

Abstract
The structure-activity relationship (SAR) study of two chemotypes identified as inhibitors of the human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (HPGD, 15-PGDH) was conducted. Top compounds from both series displayed potent inhibition (IC50 <50 nM), demonstrate excellent selectivity towards HPGD and potently induce PGE2 production in A549 lung cancer and LNCaP prostate cancer cells.
Keywords: HPGD, 15-PGDH, prostaglandin, PGE2, inhibitor
Introduction
Prostaglandins are members of the eicosanoid family of signaling molecules, along with lipoxins and leukotrienes. They are synthesized from arachidonic acid (AA), which following its release from membrane phospholipids, undergoes a series of transformations mediated by enzymes such as cyclooxygenases (COX) and prostaglandin synthases. Prostaglandins are also inactivated through chemical modification. One key metabolizing enzyme is 15-hydroxyprostaglandin dehydrogenase (HPGD, also referred to as 15-PGDH), which catalyzes the oxidation of the prostaglandin hydroxyl group at C-15 to the corresponding ketone.1
A number of prostaglandins of particular biological interest have been identified as substrates of HPGD. Prostaglandin E2 (PGE2), a product of COX-2-mediated AA metabolism via prostaglandin H2 (PGH2) is converted by HPGD to its biologically inactive metabolite, 15-keto-PGE2. Over-production of PGE2 has been reported to promote tumor-associated neovascularization, cell proliferation, and tumor growth.2 It has also been suggested that decreased levels of PGE2 resulting from amplified HPGD activity through pharmacological means or enhanced expression may mitigate these phenotypes and result in a chemoprevention strategy.3 Conversely, HPGD was found to be highly expressed in androgen receptor-overexpressing advanced tumors, metastatic prostate cancers,4 and metastatic breast cancer, where it has been found to contribute to epithelial-mesenchymal transition (EMT). 5 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) is a product of albumin-mediated transformation of prostaglandin D2 (PGD2), itself derived from PGH2.6 15d-PGJ2 was shown to inhibit IKKβ, a positive regulator of NF-κB,7,8 and to covalently bind Keap1, a negative regulator of Nrf2.9 As such, the metabolism of PGD2 by HPGD provides a possible link between HPGD activity and levels of 15d-PGJ2. Moreover, Blair et al. recently demonstrated that HPGD catalyzed the transformation of 11-hydroxy-eicosatetranoic acid (11-HETE), a product of COX-2/HPGD-mediated AA transformation, into 11-oxo-eicosatetranoic acid (11-oxo-ETE),10 and that 11-oxo-ETE inhibits NF-κB signaling through inhibition of IKKβ.11 Therefore, down regulation of HPGD may lead to a decrease in the NF-κB signaling pathway and to an increase in Nrf2 signaling.
As a consequence of prostaglandins' roles in a number of processes such as inflammation, differentiation, and cellular signaling, their cellular levels are tightly regulated. The central role of HPGD to inactivate prostaglandins such as PGE2 or PGD2 makes it an important target for pharmacological intervention. Prior to our efforts, few inhibitors of HPGD have been identified, and all are based off ciglitazone, a PPARγ inhibitor possessing a thiazolidinedione scaffold. The parent compound was found to have an IC50 against HPGD of 2.7 μM12 whereas an optimized analogue, CT-8, displayed an IC50 of 51 nM.13 More recently, the same group continued their SAR studies around ciglitazone and related analogues, with demonstrated activity in vivo for their top compounds,14 but the selectivity of CT-8 toward other dehydrogenases was not reported. In addition, the thiazolidinedione chemotype is likely to have undesired off-target effects given the well documented PPARγ activity for this class of molecules.
In an effort to identify selective inhibitors of HPGD, we conducted a quantitative high throughput screen (qHTS) of ∼160,000 compounds as part of the Molecular Libraries Small Molecule Repository (MLSMR). These studies led to the identification of three structurally distinct small molecule chemical probes as potent and selective inhibitors of HPGD (denoted as ML147, ML148, and ML149, Fig. 1).15 The first scaffold, ML147, is characterized by an azabenzimidazole core appended with a thiobenzyl group at C-1. The second compound, ML148 possesses a benzimidazole core substituted with a cyclohexylamide moiety and 3-methyl-phenyl group. Finally, ML149 contains a triazoloazepine core with 2,5-dimethylpyrrole group linked to the triazole ring. Given the potential liabilities (e.g. glutathione reactivity) of the thiol ether linkage on ML147 we decided to focus our initial SAR efforts on the optimization of ML148 and ML149. In addition to our SAR efforts, we also sought to define key in vitro ADME properties, cellular efficacy (PGE2 levels) and selectivity against several related dehydrogenases (vide infra).
