

Abstract
Autophagy and apoptosis control the turnover of organelles and proteins within cells, and of cells within organisms, respectively, and many stress pathways sequentially elicit autophagy, and apoptosis within the same cell. Generally autophagy blocks the induction of apoptosis, and apoptosis-associated caspase activation shuts off the autophagic process. However, in special cases, autophagy or autophagy-relevant proteins may help to induce apoptosis or necrosis, and autophagy has been shown to degrade the cytoplasm excessively, leading to ‘autophagic cell death’. The dialogue between autophagy and cell death pathways influences the normal clearance of dying cells, as well as immune recognition of dead cell antigens. Therefore, the disruption of the relationship between autophagy and apoptosis has important pathophysiological consequences.
Two distinct self-destructive processes, autophagy (‘self-eating’) (BOX 1) and apoptosis (‘self-killing’) (BOX 2), determine the turnover of cytoplasmic organelles and entire cells, respectively. Loss and gain of either autophagy or apoptosis influence numerous pathological processes, and these phenomena affect each other1.
Box 1. Autophagy — the basics.
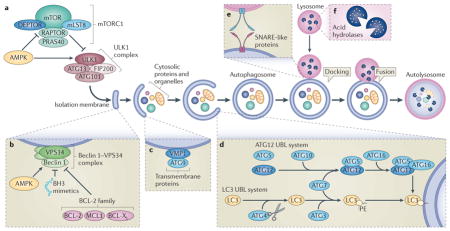
The core autophagy pathway starts with the formation of an isolation membrane (also called a phagophore), most often at the contact sites between mitochondria and the endoplasmic reticulum111. However, plasma membranes or other cytoplasmic organelles, including the Golgi, may constitute additional membrane sources for the generation of autophagosomes. As shown in the figure, autophagy involves the spatially and temporarily coordinated activation of multiple molecular components, including the ULK1 (UNC-51-like kinase 1)–FIP200 (FAK family kinase-interacting protein of 200 kDa) ATG13 ATG101 complex, which is functionally coupled to the negative autophagy regulator, mTOR complex 1 (mTORC1; see the figure, part a), and initiates autophagy; the lipid kinase vacuolar protein sorting 34 (VPS34) Beclin 1 complex, which is usually inactivated by anti-apoptotic proteins from the BCL-2 family and by other signalling compounds, but when active drives the nucleation of the isolation membrane (see the figure, part b); two transmembrane proteins, ATG9 and vacuole membrane protein 1 (VMP1), which recycle between the Golgi, endosomes and autophagosomes, probably participating in the recruitment of lipids to the isolation membrane (see the figure, part c); two ubiquitin-like (UBL) protein conjugation systems (ATG12 and protein light chain 3 (LC3)) that between them involve one protease (ATG4, which cleaves LC3 at its carboxyl terminus), the E1-like enzyme ATG7 (common to both conjugation systems), and the E2-like enzymes ATG10 (ATG12 system), and ATG3 (LC3 system), which together catalyse the covalent conjugation of ATG12 to ATG5 (which together with ATG16 forms the E3-like ligase of LC3) and that of phosphatidylethanolamine (PE) to LC3 (see the figure, part d); several SNARE-like proteins that mediate fusion between autophagosomes and lysosomes (see the figure, part e); and various lysosomal enzymes that hydrolyse proteins, lipids and nucleic acids at a low optimum pH14 (see the figure, part f). Note that LC3 remains associated with autophagosomes and autolysosomes, facilitating their identification.
Most assays for autophagy evaluate the redistribution of LC3 and its homologues (such as GABARAP (GABA receptor-associated protein)) to autophagosomes and autolysosomes by immunohistochemical labelling, or by imaging them in cells after fusion to fluorescent proteins such as GFP. Alternatively, autophagy assays quantify the lipidation of these proteins, which causes an increase in their electrophoretic mobility that is detectable by standard immunoblots11. Autophagic cargo is often recognized by the presence of linear Lys63 ubiquitylation, which can tag cargo for uptake by autophagosomes. Organelles or proteins that are marked with Lys63-linked ubiquitin chains interact with a series of adaptors, which possess a LC3-interacting region (LIR) that specifically interacts with LC3-like proteins, thus targeting the cargo to autophagosomes. Such adaptors, which include sequestosome 1 (SQSTM1), are destroyed during autophagy, hence a reduction of their abundance enables an indirect measurement of autophagy11.
AMPK, AMP-activated protein kinase; BCL-XL, BCL extra large; BH3, BCL-2 homology 3; DEPTOR, DEP domain-containing mTOR-interacting protein; MCL1, myeloid cell leukaemia sequence 1; mLST8, mammalian lethal with SEC13 protein; PRAS40, 40 kDa Pro-rich AKT substrate; RAPTOR, regulatory-associated protein of mTOR.
Box 2. Apoptosis and other cell death modalities.
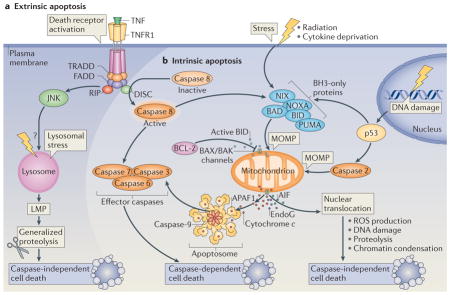
The morphological classification of cell death modalities is being progressively replaced by biochemical definitions of the underlying pathways79.
Extrinsic apoptosis
This occurs in response to ligation of the so-called death receptors, which are CD95 (also known as FAS), tumour necrosis factor receptor 1 (TNFR1; see the figure, part a) or TNF-related apoptosis-inducing ligand receptor (TRAILR). This results in the recruitment of several proteins, including FAS-associated death domain (FADD), TNFR1-associated death domain (TRADD) and caspase 8. Activated caspase 8 then proteolytically activates downstream effector caspases or truncates the BH3 (BCL-2 homology 3)-only protein BID (BH3-interacting domain death agonist), which co-activates the intrinsic pathway of apoptosis by translocating to mitochondria.
Intrinsic apoptosis
This is marked by one central event — mitochondrial outer membrane permeabilization (MOMP) — which results in the release of cytochrome c from the mitochondrial intermembrane space (see the figure, part b). Cytosolic cytochrome c then triggers the assembly of a caspase-activating complex between caspase 9 and apoptotic protease-activating factor 1 (APAF1; the apoptosome). MOMP can be triggered by the activation of BH3-only proteins following their mobilization from other subcellular compartments and their post-translational modification (such as phosphorylation or proteolysis) or transcriptional upregulation (for instance in response to p53 activation)34. BH3-only proteins generally stimulate MOMP by inducing the oligomerization of BCL-2-associated X protein (BAX) and/or BCL-2 antagonist or killer (BAK) in the outer mitochondrial membrane, thereby forming supramolecular channels that mediate the liberation of soluble proteins from the mitochondrial intermembrane space112–114. DNA damage can stimulate the transactivation of genes encoding pro-apoptotic proteins (such as the BH3-only proteins p53 upregulated modulator of apoptosis (PUMA) and NOXA) by p53, or activate caspase 2 in a p53-dependent manner. Caspase 2 may then induce MOMP and/or activate other caspases, such as caspase 3, directly. Among the mitochondrial proteins that are released as a result of MOMP, apoptosis-inducing factor (AIF) and endonuclease G (EndoG) can promote caspase-independent cell death115, which can also result from stimuli that cause lysosomal membrane permeabilization (LMP) to release cathepsin proteases into the cytosol116. Cathepsins can also trigger MOMP, thereby stimulating the intrinsic apoptotic pathway.
