs exhibit broad substrate specificity (e.g., l-AAOs from snake venoms) [4], whereas some other l-AAOs, including l-AAO/MOG and TMO, are specific to one or a few amino acids. l-AAOs generally have a relatively hydrophobic pocket at its active site, but their size, shape, and characteristics are different since they show only 16–21% amino acid sequence identity (Fig. S4). l-AAO/MOG is active for only a few amino acids with a long positively charged side chain (l-Lys, l-Orn, and l-Arg) [11], whereas TMO prefers l-Trp and l-Phe [45]. Fig. 5 shows a structural comparison of l-AAO/MOG with TMO complexed with indole-3-acetamide at the active site [9]. The residues involved in fixing the carboxylate of the substrate (Arg-102 and Tyr-416) and the three aromatic residues forming the aromatic cage (Trp-418, Phe-473, and Trp-516) are conserved while the residues recognizing the side chain of the substrate are not. In TMO, Phe-244, Val-247, Met-258, and Leu-478 are involved in forming a wide hydrophobic pocket for the indole ring of the substrate. In l-AAO/MOG, these residues are not conserved, but Asp-238 is located at the position of Val-247 of TMO, forming a ceiling of a long hydrophobic pocket. This structural feature is consistent with the substrate preference of l-AAO/MOG.
Fig. 5.
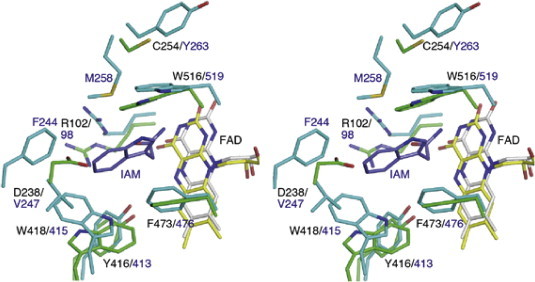
A structural comparison with tryptophan-2-monooxygenase (TMO) from P. savastanoi at the active site. Stereoview of superimposition of the structures of l-AAO/MOG (green, FAD yellow) and TMO (cyan, FAD grey) complexed with 2-indoleacetamide (IAM, blue). Labels indicate the residues and numbers of l-AAO/MOG (black)/TMO (blue). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The key residue for the activity conversion of l-AAO/MOG, Cys-254, is located near the aromatic cage (Fig. 4C). The location of Cys-254 indicate that it is not directly involved in substrate binding, but mutations or chemical modification of this residue would have a significant impact on substrate binding via the side chain of Trp-516. A slight difference of the binding position of substrate can dictate the activity of this type of enzymes for oxidase or monooxygenase. In the case of PAO, a proper positioning of the Cα atom of the substrate and its distance from a water molecule in the active site are very important factors for determining the enzyme activity [46]. In the case of TMO, mutations at residues interacting with the carboxylate of the substrate (Y413F, Y413A, R98K, and R98A) convert the enzyme to an oxidase [47,48], probably because loss of the anchoring disrupted the correct positioning of the substrate. However, l-AAO/MOG has a unique characteristic that its activities can be controlled by modification of Cys-254, and the Cys residue is not conserved in TMO or PAO. A detailed mechanism underlying the activity conversion of l-AAO/MOG remains to be elucidated. Structural study of substrate complex of l-AAO/MOG and the C254I mutant are currently in progress.
The phylogenetic analysis indicated that l-AAO/MOG, PAO, and TMO are located in a distinct branch from other MAO family enzymes and FMOs (Fig. S4). Importantly, all of these enzymes have been shown to exhibit both of oxidase (oxidative deamination) and monooxygenase (oxidative decarboxylation) activities. However, the conditions for exhibiting the two activities and their ratio are different among these enzymes. As described above, the wild-type l-AAO/MOG exhibited higher monooxygenase activity than oxidase activity toward l-Lys whereas the C254I mutation and chemical modification by p-CMB induced an opposite characteristics. PAO catalyzes both the oxidase and monooxygenase activities depending on the substrate used [49]. TMO is basically a monooxygenase devoid of oxidase activity [45] but mutations at the active site convert the enzyme to an oxidase [47,48]. Therefore we propose that these enzymes form a distinct subfamily of the MAO family, which exhibits both of the oxidase and monooxygenase activities depending on conditions, substrates, mutations, and chemical modifications.