Figure 1.
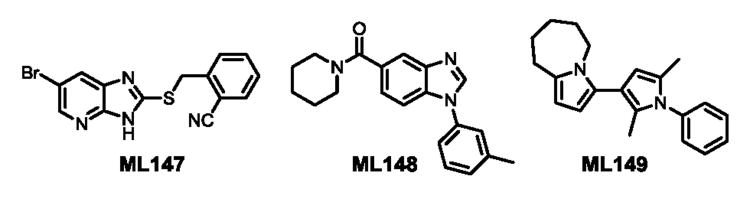
Structures of chemotypes ML147, ML148, and ML149.
Optimization of ML149
In order to support compound scale-up for more detailed biological testing and to facilitate SAR explorations, we designed concise synthetic routes to ML148, ML149 and related analogues. Our initial efforts were focused on ML149 as shown in Scheme 1.
Scheme 1.

Reagents and conditions: (a) CDI, THF, then hydrazine-hydrate, 23 °C, 3.5 h, 65%; (b) 1-aza-2-methoxy-1-cycloheptene, PhCl, MW, 180 °C, 45 min, 71%; (c) PhI, Cs2CO3, Cu2O, 4,7-dimethoxy-1,10-phenanthroline, NMP-PEG, 100 °C, 16 h, 44%.
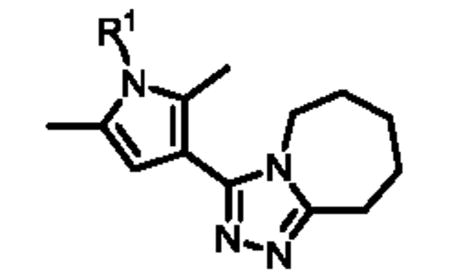
The carboxylic acid functionality of commercially available pyrrole 1 (Scheme 1) was activated using carbonyldiimidazole (CDI) in THF and the resulting acylimidazole was then treated with hydrazine monohydrate to afford hydrazide 2. This reaction was performed on a two-gram scale, and the product was isolated from the imidazole (reaction by-product) by recrystallization in ethyl acetate. Treatment of hydrazide 2 with 1-aza-2-methoxy-1-cycloheptene in chlorobenzene in a microwave (MW) reactor led to formation of triazole 3 in 71% yield. Finally, Ullman-type coupling between 3 and iodobenzene afforded ML149. The use of 4,7-dimethoxy-1,10-phenanthroline (L) as a copper ligand allowed for the efficient activation of the air-stable metal catalyst;16 furthermore, using polyethylene glycol (PEG) as a co-solvent improved the solubilization of the inorganic base and of the copper salt, accounting for the improved performance of the reaction.17 Using this synthetic sequence, eighteen analogues (4a-r) with differentially substituted N-pyrrole aromatic rings were synthesized (Table 1).
Table 1.
SAR of ML149 and related analogues.
|
|
||||
|---|---|---|---|---|---|
|
| |||||
| Compound | R1 | IC50 (nM)a | Compound | n | IC50 (nM)a |
| ML149 | phenyl | 54 | 6a | 1 | 3040 |
| 4a | 3,4-Cl-phenyl | 22 | 6b | 2 | 192 |
| 4b | 3-Cl-phenyl | 34 | 6c | 4 | 242 |
| 4c | 4-Cl-phenyl | 38 | 6d | 5 | 383 |
|
|
|||||
| 4d | 4-Br-phenyl | 48 |
|
||
| 4e | 4-F-phenyl | 121 | |||
| 4f | 3-CF3-phenyl | 48 | |||
| 4g | 4-CF3-phenyl | 68 | |||
| 4h | 4-Me-phenyl | 34 | |||
| 4i | 3-CN-phenyl | 76 | |||
|
|
|||||
| 4j | 4-CN-phenyl | 96 | Compound | R2 | IC50 (nM)a |
|
|
|||||
| 4k | 4-OMe-phenyl | 86 | 7a |
|
607 |
| 4l | 4-NO2-phenyl | 108 | |||
| 4m | 4-tBu-phenyl | 136 | |||
| 4n | 4-CO2Et-phenyl | 38 | 7b |
|
3827 |
| 4o | 2-pyridine | 1078 | |||
| 4p | 3-pyridine | 271 | 7c |
|
681 |
| 4q | 4-pyridine | 383 | |||
| 4r | 1-naphthalene | 108 | |||
determined using the HTS enzymatic assay and tested in triplicate.