Regulated necrosis
This is often referred to as ‘necroptosis’ and is a caspase-independent process that involves a series of other effector molecules, including receptor-interacting protein 1 (RIP1) and RIP3 kinases (see the figure, part a)117.
Mitotic catastrophe
This constitutes an onco-suppressive mechanism that is initiated by disruption of the mitotic apparatus during the M phase of the cell cycle, which is preceded by some degree of mitotic arrest and eventually causes cell death or senescence118.
Autophagic cell death
Another cell death modality that, by definition, does not involve apoptotic or necrotic effector molecules, or occurs during mitosis (BOX 3).
BAD, BCL-2 antagonist of cell death; DISC, death-inducing signalling complex; JNK, JUN N-terminal kinase; NIX, NIP3-like protein X; ROS, reactive oxygen species.
Macroautophagy (hereafter referred to as autophagy) involves the sequestration of portions of the cytoplasm within double-membrane vesicles (autophagosomes), which fuse with lysosomes to form autolysosomes in which autophagic cargo is degraded2–4. This catabolic process, which is mediated by a number of autophagy and autophagy-related proteins (BOX 1), is primarily used for cellular adaptation to dwindling nutrient resources5,6 and is generally considered a protective response. However, in some cases it may also participate in cell death7,8, and it has sometimes been viewed as a separate modality of programmed cell death (PCD) (BOX 3). As one of the first steps of autophagy is the formation of autophagosomes9,10, its activity is usually assessed on the basis of autophagosome accumulation. Two opposing mechanisms can promote autophagosome accumulation, namely an increase in the rate of autophagosome formation (and hence an increase in autophagic flux) or a decrease in the rate of autophagosome clearance that follows their fusion with lysosomes (and hence a decrease in autophagic flux)11, which complicates the interpretation of autophagy assays12.
Box 3. Autophagic cell death.
The mere presence of autophagosomes in dying cells, especially if the cytoplasmic volume is not reduced, cannot be interpreted as a sign of autophagic cell death (ACD), and we suspect that many descriptive cases of so-called ACD actually reflect death accompanied by the block of autophagic flux rather than death mediated by autophagy. A strict definition of ACD restricts the use of this term to cell death by autophagy (rather than cell death with autophagy), in which the specific suppression of autophagy prevents cell death and the final cell death process is mediated by enhanced autophagic flux rather than by apoptosis or necroptosis18,77,78,79.
Much of the evidence in favour of true ACD has been produced in model organisms, including nematodes119,120 and the fruitfly Drosophila melanogaster. During D. melanogaster development, a rise in the hormone ecdysone triggers the programmed cell death (PCD) of obsolete larval tissues, including the salivary glands and the midgut, which both sequentially manifest characteristics of autophagy and caspase-dependent apoptosis. The combined inhibition of autophagy (by mutation or knockdown of multiple Atg genes) and caspases blocks salivary gland degradation more efficiently than blockade of either of these processes alone121,122. The suppression of the caspase activation pathway has no effect on midgut removal123. By contrast, genetic impairment of autophagy by mutations in either Atg2 or Atg18, or knockdown of either Atg1 or Atg18, delays midgut PCD123. Autophagy in the midgut does not require the core E1 and E2 conjugating enzyme-encoding autophagy genes Atg7 and Atg3 (REF. 124). Rather, these cells depend on ubiquitin-activating enzyme 1 (Uba1), the E1 enzyme required for ubiquitylation, which operates downstream of Atg1 (UNC-51-like kinase 1 (ULK1) in mammals)8.
In mammals, evidence for the existence of ACD is scarce, and most reports rely on phenomenological descriptions or the use of nonspecific pharmacological autophagy inhibitors, such as 3-methyladenine or chloroquine. Moreover, the inhibition of cell death achieved by specific manipulations, such as siRNA-mediated depletion of Atg genes, is usually incomplete125. However, depletion of ATG5, ATG7 or Beclin 1 can prevent death and restore the clonogenicity of cells that would usually die following overexpression of oncogenic RAS126,127. This observation, which relies on acute RAS overexpression, contrasts with the finding that oncogenic transformation by sustained expression of oncogenic RAS actually requires an intact autophagic machinery56,57,58.
Altogether, the available evidence indicates that ACD does not have an essential role in mammalian development and physiological PCD; however, it may contribute to pathological or drug-induced cell loss. Pyramidal neuronal death in the hippocampus after hypoxia or ischaemia is reduced in mice lacking Atg7 in neurons, but proceeds normally in mice lacking caspase 3 (REF. 128). Maladaptive autophagy induced by pressure overload can precipitate cardiac failure, and both autophagy and cardiac dysfunction are avoided by heterozygous disruption of the gene coding for Beclin 1 (REF. 129). Knockdown of Atg5 or Becn1, but not the addition of the caspase inhibitor zVAD.fmk or the necroptosis inhibitor necrostatin, can protect mice from otherwise lethal infection by the H5N1 influenza virus130. Knockdown of LC3 can reduce the susceptibility of tumour cells to C18 ceramide-induced growth inhibition in vivo. C18 ceramide causes dynamin-related protein 1 (DRP1)-dependent mitochondrial fission, as it incorporates into mitochondrial membranes and directly interacts with LC3, thereby stimulating lethal mitophagy131.
Apoptosis is one of several types of PCD (BOX 2) and is characterized by a series of morphological changes, including nuclear condensation (pyknosis) and fragmentation (karyorrhexis), as well as plasma membrane blebbing, which lead to the formation of apoptotic bodies. Apoptosis is accompanied by a sequence of characteristic biochemical changes, including mitochondrial outer membrane permeabilization (MOMP), activation of the effector caspases caspase 3, caspase 6 and caspase 7, and the activation of catabolic hydrolases that degrade most of the macromolecules of the cell, which includes DNA13. These morphological and biochemical hallmarks facilitate the detection of apoptosis, although cells that die in vivo are usually engulfed and degraded by healthy cells before they acquire the full apoptotic phenotype, which means that the incidence of apoptosis is usually underestimated13.
Autophagy and apoptosis often occur in the same cell, mostly in a sequence in which autophagy precedes apoptosis1. This is because stress often stimulates an autophagic response, especially if the level of stress is not lethal. Apoptotic and non-apoptotic lethal programmes are activated when stress exceeds a critical duration or an intensity threshold. In many cases, autophagy constitutes a strategy to adapt to and cope with stress14. Nonetheless, if the cell commences apoptosis, autophagy can be inactivated, in part owing to the caspase-mediated cleavage of essential autophagy proteins. Beyond this general scenario, in particular circumstances autophagy or essential proteins involved in the autophagic process may promote cellular demise, either by catabolizing indispensable portions of cells or facilitating the activation of apoptotic or necrotic programmes, respectively13 (BOX 3).
In this Review, we discuss the complex relationship between autophagy and apoptosis, with a focus on the impact of intersecting signal transduction pathways on the final fate of animal cells in health and disease.
Autophagy precedes apoptosis
Many stimuli that ultimately cause cell death also trigger autophagy. In such cases, autophagy usually manifests well before apoptosis dismantles the cell. Autophagy induction is exacerbated if apoptosis is suppressed, for instance, by removing pro-apoptotic proteins such as BCL-2-associated X protein (BAX) and BCL-2 antagonist or killer (BAK) or by adding caspase inhibitors, including those encoded by viruses15,16. A rapid induction of autophagy, reflecting the instinct of the cell to adapt to stress, is followed by the activation of cell death pathways in response to multiple external signals such as ionizing radiation, chemotherapeutic anticancer agents, the inhibition of growth factor receptors or a scarcity of essential nutrients17,18. The fact that many signal transduction pathways that are elicited by cell-intrinsic stress regulate both autophagy and apoptosis might explain the sequential activation of both processes. This applies to transcription factors such as p53 and to other key signal-transducing molecules, as detailed below.