From the genomes of Pseudomonas species including P. putida KT2440 [50], P. fluorescens F113 [51], and P. syringae pv. tomato DC30000, a large number of close homologs of l-AAO/MOG (amino acid sequence identities >89%) are found (Fig. S4). These ORFs are annotated as a (mono)amine oxidase or a 2-monooxygenase. From the present study, we can predict that these putative enzymes have dual activities and are specific to l-Lys because Asp-238 and Cys-254 are conserved. More than 40 years ago, Hayaishi and colleagues reported a series of biochemical characterizations of an l-Lys monooxygenase from P. fluorescens ATCC 11250 [currently P. putida (Trevisan) Migula] that has very similar characteristics to l-AAO/MOG such as increase of oxidase activity by thiol modification [44,52]. Therefore, we presume that the l-Lys monooxygenase from P. fluorescens studied earlier was a close homolog of l-AAO/MOG and is a member of the subfamily exhibiting the dual activities. In this study, we could determine the key residue for the thiol modification and locate the position in the three-dimensional structure.
Acknowledgments
This work was financially supported by the Hokuriku Innovation Cluster for Health Science, Ministry of Education, Culture, Sports, Science, and Technology (to Y.A.) and Asano Active Enzyme Molecule Project, ERATO, JST (to Y.A.). We thank S. Tatta (Toyama Prefectural University, Japan) for help in the screening of the mutant.
Appendix A. Supplementary data
Supplementary material.
References
- 1.Le K.H., Villanueva V.R. Purification and characterization of epsilon-N-trimethyllysine l-amino oxidase from Neurospora crassa. Biochim. Biophys. Acta. 1978;524:288–296. doi: 10.1016/0005-2744(78)90165-1. [DOI] [PubMed] [Google Scholar]
- 2.Piedras P., Pineda M., Muñoz J., Cárdenas J. Purification and characterization of an l-amino-acid oxidase from Chlamydomonas reinhardtii. Planta. 1992;188:13–18. doi: 10.1007/BF00198934. [DOI] [PubMed] [Google Scholar]
- 3.Coudert M., Vandecasteele J.P. Characterization and physiological function of a soluble l-amino-acid oxidase in Corynebacterium. Arch. Microbiol. 1975;102:151–153. doi: 10.1007/BF00428360. [DOI] [PubMed] [Google Scholar]
- 4.Guo C., Liu S., Yao Y., Zhang Q., Sun M.Z. Past decade study of snake venom l-amino acid oxidase. Toxicon. 2012;60:302–311. doi: 10.1016/j.toxicon.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Pawelek P.D., Cheah J., Coulombe R., Macheroux P., Ghisla S., Vrielink A. The structure of l-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faust A., Niefind K., Hummel W., Schomburg D. The structure of a bacterial (l)-amino acid oxidase from Rhodococcus opacus gives new evidence for the hydride mechanism for dehydrogenation. J. Mol. Biol. 2007;367:234–248. doi: 10.1016/j.jmb.2006.11.071. [DOI] [PubMed] [Google Scholar]
- 7.Arima J. Structural characterization of l-glutamate oxidase from Streptomyces sp. X-119-6. FEBS J. 2009;276:3894–3903. doi: 10.1111/j.1742-4658.2009.07103.x. [DOI] [PubMed] [Google Scholar]
- 8.Ida K., Kurabayashi M., Suguro M., Hiruma Y., Hikima T., Yamomoto M., Suzuki H. Structural basis of proteolytic activation of l-phenylalanine oxidase from Pseudomonas sp. P-501. J. Biol. Chem. 2008;283:16584–16590. doi: 10.1074/jbc.M800366200. [DOI] [PubMed] [Google Scholar]
- 9.Gaweska H.M., Taylor A.B., Hart P.J., Fitzpatrick P.F. Structure of the flavoprotein tryptophan 2-monooxygenase, a key enzyme in the formation of galls in plants. Biochemistry. 2012;52:2620–2626. doi: 10.1021/bi4001563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitzpatrick P.F. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isobe K., Sugawara A., Domon H., Fukuta Y., Asano Y. Purification and characterization of an l-amino acid oxidase from Pseudomonas sp. AIU 813. J. Biosci. Bioeng. 2012;114:257–261. doi: 10.1016/j.jbiosc.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Kusakabe H., Kodama K., Kuninaka A., Yoshino H., Misono H., Soda K. A new antitumor enzyme, l-lysine α-oxidase from Trichoderma viride. purification and enzymological properties. J. Biol. Chem. 1980;255:976–981. [PubMed] [Google Scholar]