Overall, the substitution at the meta or para position of the N-pyrrole phenyl ring with small substituents provided analogues with activities comparable to that of ML149, however, some subtle SAR trends emerged. Modification of the para position with a halogen group led to analogues with slightly improved potencies [4a (3,4-dichloro-phenyl), 4c (4-Cl-phenyl and 4d (4-Br-phenyl)], with bis-substituted analogue 4a being the most potent (IC50 = 22 nM). However, introduction of other electron-withdrawing groups at the para position such as F (4e), CF3 (4g), CN (4j) and NO2 (4l), resulted in a slight loss of potency. We did find that the phenyl ring could not be replaced by a heterocycle, such as a pyridine ring (compounds 4o-q) without a more significant drop in potency. Of note, a number of phenyl analogues bearing ortho substituents could not be successfully synthesized, likely because the steric hindrance of the two pyrrole methyl groups prevent the Ullman-type coupling reaction from taking place.
The synthetic sequence outlined in Scheme 1 is ideal for late-stage diversification of the pendant aryl ring but is less optimal for investigations of the triazoloazepine core. For facile preparation of these analogues we utilized common intermediate 2, followed by cyclization with various 1-aza-2-methoxy derivatives 5a-e to ultimately provide five analogues (6a-d) with rings of various sizes fused to the triazole moiety (Scheme 2).
Scheme 2.

Reagents and conditions: (a) PhCl, MW, 180 °C, 45 min; (b) PhI, Cs2CO3, Cu2O, L, NMP-PEG, 80-100 °C, 16 h.
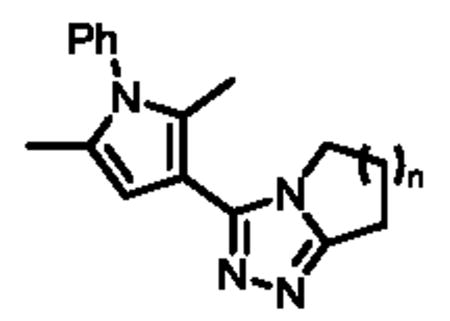
Upon biological testing of the compounds, no direct correlation was found between the sizes of the ring fused to the triazole moiety and HPGD activity (Table 1). However, the five-membered ring analogue 6a resulted in a significant loss of potency (IC50 = 3.04 μM) suggesting the importance of having at least a 6-membered fused ring. Whereas, the 6-(6b), 8-(6c) and 9-membered (6d) ring analogues were less active than ML149. Having completed our initial SAR investigations of the core and pendant phenyl ring, we then wanted to probe the influence of the substituents on the pyrrole ring (Table 1). The synthesis of these compounds was achieved in a manner similar to the analogues described previously, except the starting carboxylic acid was varied (Scheme 3).
Scheme 3.

Reagents and conditions: (a) CDI, THF, then hydrazine-hydrate, 23 °C; (b) (1) 1-aza-2-methoxy-1-cycloheptene, PhCl, MW, 180 °C; (2) PhI, Cs2CO3, Cu2O, L, NMP-PEG, 100 °C.
The unsubstituted pyrrole 7a displayed a six-fold drop in activity (IC50 = 607 nM) as compared to ML149, highlighting the importance of substitution on the pyrrole. The indole derivative (7c) was less active (IC50 = 681 nM), and the addition of other heteroatoms were not tolerated [e.g. pyrrazole derivative 7b (IC50 = 3.83 μM)].
Optimization of ML148
Following the optimization of ML149, which led to the identification of compounds 4a (IC50 = 22 nM) and 4b (IC50 = 34 nM), we turned our attention to benzimidazole-based chemotype ML148. The synthesis route was optimized in order to provide rapid access to a variety of analogues as shown in Scheme 4.
Scheme 4.