The p53 protein
The tumour suppressor protein p53 functions as a signal transduction integrator that is activated by various stressors as diverse as DNA damage, ischaemia–reperfusion and nutrient stress. p53 is usually present in the cytosol but translocates to the nucleus upon DNA damage following its phosphorylation by a number of distinct stress-activated kinases19. The available evidence indicates that the cytosolic pool of p53 represses autophagy and that the nuclear translocation of p53 leads to a decrease in this p53 pool, thereby facilitating the induction of autophagy20 (FIG. 1a). Cytosolic p53 represses autophagy by interacting with the autophagy protein FIP200 (FAK family kinase-interacting protein of 200 kDa; Atg17 in yeast), thus blocking the activation of the ULK1 (UNC-51-like kinase 1)–FIP200–ATG13–ATG101 complex and inhibiting autophagosome formation21,22 (BOX 1). Moreover, in conditions of cellular stress, a portion of cytosolic p53 can translocate to the mitochondrial matrix, where it interacts with cyclophin D and promotes opening of the permeability transition pore (PTP; a multiprotein pore that permeabilizes the inner mitochondrial membrane)23. This dissipates the proton gradient that is required for oxidative phosphorylation24. At a low intensity, PTP opening can stimulate the autophagic removal of dysfunctional mitochondria25. However, PTP opening beyond a critical threshold, which is one of the mechanisms causing MOMP, constitutes a lethal event24. Once in the nucleus, p53 transactivates a myriad of genes, influenced by its cofactors and by epigenetic marks (FIG. 1a). These transcriptional programmes then cause adaptation to stress, cell cycle arrest, autophagy and/or apoptosis26. p53 binds to the promoters of multiple genes, the protein products of which regulate autophagy, including upstream regulators, such as the β1, β2 and γ-subunits of AMP-activated protein kinase (AMPK), several regulators of the AMPK and mTOR1 complexes, proteins of the autophagy core pathway and lysosomal proteins27,28,29. Another member of the p53 family, p73, also transactivates pro-autophagic genes, in particular ATG5 (REF. 30). DNA damage-regulated autophagy modulator 1 (DRAM1) exemplifies a p53-induced gene product that stimulates autophagic flux through as yet elusive mechanisms31,32. However, DRAM1 may induce lethal lysosomal membrane permeabilization (LMP) in specific circumstances, for instance in HIV-1 infected CD4+ T cells33. In addition, p53 transactivates multiple pro-apoptotic genes, including genes encoding for members of the BCL-2 family such as BAX, BCL-2-interacting mediator of cell death (BIM; also known as BCL2L11) and p53 upregulated modulator of apoptosis (PUMA; also known as BBC3)34.
Figure 1. Signals that induce both apoptosis and autophagy.

a | p53. The tumour suppressor p53 exerts both pro- and anti-autophagic functions, which depends on its subcellular localization and activation status. When present in the cytoplasm, p53 inhibits autophagy by interacting with FIP200 (FAK family kinase-interacting protein of 200 kDa) and thus interfering with ULK1 (UNC-51-like kinase 1) complex activity. In response to stress, p53 translocates to the nucleus, where it binds to the promoter region of multiple pro-autophagic genes, including the β1, β2 and γ-subunits of AMP-activated protein kinase (AMPK), damage-regulated autophagy modulator 1 (DRAM1), sestrin 1, sestrin 2 and phosphatase and tensin homologue (PTEN). Nuclear p53 also induces transcription of multiple pro-apoptotic genes. Under certain circumstances, p53 translocates to mitochondria, where it triggers mitochondrial outer membrane permeabilization (MOMP), and thus induces both mitophagy (at first) and apoptosis (when the intensity of the pro-apoptotic stimuli overcomes the protective ability of mitophagy). b | BH3 (BCL-2 homology 3)-only proteins and BH3 mimetics. Through its BH3 domain, Beclin 1 interacts with anti-apoptotic proteins from the BCL-2 family. This inhibits the pro-autophagic function of Beclin 1, but does not interfere with the anti-apoptotic activity of the BCL-2 family proteins. BH3-only proteins, as well as BH3 mimetics, disrupt this interaction, which enables Beclin 1 to increase autophagic activity. One particular BH3-only protein, BCL-2-interacting mediator of cell death (BIM), has a peculiar effect on autophagy. BIM interacts with Beclin 1 and mislocalizes it to dynein light chain 1 (DLC1; not shown), thereby preventing autophagy. Moreover, another BH3-only protein, NIX (NIP3-like protein X), which localizes to mitochondria, favours mitophagy. c | Death associated protein kinase (DAPK). This kinase can stimulate autophagy by phosphorylating Beclin 1, which enables its dissociation from BCL-2 and its association with vacuolar protein sorting 34 (VPS34). DAPK can also activate a second kinase, protein kinase D (PKD), which phosphorylates and activates VPS34. d | JUN N-terminal kinase (JNK). JNK induces autophagy by phosphorylating BCL-2 or BIM, which disrupts their inhibitory interaction with Beclin 1. This leads to an active Beclin 1–VPS34 complex and autophagy and also prevents BCL-2 from inhibiting pro-apoptotic proteins, thus promoting apoptosis. BAD, BCL-2 antagonist of cell death; BCL-XL, BCL extra large; BID, BH3-interacting domain death agonist; PUMA, p53 upregulated modulator of apoptosis.
BH3-only proteins
Several BH3 (BCL-2 homology 3)- only proteins have the dual capacity to activate autophagy and apoptosis (FIG. 1b). To induce apoptosis, BH3-only proteins directly interact with multidomain proteins from the BCL-2 family, thereby neutralizing those that have anti-apoptotic roles and stimulating those with pro-apoptotic functions. This gives rise to a complex cascade of reactions that is dictated by the subcellular localization of these proteins, their phosphorylation status and the affinities of each of the protein–protein interactions35. Several BH3-only proteins, including BAD (BCL-2 antagonist of cell death), BID (BH3-interacting domain death agonist), BNIP3 (BCL-2 and adenovirus E1B 19 kDa protein-interacting protein 3), NIX (NIP3-like protein X; also known as BNIP3L), NOXA (also known as PMAIP1) and PUMA, can also promote autophagy by competitively disrupting the inhibitory interactions between Beclin 1, one of the central regulators of autophagy, and the anti-apoptotic proteins from the BCL-2 family (BCL-2, BCL-XL (BCL extra large) or MCL1 (myeloid cell leukaemia sequence 1)) (FIG. 1b). Pharmacological BH3 mimetics elicit a similar effect36,37,38,39. The separation of Beclin 1 from BCL-2 or BCL-XL activates the PI3K activity of the autophagy protein vacuolar protein sorting 34 (VPS34), for which Beclin 1 is a necessary co-activator40. In addition, the BH3-only protein NIX may stimulate the selective removal of damaged mitochondria (a process known as mitophagy) by directly interacting with GABA receptor-associated protein (GABARAP), which is a functional homologue of the autophagy protein light chain 3 (LC3; also known as MAP1LC3B) that is present on autophagosomes41. Moreover, BIM directly interacts with Beclin 1 and mislocalizes it to dynein light chain 1 (DLC1; also known as DYNLL1), thereby inhibiting Beclin 1 and autophagy activation42. Thus, BIM exemplifies a BH3-only protein that induces apoptosis, yet inhibits autophagy. Beyond this exception, it is noteworthy that most of the proteins interacting with Beclin 1 modulate autophagy and apoptosis in the same direction; that is, they inhibit or promote both processes (Supplementary information S1 (table)). It is not entirely clear through which mechanisms BH3-only proteins sequentially stimulate autophagy and apoptosis. One possibility is that BH3–only proteins induce autophagy at low, initial levels of stress by activating the Beclin 1–VPS34 complex, when mitochondria are still protected against lethal permeabilization. However, at a higher, more advanced level of stress, BH3-only proteins would induce MOMP, thereby setting off the apoptotic cascade43 (BOX 2).