- 13.Berezov T., Lukasheva E. Effect of substrate and product analogs on the activity of l-lysine α-oxidase from Trichoderma sp. Biochem. Int. 1988;17:529–534. [Google Scholar]
- 14.Yamauchi T., Yamamoto S., Hayaishi O. Reversible conversion of lysine monooxygenase to an oxidase by modification of sulfhydryl groups. J. Biol. Chem. 1973;248:3750–3752. [PubMed] [Google Scholar]
- 15.Isobe K., Tamauchi H., Fuhshuku K., Nagasawa S., Asano Y. A simple enzymatic method for production of a wide variety of d-amino acids using l-amino acid oxidase from Rhodococcus sp. AIU Z-35-1. Enzyme Res. 2010;2010:567210. doi: 10.4061/2010/567210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandeyar M.A., Weiner M.P., Hutton C.J., Batt C.A. A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene. 1988;65:129–133. doi: 10.1016/0378-1119(88)90425-8. [DOI] [PubMed] [Google Scholar]
- 17.Sambook J., Russell D. 3rd ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 18.Asano Y., Ueda M., Yamada H. Microbial production of d-malate from maleate. Appl. Environ. Microbiol. 1993;59:1110–1113. doi: 10.1128/aem.59.4.1110-1113.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. (Pt A) [DOI] [PubMed] [Google Scholar]
- 21.Adams P.D. Phenix: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 22.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 23.Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 26.Davis I.W. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M., Binda C., Mattevi A., Edmondson D.E. Functional role of the “aromatic cage” in human monoamine oxidase B: structures and catalytic properties of Tyr435 mutant proteins. Biochemistry. 2006;45:4775–4784. doi: 10.1021/bi051847g. [DOI] [PubMed] [Google Scholar]
- 28.Senda M., Kishigami S., Kimura S., Fukuda M., Ishida T., Senda T. Molecular mechanism of the redox-dependent interaction between NADH-dependent ferredoxin reductase and Rieske-type [2Fe–2S] ferredoxin. J. Mol. Biol. 2007;373:382–400. doi: 10.1016/j.jmb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 29.van Berkel W.J., Kamerbeek N.M., Fraaije M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 30.Murzin A.G., Brenner S.E., Hubbard T., Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto H., Nozaki M., Hayaishi O. A role of sulfhydryl groups in imidazoleacetate monooxygenase. Biochem. Biophys. Res. Commun. 1968;32:30–36. doi: 10.1016/0006-291x(68)90421-x. [DOI] [PubMed] [Google Scholar]
- 32.Neims A.H., Coffey D.S., Hellerman L. Interaction between tetraethylthiuram disulfide and the sulfhydryl groups of d-amino acid oxidase and of hemoglobin. J. Biol. Chem. 1966;241:5941–5948. [PubMed] [Google Scholar]
- 33.Neujahr H., Gaal A. Phenol hydroxylase from yeaset: purification and properties of the enzyme from Trichosporon cutaneum. Eur. J. Biochem. 1973;35:386–400. doi: 10.1111/j.1432-1033.1973.tb02851.x. [DOI] [PubMed] [Google Scholar]
- 34.Massey V., Hofmann T., Palmer G. The reaction of function and structure in lipoyl dehydrogenase. J. Biol. Chem. 1962;237:3820–3828. [PubMed] [Google Scholar]
- 35.Colman R.F., Black S. On the role of flavin adenine dinucleotide and thiol groups in the catalytic mechanism of yeast glutathione reductase. J. Biol. Chem. 1965;240:1796–1803. [PubMed] [Google Scholar]
- 36.Massey V., Williams C.H., Jr. On the reaction mechanism of yeast glutathione reductase. J. Biol. Chem. 1965;240:4470–4480. [PubMed] [Google Scholar]