Reagents and conditions: (a) o-toluidine, NMP, 60 °C; (b) hydrazine, Ra-Ni, MeOH, 23 °C; (EtO)3CH, TsOH, THF, MW, 120 °C; (d) R1R2NH or R3NH2, HATU, DIPEA, DMF, 23 °C; (e) (COCl)2, DMF, DCM, 23 °C, then piperidine-HCl, DCM, 23 °C; (f) R3NH2, DIPEA, NMP, 120 °C.
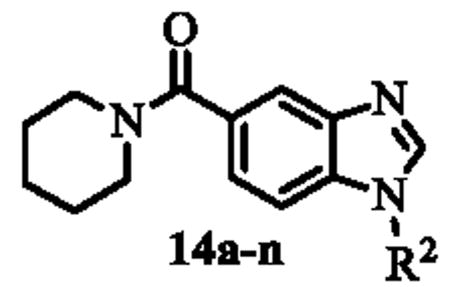
Benzoic acid 8 was treated with m-toluidine in N-methylpyrolidinone (NMP) to afford intermediate 9. The nitro group was reduced using hydrazine and Raney nickel (Ra-Ni) in methanol to afford the crude 3-amino-4-(m-tolylamino)benzoic acid which was then converted to benzimidazole 10 via treatment with triethylorthoformate and catalytic p-toluenesulfonic acid (TsOH) in THF. This synthetic sequence was carried out on a gram-scale, and no flash chromatography or HPLC purification was required to isolate any of the synthetic intermediates. Finally, treatment of carboxylic acid 10 with a range of amines (R1R2NH), using HATU and diisopropylethylamine (DIPEA) in DMF provided a library of 14 analogues of ML148 (compounds 11a-n) possessing differing amide moieties. Unfortunately, none of these compounds resulted in improved potency (Table 2); however, it did provide key information regarding the steric requirements around the cyclohexylamide functionality. A smaller ring (11i) or an exocyclic amine (11k) led to compounds with decreased activity (20-50 fold respectively). Substituents at the 3-position on the piperidine ring were well tolerated (11a and 11c-e), including the relatively bulky isopropyl group (11c). While adding a 4-Br group (11f) only led to a modest decrease in potency, the 4-CF3 group led to a drastic loss of potency (80-fold) compared to the corresponding 3-CF3 (11e). Moreover, the presence of heteroatoms (N or O) at the 4-position was also not well tolerated, with activity in the micromolar range (11m: N-Me-Piperazine) or inactivity (11n: morpholine). Finally, acyclic amides (11h and 11l) were much less active than cyclic ones, and secondary amides (11k and 11l) were less active than tertiary amides (11h). In order to explore SAR around the N-linked phenyl ring of ML148, we wanted to devise a route in which the varying phenyl groups could be introduced in the last step. As such, benzoic acid 8 was treated with oxalyl chloride and a small amount of DMF in dichloromethane, and the resulting acyl chloride was treated with piperidine-hydrochloride in dichloromethane to afford amide 12 (Scheme 4). The remainder of the synthesis proceeded as planned, starting with substitution of the aromatic fluorine with various amines (R3NH2) in the presence of DIPEA in DMF to provide anilines 13a-n. The aromatic nitro group was reduced, and the resulting anilines were treated with triethylorthoformate as described above to afford analogues 14a-n.
Table 2.
SAR of ML148 and related analogues.