Ser/Thr kinases
Ser/Thr kinases, including death-associated protein kinase (DAPK), JUN N-terminal kinase (JNK) and AKT (also known as PKB), have also been implicated in the regulation of both autophagy and apoptosis and hence may contribute to the sequential or simultaneous induction of both phenomena. DAPK phosphorylates Beclin 1 within its BH3 domain (at Thr119), which reduces its inhibitory interaction with BCL-2 or BCL-XL (REF. 44). Moreover, DAPK can activate protein kinase D (PKD), which then phosphorylates and activates VPS34 (REF. 45); in both of these cases, this promotes the induction of autophagy (FIG. 1c). DAPK also mediates autophagy-independent pro-apoptotic effects46, for instance following activation of protein phosphatase 2A (PP2A) when the UNC5H dependence receptor (a receptor that transmits lethal signals when its ligand is absent) is unligated47 or in the context of ceramide-induced anoikis48 (a cell death modality caused by detachment of adherent cells from their substrate). JNK can trigger both apoptosis and autophagy by virtue of its capacity to phosphorylate BCL-2 in its flexible loop between the BH4 and BH3 domains (at Thr69, Ser70 and Ser87), which decreases its inhibitory interaction with Beclin 1 and with pro-apoptotic members of the BCL-2 family (FIG. 1d). Cells that express phosphorylation site mutant variants of BCL-2 are resistant to the induction of autophagy and apoptosis by starvation43. In addition, mice bearing such a BCL-2 knock-in phosphorylation mutation are resistant to starvation- and exercise-induced autophagy49. JNK also phosphorylates BIM, which leads to the dissociation of BIM and Beclin 1 from DLC1 (REF. 42). Another kinase with dual autophagy–apoptosis regulatory potential is AKT. Growth-stimulatory signalling by the PI3K–AKT axis usually represses both autophagy and apoptosis, meaning that inhibition of PI3K or AKT induces both processes. AKT can phosphorylate Beclin 1 and BAD, thus stimulating their interaction with 14-3-3 to inhibit their pro-autophagic and pro-apoptotic functions, respectively50,51. Furthermore, growth factor-mediated activation of AKT represses autophagy by inducing mTOR and repressing the ULK1 complex (Atg1 complex in yeast) 52,53. Taken together, this explains why both autophagy and apoptosis are triggered when DAPK or JNK are activated or AKT is inhibited. Again, it seems plausible that cytoprotective autophagy is triggered by low levels of kinase-regulated stress, whereas more intense and protracted stress culminates in apoptotic demise.
Oncogenes
Oncogenes are an additional example of a stress that induces both autophagy and apoptosis. The sudden activation of the proto-oncogene MYC induces apoptosis in most cells as a tumour-suppressive fail-safe mechanism54. Through its stimulatory effect on global translation, MYC activation also elicits an unfolded protein response (UPR), which in turn activates cytoprotective autophagy via the endoplasmic reticulum stress kinase protein kinase RNA-like ER kinase (PERK) to favour cell survival55. Whether MYC preferentially induces apoptosis or autophagy-dependent survival is probably determined by the cellular context55. When introduced into normal cells, oncogenic H-RASV12 and K-RASV12 stimulate autophagy, which is then required for cell survival and oncogenic transformation, presumably because it maintains a functional pool of mitochondria and has other positive bioenergetic effects56,57, or because it enables the cells to prevent cellular senescence58. Conversely, in established tumours, the RAS–PI3K–mTOR signalling pathway may suppress autophagy, and inhibition of RAS then stimulates cytoprotective autophagy, limiting the desired death of cancer cells59. The molecular circuitries through which abrupt activation of RAS stimulates an autophagic stress response during oncogenic transformation — in contrast to the inhibition of autophagy by the sustained presence of RAS in malignant cells — are not fully understood. Nonetheless, these examples underscore the observation that aberrant homeostasis can stimulate autophagy as an early response that precedes (and often avoids) cell death, even when the same cellular parameter is affected in opposing directions (for example, the activation or inhibition of RAS).
Inhibitory crosstalk
Although autophagy and apoptosis are under the control of multiple common upstream signals, these processes also cross-regulate each other, mostly in an inhibitory manner. Thus, autophagy reduces the propensity of cells to undergo apoptosis, and activation of the apoptotic programme is coupled to the suppression of autophagy.
Inhibition of apoptosis by autophagy
One of the principal mechanisms through which autophagy reduces the tendency of cells to undergo apoptosis is mitophagy25. Mitochondria constitute the ‘battleground’ where survival and death signals converge to determine whether or not intrinsic apoptosis, which is marked by MOMP, is initiated24. Indeed, hundreds of distinct factors, including proteins, lipids and metabolites, affect the functional and physical integrity of the two mitochondrial membranes. During cell death, MOMP causes the release of catabolic hydrolases (such as apoptosis-inducing factor (AIF) and endonuclease G) and caspase activators (such as cytochrome c and SMAC (second mitochondria-derived activator of caspase; also known as DIABLO)) from mitochondria and causes the inner mitochondrial transmembrane potential (ΔΨm) to dissipate, which gives rise to a bioenergetic catastrophe (FIG. 2A). These changes mark the ‘point-of-no-return’ during intrinsic apoptosis24. Damaged mitochondria are particularly prone to activating the apoptotic programme (BOX 2), so their removal by autophagy can increase the threshold for apoptosis induction.
Figure 2. Effects of autophagy on lethal signalling.

A–C | Inhibition of apoptosis and anoikis by autophagy. A | Distinct types of proteins, lipids and metabolites may induce mitochondrial damage. Once established, mitochondrial outer membrane permeabilization (MOMP) leads to the release of catabolic hydrolases (apoptosis-inducing factor (AIF) or endonuclease G (EndoG)) and caspase activators (such as cytochrome c and SMAC (second mitochondria-derived activator of caspase)) into the cytoplasm. These changes usually mark the ‘point-of-no-return’ of intrinsic apoptosis. The selective autophagy of damaged mitochondria (mitophagy) delays intrinsic apoptosis by limiting the release of pro-apoptotic factors. B | The extrinsic pathway of apoptosis is triggered by the binding of trimeric ligands to so-called death receptors, which include CD95, the tumour necrosis factor receptor 1 (TNFR1, depicted in the figure) and the TRAIL (TNF-related apoptosis-inducing ligand) receptors. One key event in this pathway is the activation of caspase 8. Selective clearance of active caspase 8 by autophagy may delay the onset of extrinsic apoptosis after death receptor stimulation. As caspase 8-induced apoptosis often relies on the cleavage and activation of the BH3-only protein BID (BH3-interacting domain death agonist), which is a potent MOMP inducer, mitophagy may also inhibit lethal signalling via death receptors. C | The selective clearance of the SRC protein by autophagy, which is overactivated in response to disrupted focal adhesion kinase (FAK) signalling, delays the onset of anoikis, which is induced by cell detachment from the extracellular matrix. D, E | Role of autophagy proteins in cell death. Although it generally constitutes a cytoprotective process, autophagosomes or ATG proteins may participate in lethal signalling in some specific circumstances. Da | Autophagosomes may act as platforms for caspase 8 activation, although the precise mechanism (or mechanisms) underlying this event remain poorly understood (indicated by the question mark). Db | In Drosophila melanogaster, the selective autophagy of anti-apoptotic proteins, such as Bruce (BIR-containing ubiquitin-conjugating enzyme; an orthologue of the mammalian inhibitor of apoptosis proteins (IAPs)), contributes to the induction of apoptosis. Dc | Autophagosome accumulation, due to the saturation of the degradative capacity of lysosomes, may facilitate PTP opening and thus lethal MOMP by hitherto unknown mechanisms (indicated by the question mark). Ea | Certain pro-apoptotic stimuli, such as C6 ceramide or tunicamycin, induce the interaction of ATG12 with anti-apoptotic BCL-2 or MCL1 (myeloid cell leukaemia sequence 1) proteins, thereby inhibiting their function and increasing apoptosis. Eb | ATG7 facilitates lysosomal membrane permeabilization, one of the entry points of the apoptotic pathway, after lysosomal photodamage. DISC, death-inducing signalling complex; PTP, permeability transition pore.