- 37.Thelander L. Studies onthioredoxin reductase from Escherichia coli B: the relation of structure and function. Eur. J. Biochem. 1968;4:407–422. doi: 10.1111/j.1432-1033.1968.tb00227.x. [DOI] [PubMed] [Google Scholar]
- 38.Sutton W.B. Sulfhydryl and prosthetic groups of lactic oxidative decarboxylase from Mycobacterium phlei. J. Biol. Chem. 1955;216:749–761. [PubMed] [Google Scholar]
- 39.Sullivan P.A. Crystallization and properties of l-lactate oxidase from Mycobacterium smegmatis. Biochem. J. 1968;110:363–371. doi: 10.1042/bj1100363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto S., Katagiri M., Maeno H., Hayaishi O. Salicylate hydroxylase, a monooxygenase requiring flavin adenine dinucleotide: I. Purification and general properties. J. Biol. Chem. 1965;240:3408–3413. [PubMed] [Google Scholar]
- 41.White-Stevens R.H., Kamin H. Studies of a flavoprotein, salicylate hydroxylase: I. Preparation, properties, and the uncoupling of oxygen reduction from hydroxylation. J. Biol. Chem. 1972;247:2358–2370. [PubMed] [Google Scholar]
- 42.Levy C.C., Frost P. The metabolism of coumarin by a microorganism: V. Melilotate hydroxylase. J. Biol. Chem. 1966;241:997–1003. [PubMed] [Google Scholar]
- 43.Maki Y., Yamamoto S., Nozaki M., Hayaishi O. Studies on monooxygenases II. Crystallization and some properties of imidazole acetate monooxygenase. J. Biol. Chem. 1969;244:2942–2950. [PubMed] [Google Scholar]
- 44.Takeda H., Yamamoto S., Kojima Y., Hayaishi O. Studies on monooxygenases I. General properties of crystalline l-lysine monooxygenase. J. Biol. Chem. 1969;244:2935–2941. [PubMed] [Google Scholar]
- 45.Emanuele J.J., Heasley C.J., Fitzpatrick P.F. Purification and characterization of the flavoprotein tryptophan 2-monooxygenase expressed at high-levels in Escherichia coli. Arch. Biochem. Biophys. 1995;316:241–248. doi: 10.1006/abbi.1995.1034. [DOI] [PubMed] [Google Scholar]
- 46.Ida K., Suguro M., Suzuki H. High resolution X-ray crystal structures of l-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. Structures of the enzyme-ligand complex and catalytic mechanism. J. Biochem. 2011;150:659–669. doi: 10.1093/jb/mvr103. [DOI] [PubMed] [Google Scholar]
- 47.Sobrado P., Fitzpatrick P.F. Analysis of the role of the active site residue Arg98 in the flavoprotein tryptophan 2-monooxygenase, a member of the l-amino oxidase family. Biochemistry. 2003;42:13826–13832. doi: 10.1021/bi035299n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sobrado P., Fitzpatrick P.F. Identification of Tyr413 as an active site residue in the flavoprotein tryptophan 2-monooxygenase and analysis of its contribution to catalysis. Biochemistry. 2003;42:13833–13838. doi: 10.1021/bi035300i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyama H. Oxidation and oxygenation of l-amino acids catalyzed by a l-phenylalanine oxidase (deaminating and decarboxylating) from Pseudomonas sp. P-501. J. Biochem. 1984;96:421–427. doi: 10.1093/oxfordjournals.jbchem.a134853. [DOI] [PubMed] [Google Scholar]
- 50.Nelson K.E. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002;4:799–808. doi: 10.1046/j.1462-2920.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- 51.Redondo-Nieto M. Genome sequence reveals that Pseudomonas fluorescens F113 possesses a large and diverse array of systems for rhizosphere function and host interaction. BMC Genomics. 2013;14:54. doi: 10.1186/1471-2164-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakazawa T., Hori K., Hayaishi O. Studies on monooxygenases: V. Manifestation of amino acid oxidase activity by l-lysine monooxygenase. J. Biol. Chem. 1972;247:3439–3444. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.