|
|
||||
|---|---|---|---|---|---|
|
| |||||
| Compound | R1 | IC50 (nM)a | Compound | R2 | IC50 (nM)a |
| ML148 |
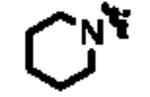
|
19 | 14a | phenyl | 22 |
| 11a |
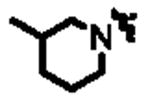
|
22 | 14b | 2-OMe-phenyl | 27 |
| 11b |
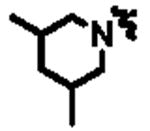
|
271 | 14c | 3-Cl-phenyl | 12 |
| 11c |
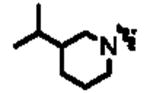
|
48 | 14d | 3-OMe-phenyl | 19 |
| 11d |

|
68 | 14e | 3-CF3-phenyl | 48 |
| 11e |

|
68 | 14f | 3-iPr-phenyl | 68 |
| 11f |
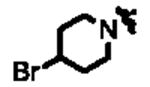
|
76 | 14g | 3-tBu-phenyl | 171 |
| 11g |
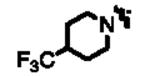
|
1524 | 14h | 4-Me-phenyl | 19 |
| 11h |

|
383 | 14i | 4-Cl-phenyl | 22 |
| 11i |
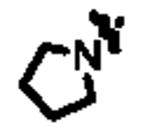
|
383 | 14j | 4-OMe-phenyl | 19 |
| 11j |
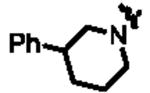
|
429 | 14k | 4-CF3-phenyl | 86 |
| 11k |
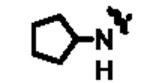
|
961 | 14l | 4-tBu-phenyl | 54 |
| 11l |
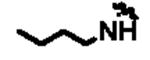
|
1358 | 14m | tBu | 34 |
| 11m |
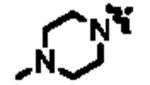
|
2710 | 14n | nBu | 68 |
| 11n |
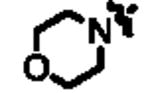
|
3040 | |||
determined using the HTS enzymatic assay and tested in triplicate.
Analogues 14a-n displayed activities similar to that of ML148 (Table 2) with relatively flat SAR being observed. Introduction of a small substituents in para- and in meta-position afforded compounds with similar activities, whereas electron-withdrawing groups (14e and 14k) and bulkier substituents (14f,g,l) in para- and in meta-position resulted in a loss of potency.
Inporation of non-aromatic groups (14m,n) had minimal effect on the potency. These data suggest that this region could be exploited for future modulation of ADME properties since it appears very tolerant to change. However, it remains unclear whether introduction of a hydrophilic group will be tolerated since all analogues prepared thus far are generally lipophilic in this region.
ADME profile
To assess the potential for these compounds to be used in vivo, we obtained in vitro ADME properties of a few selected analogues of the ML149 and ML148 chemotypes (Table 3).
Table 3.
In vitro ADME properties of selected analogues of ML149 and ML148.
| Compound | IC50 (nM)a | Permeability (10-6 cm/s)b | Kinetic solubility (μM)c | RLM T1/2 (min)d |
|---|---|---|---|---|
| ML149 | 112 | 599 | >45 | n.d. |
| 4a | 22 | 1646 | 32 | 1.9 |
| 4b | 34 | 1100 | 37 | 2.0 |
| 4f | 48 | 1512 | 33 | 1.9 |
| 4h | 34 | 1330 | 28 | 1.8 |
| 4i | 76 | 229 | >49 | 8.1 |
| ML148 | 19 | 1196 | >47 | 7.3 |
| 11d | 68 | 2265 | 5.6 | 1.8 |
| 14c | 12 | 895 | >50 | 6.4 |
| 14e | 48 | 1346 | >55 | 4.4 |
| 14j | 19 | 624 | >50 | 23 |
represents the IC50 against HPGD in the HTS enzymatic assay.
represents the PAMPA permeability at pH 7.4.
represents the kinetic aqueous solubility in PBS buffer (pH 7.4) as measured by UV quantification.
represents the T1/2 in rat liver microsomes (RLM) in the presence of NADPH.
In general, ML149 and related analogues showed good kinetic solubility (>28 μM in phosphate buffered saline (PBS) at pH 7.4). Results from the PAMPA assay, which measures passive (artificial) membrane permeability, showed that halogen substituents seem to improve permeability (e.g. compounds 4a and 4b). On the other hand, substitution with a nitrile moiety led to a ten-fold drop in permeability (4i). Unfortunately, ML149 and its analogues degraded quickly in the presence of rat liver microsomes, as evidenced by their short half-life (<10 min). ML148 and its analogues also displayed good kinetic solubility (>47 μM), except for compound 11d (5.6 μM). PAMPA permeability of ML148 and related analogues was similar to the ML149 series. As with ML149, the rat liver microsomal stability remained undesirable, except for compound 14j (T1/2 = 23 min).
Selectivity toward HPGD
In order to evaluate the selectivity of the prepared compounds toward HPGD, analogues of ML148 and of ML149 were tested for their inhibitory activities versus a panel of five other dehydrogenases (Supplemental Table 1).18 Encouragingly, none of the compounds tested showed any activity against GAPDH, LDHA, LDHB and HSD17β4. Some inhibition was observed against ALDH1 A1, though all compounds displayed considerable selectivity toward HPGD (greater than 28-fold selectivity). Analogues 4b (142-fold selectivity), 14c (160-fold selectivity), and 14e (142-fold selectivity) were the most selective HPGD inhibitors.