How can autophagy selectively target damaged mitochondria for degradation? A reduction in ΔΨm, which indicates a loss-of-function of the respiratory chain or the permeabilization of the inner membrane, stimulates the Lys63-linked ubiquitylation of proteins anchored in the outer mitochondrial membrane, including voltage-dependent anion-selective channel 1 (VDAC1), mitofusin 1 (MFN1) and MFN2, which tags the organelle for mitophagy. This is possible because dissipation of the ΔΨm shuts off a protease that usually destroys the kinase PINK1 (PTEN-induced putative kinase 1), causing PINK1 to accumulate at the surface of damaged mitochondria. PINK1 then recruits and phosphorylates PARKIN (Parkinson’s disease protein), an E3 ubiquitin ligase that ubiquitylates substrates in the outer mitochondrial membrane, targeting the organelle for autophagic destruction25. In addition to ΔΨm dissipation, mitochondrial fragmentation through fission is required to cause mitophagy, which constitutes a fail-safe mechanism against the unwarranted removal of functional mitochondria60. Mitochondria from starved cells undergo ΔΨm dissipation but do not fragment, meaning that they do not experience mitophagy60. By contrast, mitochondria that undergo opening of the PTP show ΔΨm dissipation and are fragmented, which enables their removal by autophagy61.
Autophagy also seems to have a role in the suppression of unwarranted cell death in the pancreas. Pancreatic acinar cells contain inactive enzyme precursors (zymogens), such as trypsinogen (the precursor of trypsin), that are sequestered in granules but may leak into the cytosol to cause cell death. In a model of cerulein-induced pancreatitis, transgenic overexpression of the autophagy-promoting protein vacuole membrane protein 1 (VMP1) stimulated the autophagic removal of granules containing aberrantly activated trypsin62. VMP1 overexpression reduces enzymological, histopathological and macroscopic signs of pancreatitis62, whereas blocking autophagic flux by depleting VMP1 exacerbates pancreatitis that has been induced by a combination of ethanol and bacterial lipopolysaccharide63. Thus, VMP1 promotes the autophagic removal of granules to prevent cell death.
Autophagy can also attenuate cell death by selectively reducing the abundance of pro-apoptotic proteins in the cytosol (FIG. 2B). Indeed, autophagy can selectively target specific proteins that have been ubiquitylated; this modification enables them to interact with autophagy receptors, a series of adaptors, including sequestosome 1 (SQSTM1; also known as p62), that bind both ubiquitylated substrates and LC3 (REF. 64). As an example, colon cancer cells lacking the pro-apoptotic protein BAX are resistant to TRAIL (TNF-related apoptosis-inducing ligand)-induced cell death unless autophagy is inhibited. This is because autophagy mediates the selective removal of active caspase 8 (REF. 65). Similarly, in a model of tumour necrosis factor (TNF)-induced hepatocyte apoptosis, inhibition of autophagy by liver-specific knockout of Atg7 increased caspase 8 activity66, presumably caused by failed clearance of caspase 8. Moreover, autophagy can target the kinase SRC, thereby enabling cancer cells to avoid anoikis owing to disrupted signalling by focal adhesion kinase (FAK) (FIG. 2C). In this context, the proto-oncogene Tyr kinase SRC binds to the E3 ubiquitin protein ligase CBL, which possesses an LC3-interacting region that serves as a molecular adaptor to target SRC to LC3-positive phagophores67. Finally, autophagy decreases the abundance of SQSTM1, a protein that, if overexpressed, can stimulate the production of reactive oxygen species (ROS) and cell death68.
Inhibition of autophagy by apoptosis
When the intensity or duration of stress reaches the limit of the cell, adaptive mechanisms, including autophagy, are overwhelmed and suicidal cell death programmes are activated. Apoptosis, which is one of the most frequently occurring cell death modalities, is usually accompanied by a high degree of caspase activation (BOX 2). This results in the cleavage of multiple proteins, which promotes an apoptotic morphology. Caspases can digest several essential autophagy proteins, resulting in the inactivation of the autophagic programme, perhaps with the goal to abort its cyto-protective function and to accelerate cellular demise. Among their targets are ATG3 (REF. 69) and Beclin 1 (REF. 70,71), the degradation of which leads to the loss of their autophagy-stimulatory function. Moreover, the autophagy protein AMBRA1 (activating molecule in BECN1-regulated autophagy 1) is irreversibly degraded by the combined activity of caspases and calpains, and cells expressing an AMBRA1 mutant lacking the caspase 3 cleavage site are relatively resistant to apoptosis induction72. However, it is important to note that, theoretically, cleavage site-mutated proteins could function as pseudo-substrates and hence inhibit the enzymatic activity of caspases73, thereby causing difficulties in interpreting experiments in which a mutated protein is expressed at higher levels than its endogenous equivalent. The observed phenotype might result from general caspase inhibition rather than from the suppression of target protein cleavage.
Consistent with a role for the degradation of autophagy proteins in accelerating cell death, several autophagy protein fragments that result from caspase cleavage are thought to acquire pro-apoptotic functions. The cleavage of Beclin 1 by caspase 3, caspase 6 or caspase 9 results in the generation of a carboxy-terminal fragment, downstream of the BH3 domain, that localizes to mitochondria in cells and permeabilizes isolated mitochondria in vitro to cause the release of cyto-chrome c71. Upon its cleavage by caspase 3, ATG4D also acquires a pro-apoptotic function that may be mediated by a BH3-like domain74. On a similar note, the calpain-mediated cleavage of ATG5 results in the generation of an amino-terminal fragment that translocates to mitochondria and sensitizes cells to apoptosis. This ATG5 fragment also interacts with BCL-XL, although the precise molecular contact points and functional implications of this interaction remain elusive75. Nonetheless, these findings suggest that the upregulation of ATG4D, ATG5 and Beclin 1 expression as a result of increased transcription does not only stimulate autophagy but, in addition, generates precursors of proteins that, upon caspase activation, can accelerate apoptotic demise.
One unique example of the regulation of autophagy by caspases is illustrated by the degradation of BCL-2-associated transcription factor 1 (BCLAF1) by caspase 10 (REF. 76). Caspase 10 is an apical caspase that, like caspase 8, can contribute to the induction of apoptosis in response to TNF binding to death receptors. However, in multiple myeloma cells, caspase 10 is constitutively active to degrade BCLAF1, which prevents this molecule from interacting with BCL-2 and competitively disrupting the BCL-2–Beclin 1 complex. Inhibition of caspase 10 causes an uncontrolled increase in autophagic flux that culminates in cell death without the features of apoptosis76. This case illustrates the connection between two unusual phenomena — the cytoprotective action of a caspase (because caspase activation usually results in cell death) and excessive, lethal autophagy. Whether this paradigm constitutes an exception or the first example of a new caspase paradox remains to be determined.
Autophagy activates apoptosis
Autophagy can mediate cell death as an effector mechanism, in a process known as autophagic cell death (ACD) (BOX 3). ACD refers to cell death by autophagy (rather than cell death with autophagy) in which the specific suppression of autophagy by inhibiting, depleting or deleting several distinct essential autophagy genes and/or proteins prevents cell death; the final cell death process is mediated by autophagy rather than another cell death modality such as apoptosis or necroptosis18,77,78,79. However, there are also several examples in which the induction of autophagy facilitates the activation of apoptosis. Moreover, individual proteins that have an essential role in autophagy may have additional roles in pro-apoptotic signalling. In this context, it is important to note that some ATG genes are not exclusively involved in autophagy but are also implicated in various other important cellular processes, including endocytosis and protein secretion80,81. Therefore, it is important to distinguish the function of genes and/or proteins from the process.
Through which mechanisms can autophagy favour the induction of apoptosis? Autophagosome formation, rather than the degradative process, has been implicated in the activation of caspase 8 after treatment of cells with SKI-I, a pan-sphingosine kinase inhibitor, and bortezomib (Velcade; Millennium Pharmaceuticals), a proteasome inhibitor (FIG. 2Da). In this context, caspase 8 forms a complex with the death receptor adaptor protein FAS-associated death domain (FADD) and ATG5, colocalizes with ATG5, LC3 and SQSTM1, and becomes activated in an ATG5-, FADD- and SQSTM1-dependent manner82. In cells treated with SKI-I or bortezomib, inhibition of the early steps of autophagy (by knockout of Atg3 or Atg5) reduced the activation of caspase 8 and that of the effector caspase 3, whereas inhibition of the late steps of autophagy (by bafilomycin A1) increased caspase-dependent cell death82. These data indicate that autophagosome formation, rather than the complete process of autophagy, generates a platform for the activation of caspase 8. However, caspase 8 activation usually does not occur when autophagy is induced and liver-specific knockout of ATG7, a protein that acts at early steps of autophagy, can stimulate caspase 8 activation that has been elicited by TNF in vivo66. Which factors determine whether autophagosomes can stimulate caspase 8 activation remains elusive.
Autophagy may also stimulate apoptosis by depleting endogenous inhibitors of this cell death pathway. In Drosophila melanogaster, apoptosis is mostly regulated by the interplay between caspases and inhibitor of apoptosis proteins (IAPs). One of these IAPs, Bruce (BIR-containing ubiquitin-conjugating enzyme), is degraded by autophagy, which explains why the genetic inhibition of autophagy (by mutation of atg1, atg13 or vps34) prevents the developmental apoptosis of nurse cells in late oogenesis83 (FIG. 2Db).
In addition to stimulating cell death by apoptosis, autophagy can trigger necrosis, a modality of cell death that lacks salient features of apoptosis (such as pyknosis and karyorrhexis) and culminates in early rupture of the plasma membrane79. A study carried out in mouse L929 fibrosarcoma cells showed that autophagy can degrade catalase, the enzyme that detoxifies hydrogen peroxide, thereby promoting necrotic cell death84 (FIG. 2Dc). In myocardial ischaemia, autophagy is considered to be cardioprotective, but accumulating autophagosomes can become toxic when the blood supply (and thus oxygen) is restored (known as reperfusion). Indeed, upon reperfusion of hypoxic cardiomyocytes, autophagosomes are formed but fail to be removed. This may be explained by the reduced expression of lysosome-associated membrane protein 2 (LAMP2) as a result of tissue damage; LAMP2 is required for autophagosome–lysosome fusion85. Notably, restoring LAMP2 levels synergizes with partial BECN1 (which encodes Beclin 1) knockdown (which reduces, but does not fully block, the formation of autophagosomes) to restore autophagosome processing and to attenuate cell death86. When autophagosomes accumulate, cardiomyocytes succumb to a mitochondrial pathway of necrosis, which is blocked by cyclosporin A, an inhibitor of the mitochondrial PTP86 (FIG. 2Dc). It remains to be determined through which mechanisms accumulating autophagosomes may stimulate opening of the mitochondrial PTP86 and hence trigger apoptosis.
ATG proteins may also contribute to lethal signalling independently of the autophagic process. ATG12 is thought to have a role in activating caspases through the mitochondrial pathway (BOX 2), as its depletion (but not that of other essential ATG proteins) reduces caspase activation in response to various apoptotic stresses, including C6 ceramide, etoposide, paclitaxel, stauro-sporine or tunicamycin (FIG. 2Ea). This effect involves a BH3 domain in ATG12 that mediates its interaction with BCL-2 and MCL1, as well as its pro-apoptotic function, but is not required for its autophagic activity87. Similarly, ATG7 (but not ATG5) facilitates the induction of apoptosis after lysosomal photodamage, presumably by triggering lysosomal membrane permeabilization88, which, similarly to MOMP, can trigger apoptosis89 (FIG. 2Eb). These examples illustrate how individual ATG proteins can contribute to lethal signalling.
Intercellular crosstalk
Beyond their crosstalk within the same cell, autophagy and cell death shape the relationship among distinct cells in tissues. For example, soluble products released from necrotic cells may induce autophagy in neighbouring cells. This has been documented for a whole series of danger-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), S100 proteins and DNA complexes, which act on a series of pattern recognition receptors (PRRs), thereby stimulating autophagy90,91. The abundance of these DAMPs is expected to be particularly high in the context of uncontrolled pathological death, when necrosis affects vast tissue areas and the clearance of cellular debris is overwhelmed92. It is tempting to speculate that the induction of autophagy, which is presumably cytoprotective, by dead cell products can minimize cellular demise adjacent to areas in which cells are dying. Indeed, autophagy has been shown to avoid the loss of cardiomyocytes and neurons in the context of ischaemia93,94.
Autophagy occurring in cells before they undergo apoptosis (‘pre-mortem autophagy’) has a major effect on the fate of dead cells, with regard to their recognition and heterophagy by neighbouring or mobile phagocytic cells. In the context of developmental apoptosis, pre-mortem autophagy is required for the avoidance of inflammation by dead cells95 (FIG. 3a). This is because autophagy facilitates the secretion of the chemotactic signal lyso-phosphatidylcholine (LPC), which mediates the recruitment of phagocytic cells. Autophagy also promotes the cell surface expression of the ‘eat-me’ signal phosphatidyl-serine (PtdSer), which facilitates the engulfment of dead cells by phagocytes. When autophagy is inactivated in mouse embryonic tissues, dead cells accumulate and signs of inflammation become apparent95. Defective LPC secretion and PtdSer exposure on the surface of dying cells in Atg5−/− and Becn1−/− embryoid bodies could be restored by exogenously supplying methylpyruvate, a cell permeable tricarboxylic acid substrate that restores the reduced ATP levels of autophagy-deficient cells95. These findings suggest that pre-mortem autophagy may constitute a mechanism to sustain high levels of ATP in cells that undergo developmental apoptosis, thereby facilitating LPC secretion, PtdSer exposure and efficient removal of dead cells, and thereby preventing unwarranted inflammatory reactions.