PGE2 production
A selection of top actives around the ML148 and ML149 chemotypes were tested for their ability to increase PGE2 levels. Lung cancer A549 cells and androgen-sensitive human prostate adenocarcinoma LNCaP cells were titrated with the selected HPGD inhibitors; the half-maximal activation concentration (AC50) and percentage induction (% Induction) of PGE2 production are reported in Table 4.
Table 4.
PGE2 produced in A549 and LNCaP cells.
| A549 cells | LNCaP cells | ||||
|---|---|---|---|---|---|
|
| |||||
| Compound | IC50 (nM)a | AC50 (μM)b | % Inductionc | AC50 (μM)b | % Inductionc |
| ML149 | 54 | 0.743 | 106 | 2.80 | 73 |
| 4a | 22 | 0.296 | 110 | 0.352 | 105 |
| 4b | 34 | 0.590 | 114 | 0.558 | 68 |
| 4f | 48 | 2.09 | 145 | 2.22 | 44 |
| 4h | 34 | 0.166 | 107 | 0.703 | 71 |
| 4i | 76 | 2.64 | 350* | 1.77 | 63 |
| ML148 | 19 | 0.469 | 129 | 0.884 | 62 |
| 11d | 68 | 0.743 | 127 | 4.43 | 53 |
| 14c | 12 | 0.372 | 170 | 0.222 | 85 |
| 14e | 48 | 2.64 | 173 | 2.80 | 70 |
| 14j | 19 | 0.662 | 174 | 0.884 | 69 |
represents the IC50 against HPGD in the HTS enzymatic assay;
represents the half-maximal concentration in the PGE2 production assay;
represents the percentage induction of PGE2 production;
the titration curve was incomplete.
All the compounds tested increased PGE2 production levels when compared to a DMSO control, in both A549 cells and LNCaP cells. The percent induction was consistently higher in A549 cells (from 106% for ML149 to 174% for 14j) than in LNCaP cells (from 44% for 4f to 105% for 4a). The majority of the compounds tested displayed sub-micromolar activity in both cell lines, and a good correlation in AC50 was observed between the two cell lines: low AC50 values measured in A549 cells correlated with low AC50 values in LNCaP cells, with the exception of ML149 [AC50 = 0.743 μM (A549) and AC50 = 2.80 μM (LNCaP)] and of 11d [AC50 = 0.743 μM (A549) and AC50 = 4.43 μM (LNCaP)]. Unfortunately, no tractable SAR emerged from the PGE2 production assay, a likely consequence of the often diverging enzymatic potency and cell permeability properties of the inhibitors. Indeed, compound 4i displayed particularly low cell permeability in the PAMPA assay (229 × 10-6 cm/s), probably accounting for the low activity measured in the A549 (AC50 = 2.64 μM) and LNCaP (AC50 = 1.77 μM) cell lines. It is also possible that enzyme occupancy of different agents and the kinetics of the sequential enzymatic events adds a level of complexity that may account for these differences. Overall, the data obtained in the PGE2 production assay showed that treatment with our inhibitors leads to increased PGE2 production in A549 lung cancer and in LNCaP prostate cancer cells, consistent with inhibition of HPGD in the cell.
Conclusion
The structure-activity relationship (SAR) study of two small molecule inhibitors of the human HPGD, ML148 and ML149, was carried out. These studies revealed that while the fused seven-membered ring and the presence of a 2,5-dimethylpyrrole were required for activity of ML149, a certain tolerance in the substitution pattern of the phenyl ring was observed. As a result compound 4h was found to have improved potency toward HPGD, improved cell permeability and induced PGE2 production both in A549 and in LNCaP cells, when compared to ML149. For the benzimidazole chemotype (ML148), the cyclohexylamide group was found to be optimal for enzymatic activity, although small substituents at the 3-position of the cyclohexyl amine group were tolerated. Moreover, substitution at the meta-position of the phenyl ring was also well-tolerated, leading to analogues 14c and 14j. Both compounds displayed similar ADME properties, although 14j was more stable in the RLM assay, and both analogues had comparable effects on PGE2 levels in A549 and in LNCaP cells. Unfortunately, none of the HPGD inhibitors tested led to any observable down regulation of NF-κB, as measured in our assay (Supplemental Figure 1), perhaps due to compensatory mechanisms counteracting the effect of modulation of HPGD activity on NF-κB regulation. In conclusion, we have identified potent and selective inhibitors of HPGD (e.g. 4h and 14j) and hope these compounds help better define the role of HPGD in a variety of diseases. As such, we make these compounds freely available to the research community upon request.