Figure 3. Impact of autophagy on the removal of dead cells in tissues.

a | During the course of developmental cell death, pre-mortem autophagy is required for the release of lysophosphatidylcholine (LPC), a potent chemotactic signal, and for the exposure of the ‘eat-me’ signal phosphatidylserine (PtdSer), on the cell surface. Without these two events, recruitment of phagocytic cells is impaired, which results in the defective clearance of dead cells, increased inflammation and compromised development. b | Pre-mortem autophagy induction is essential for ATP release from dying cancer cells. Inhibition of ATP release compromises the recruitment of immune cells, and thus the activation of the immune system. This prevents the immune system from recognizing tumour-specific antigens, a process that is essential for the effectiveness of anti-tumour therapies.
In human or mouse cancer cells, pre-mortem autophagy is also required for the release of ATP, which is an important chemotactic signal for phagocytes96, from cells that are undergoing apoptosis97. This is in part because autophagy maintains high ATP levels in lysosomes and autophagolysosomes, from which ATP is extruded by lysosomal exocytosis98. Lysosomal exocytosis involves the fusion of lysosomes with the plasma membrane, through a process that requires LAMP1 (REF. 99) and vesicle-associated membrane protein 7 (VAMP7)100, and which results in the release of lysosomal content. Although autophagy is dispensable for lysosomal exocytosis, it is required for ATP release from cells through this mechanism98. Disabled autophagy in dying cells compromises the immune recognition of dead-cell antigens, as documented for both viral antigens101 and tumour-associated antigens97. The failure of dying autophagy-deficient cells to release ATP compromises the recruitment of myeloid and lymphoid cells to the tumour bed after either chemotherapy or radiotherapy, and therefore reduces immunosurveillance97 (FIG. 3b). Thus, chemical inhibitors of ecto-ATPases, which are extracellular enzymes that degrade ATP, can increase the ATP concentration in the interstitial fluid and enhance the recruitment of immune cells into the tumour after chemotherapy97 or radiotherapy102. These examples illustrate how pre-mortem autophagy affects the clearance of dead cells in vivo, thereby influencing inflammatory and immune responses103.
Concluding remarks and speculations
The mutual relationship between autophagy and apoptosis is highly context-dependent. Beyond rare cases of ACD, in which autophagy truly mediates cell death, there are a few examples in which autophagy or elements of the autophagic machinery may favour the activation of apoptotic or necrotic cell death programmes. However, in the majority of cases, it seems that apoptosis and autophagy are mutually inhibitory. In most instances, autophagy suppresses apoptosis (or at least raises the threshold of stress required to induce it), which means that autophagy tends to be anti-apoptotic rather than pro-apoptotic. As many cellular stress pathways sequentially induce autophagy (at early time points and low doses of stress) and apoptosis (at late time points and high doses of stress), the overall design of autophagy–apoptosis crosstalk reduces the propensity of cells to die (FIG. 4a, b). In addition, it seems that apoptotic caspase activation degrades essential autophagy proteins, shutting down the autophagic process and converting pro-autophagic proteins into pro-apoptotic ones (FIG. 4c). This general design of autophagy–apoptosis crosstalk has broad pathophysiological implications.
Figure 4. Functional relationship between autophagy and apoptosis.

a | Hypothetical scenario of an absent interplay between autophagy and apoptosis. If no functional connection between apoptosis and autophagy existed, the percentage of cells activating the autophagic pathway or instead succumbing to apoptosis would be solely determined by stress intensity over time. b | Effect of cytoprotective autophagy on a dose-response curve. If autophagy inhibits apoptosis, the cell population becomes relatively resistant to cell death, unless the intensity of the stressor overcomes the protective barrier of autophagy. c | Cytoprotective autophagy, followed by lethal subversion of the apoptotic machinery. In this scenario, autophagy would constitute an initial barrier against apoptosis when stress intensity is low (1). As stress intensity increases (2), the induction of apoptosis results in the subversion of cytoprotective mechanisms, including autophagy, and the conversion of cytoprotective molecules into cytotoxic ones. For example, cleavage of essential ATG proteins does not only inactivate the autophagic machinery but also leads to the generation of protein fragments with novel pro-apoptotic properties.
The mutually negative regulation of autophagy and apoptosis can be expected to increase the contrast between both processes, hence generating a steeper gradient that differentiates non-lethal from lethal stress levels (FIG. 4c). This may be important in the context of severe tissue damage, for instance, during ischaemia, in which autophagy is induced to limit tissue damage93,94. Ischaemic infarction of tissues is typically characterized by a central necrosis that is surrounded by a penumbra in which both apoptosis and autophagy are detectable. Within this zone, the mutually inhibitory cytoprotective and self-destructive mechanisms may help to delimit the infarcted area. Along the same lines, autophagy induction by soluble products released from dead necrotic cells, or apoptotic cells that have not been cleared, may constitute a strategy to limit tissue damage within the organism.
Usually, especially at low doses of damage (FIG. 4b, c), the cytoprotective function of autophagy prevails over its potential cytotoxic effects, and autophagy may be considered as one of the mechanisms of hormesis, the phenomenon in which mild, sublethal stress preconditions against the fatal manifestation of a later, normally lethal stress104. Thus, autophagy constitutes one of the mechanisms that mediates ischaemic preconditioning, that is, the protection of the heart or the brain against infarction by prior short episodes of sublethal ischaemia93,94. Similarly, autophagy is the mechanism through which chronic mild endoplasmic reticulum stress protects against degenerative diseases of the central nervous system, such as amyotrophic lateral sclerosis (ALS) in mouse models105. Although the precise molecular mechanisms through which autophagy mediates hermetic stress and prevents apoptosis or necrosis in these in vivo models have not been addressed, it is tempting to speculate that they involve the same mechanisms as those elucidated in vitro, including the autophagic removal of damaged or degenerating organelles and potentially lethal protein aggregates, hence reducing the propensity of cells to die.
In the context of cancer, autophagy is inhibited by multiple oncogene products (such as PI3K, AKT, BCL-2 and mutant p53), and inhibition of autophagy is a potentially oncogenic event106. It has been popular to explain this fact by the failure of autophagy-deficient cancer cell precursors to undergo ACD. Nonetheless, little or no evidence has been produced in favour of this possibility, and alternative explanations must be put forward. In one scenario the suppression of autophagy perturbs cellular metabolism106, provokes genome instability (for instance, by the failure to destroy micronuclei containing lagging chromosomes)107, interferes with differentiation (owing to the persistence of the midbody remnant, which is usually removed by autophagy)108 and subverts anticancer immunosurveillance97. At late stages of oncogenesis, autophagy is reinstated and increases the resistance of cancer cells to chemotherapy or radiotherapy. Multiple trials using relatively nonspecific autophagy inhibitors, such as chloroquine and hydroxychloroquine, have been launched for the treatment of cancer. It remains to be seen whether these trials will be successful and, if so, whether the anti-neoplastic mode of action of chloroquine derivatives can truly be explained by autophagy inhibition.