Supplementary Material
Acknowledgments
We thank Jim Bougie, Thomas Daniel and William Leister for compound purification, as well as Danielle van Leer, Misha Itkin, Crystal Mcknight, Christopher LeClair and Paul Shinn for assistance with compound management. We also thank Udo Oppermann at the Structural Genomics Consortium (SGC), Oxford, UK for providing the ALDH1A1 and HSD17β4 enzymes. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research grant U54HG005033 and the Intramural Research Program of the National Center for Advancing Translational Sciences at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tai HH, Cho H, Tong M, Ding Y. Curr Pharm Design. 2006;12:955. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 2.Wang D, DuBois RN. Gut. 2006;55:115. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Na HK, Park JM, Lee HG, Myung SJ, Surh YJ. Biochem Pharmacol. 2011;82:1352. doi: 10.1016/j.bcp.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Vainio P, Gupta S, Ketola K, Mirtti T, Mpindi JP, Kohonen P, Fey V, Perälä M, Smit F, Verlaegh G, Schalken J, Alanen KA, Kallioniemi O, Iljin K. Am J Pathol. 2011;178:525. doi: 10.1016/j.ajpath.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehtinen L, Vainio P, Wikman H, Reemts J, Hilvo M, Issa R, Pollari S, Brandt B, Oresic M, Pantel K, Kallioniemi O, Iljin K. J Pathol. 2012;226:674. doi: 10.1002/path.3956. [DOI] [PubMed] [Google Scholar]
- 6.Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, Kim DD. Biochem Pharmacol. 2011;82:1335. doi: 10.1016/j.bcp.2011.07.100. [DOI] [PubMed] [Google Scholar]
- 7.Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G, Glass CK. Proc Natl Acad Sci USA. 2000;97:4844. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trivedi SG, Newson J, Rajakariar R, Jacques TS, Hannon R, Kanaoka Y, Eguchi N, Colville-Nash P, Gilroy DW. Proc Natl Acad Sci USA. 2006;103:5179. doi: 10.1073/pnas.0507175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh JY, Giles N, Landar A, Darley-Usmar V. Biochem J. 2008;411:297. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Zhang S, Arora JS, Snyder NW, Shah SJ, Blair IA. Chem Res Toxicol. 2011;24:2227. doi: 10.1021/tx200336f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snyder NW, Revello SD, Liu X, Zhang S, Arora JS, Blair IA. Presented at the AACR 103rd Annual Meeting; Chicago, IL. March 31 – Apr 4, 2012. [Google Scholar]
- 12.Cho H, Tai HH. Prostaglandins Leukot Essent Fatty Acids. 2002;67:461. doi: 10.1054/plef.2002.0457. [DOI] [PubMed] [Google Scholar]
- 13.Cho H, Tai HH. Arch Biochem Biophys. 2002;405:247. doi: 10.1016/s0003-9861(02)00352-1. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y, Karna S, Choi CH, Tong M, Tai HH, Na DH, Jang CH, Cho H. J Med Chem. 2011;54:5260. doi: 10.1021/jm200390u. [DOI] [PubMed] [Google Scholar]
- 15.Niesen FH, Shultz L, Jadhav A, Bhatia C, Guo K, Maloney DJ, Pilka ES, Wang M, Oppermann U, Heightman TD, Simeonov A. Plos One. 2010;5:e13719. doi: 10.1371/journal.pone.0013719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altman RA, Buchwald SL. Org Lett. 2006;8:2779. doi: 10.1021/ol0608505. [DOI] [PubMed] [Google Scholar]
- 17.Altman RA, Koval ED, Buchwald SL. J Org Chem. 2007;72:6190. doi: 10.1021/jo070807a. [DOI] [PubMed] [Google Scholar]
- 18.The activity data of ML148 and of ML149 versus ALDH1A1, HADH2 and HSD17β4 were reported in reference 15.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.