There is accumulating evidence that ageing is associated with a progressive dysfunction of autophagy, a condition that may favour the accumulation of dysfunctional mitochondria, nuclear genome instability, stem cell attrition, enhanced loss of post-mitotic cells, immunosenescence, unwarranted inflammatory reactions and, in the end, the reduction of hormetic responses that is required for the adaptation to stressful conditions109. Moreover, autophagy is induced by, and required for, the positive influences of longevity-extending regimens, including caloric restriction, inhibition of insulin and insulin-like growth factor signalling, sirtuin 1 activation or long-term treatments with rapamycin, resveratrol or spermidine110. As autophagy is also required for the anti-diabetic effects of physical exercise49, it seems that most if not all recommended health-improving behaviours mediate their effects through the induction of autophagy. In this context, it will be interesting to explore age-associated alterations in the two modes of cellular self-consumption, autophagy and apoptosis, with the scope of gaining mechanistic insights in their functional articulation as well as potential new targets for therapeutic intervention.
Supplementary Material
Acknowledgments
The authors are supported by the Ligue contre le Cancer (équipe labellisée), Agence National de la Recherche, AXA Chair for Longevity Research, Association pour la Recherche sur le Cancer, Cancéropôle Ile-de-France, Institut National du Cancer (INCa), Fondation Bettencourt-Schueller, Fondation de France, Fondation pour la Recherche Médicale, the European Commission (ArtForce), the European Research Council, the LabEx Immuno-Oncology, the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (Socrate), Cancer Research and Personalized Medicine (Carpem), the Paris Alliance of Cancer Research Institutes, the National Institutes of Health and the Ellison Medical Foundation.
Glossary
- Autophagosomes
Vesicles characterized by two membranes that contain autophagic cargo
- Autolysosomes
Fusion proteins of autophagosomes and lysosomes, also called autophagolysosomes. Autolysosomes only have one membrane
- Programmed cell death
(PCD). Cell death that occurs during development or adult tissue homeostasis for the removal of superfluous cells
- Mitochondrial outer membrane permeabilization
(MOMP). An apoptosis-associated process that results in the release of apoptosis-inducing proteins (such as cytochrome c, AIF (apoptosis-inducing factor), SMAC (second mitochondria-derived activator of caspase) and others) that are normally retained in the mitochondrial intermembrane space, through the outer membrane into the cytosol
- Caspases
A family of Cys proteases that cleave after Asp residues. Initiator caspases are typically activated in response to a particular triggering event (for example, caspase 8 upon death receptor ligation, caspase 9 upon apoptosome activation and caspase 2 upon DNA damage), whereas effector caspases (mainly caspase 3, caspase 6 and caspase 7) are particularly important for the ordered dismantling of vital cellular structures
- p53
A central tumour suppressor protein that has multiple functions, both in the cytoplasm and in the nucleus. As a transcription factor, p53 transactivates genes that induce cell cycle arrest, cellular senescence, autophagy and apoptosis
- Lysosomal membrane permeabilization
(LMP). A disruption of lysosomal membrane function that leads to the translocation of lysosomal hydrolases, including cathepsins, from the lysosomal lumen to the rest of the cell. LMP can be induced by endogenous signal transducers (such as reactive oxygen species and sphingosine), as well as by lysosomotropic drugs
- BCL-2 family
Each member of this protein family contains at least one BCL-2 homology (BH) region. The family is divided into anti-apoptotic multidomain proteins (such as BCL-2, BCL-XL (BCL extra large) and MCL1 (myeloid cell leukaemia sequence 1)), which contain four BH domains (BH1, BH2, BH3 and BH4), pro-apoptotic multidomain proteins (for example, BAX (BCL-2- associated X protein) and BAK (BCL-2 antagonist or killer)), which contain BH1, BH2 and BH3, and the pro-apoptotic BH3-only protein family
- Unfolded protein response
(UPR). The UPR is activated in response to stress in the endoplasmic reticulum (ER). It enables cells to adapt to ER stress by reducing the quantity of misfolded proteins in this organelle
- Senescence
An irreversible G1 cell cycle arrest that is accompanied by morphological changes (flattening of the cells), metabolic alterations and transcriptional reprogramming that leads to the expression of cell cycle blockers, such as p16 and p21, and senescence-associated β-galactosidase
- Cytochrome c
A haem protein exclusively present in the mitochondrial intermembrane space. During the initiation of apoptosis, cytochrome c is released from mitochondria and triggers the assembly of the apoptosome, which is a caspase activation complex
- Reactive oxygen species
(ROS). Classic oxygen radicals and peroxides that are formed within cells
- Death receptors
Cell surface receptors that activate the extrinsic pathway of apoptosis upon ligand-induced trimerization. The family of death receptors includes CD95 (which binds CD95 ligand), tumour necrosis factor receptor 1 (TNFR1) and two receptors for TNF-related apoptosis-inducing ligand (TRAILR1 and TRAILR2)
- Sphingosine kinase
A conserved lipid kinase that catalyses the formation of the bioactive sphingolipid metabolite sphingosine 1- phosphate. This acts as a second messenger, with important roles in numerous physiological processes, including cell growth, motility and survival
- Danger-associated molecular patterns
(DAMPs). These molecules are released by or exposed on the surface of stressed and dying cells. They initiate and perpetuate sterile inflammatory responses. Pattern recognition receptors, (PRRs). A series of intracellular or surface receptors that sense danger-associated molecular patterns (DAMPs) or foreign structures from infectious pathogens, triggering inflammatory and immune responses
- Heterophagy
Phagocytosis of a cell by another cell. Heterophagy has a major role in the efficient removal of apoptotic cells. Efficient heterophagy is indispensable for avoiding inflammatory responses triggered by apoptotic material
- Amyotrophic lateral sclerosis
(ALS). A progressive and debilitating neurodegenerative disease that is characterized by progressive muscle atrophy and other degenerative manifestations. The disease pathophysiology is complex and not yet fully understood, but it is proposed to involve the accumulation of misfolded proteins
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 5.Thumm M, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 7.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 11.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. References 11 and 12 enumerate the methods to quantify autophagy, as well as possible pitfalls in the interpretation of autophagy assays. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galluzzi L, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu L, et al. Regulation of an ATG7–beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 17.Boya P, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. Provides the first evidence that autophagy inhibits apoptosis in mammalian cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–465. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 19.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasdemir E, et al. Regulation of autophagy by cytoplasmic p53. Nature Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morselli E, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10:2763–2769. doi: 10.4161/cc.10.16.16868. [DOI] [PubMed] [Google Scholar]
- 22.Yu X, et al. Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model. J Mol Neurosci. 2013;50:586–599. doi: 10.1007/s12031-013-0012-x. [DOI] [PubMed] [Google Scholar]
- 23.Vaseva AV, et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nature Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- 25.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nature Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 27.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598–1607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenzelmann Broz D, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. A complete catalogue of autophagy-relevant genes induced by p53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He Z, et al. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ. 2013;20:1415–1424. doi: 10.1038/cdd.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crighton D, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 32.Zhang XD, Qi L, Wu JC, Qin ZH. DRAM1 regulates autophagy flux through lysosomes. PloS ONE. 2013;8:e63245. doi: 10.1371/journal.pone.0063245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laforge M, et al. DRAM triggers lysosomal membrane permeabilization and cell death in CD4+T cells infected with HIV. PLoS Pathog. 2013;9:e1003328. doi: 10.1371/journal.ppat.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vousden KH, Lane D. P p53 in health and disease. Nature Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 35.Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol. 2012;30:3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maiuri MC, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellot G, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malik SA, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. 2011;30:3918–3929. doi: 10.1038/onc.2011.104. [DOI] [PubMed] [Google Scholar]
- 39.Andreu-Fernandez V, et al. BH3-mimetics- and cisplatin-induced cell death proceeds through different pathways depending on the availability of death-related cellular components. PloS ONE. 2013;8:e56881. doi: 10.1371/journal.pone.0056881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. Reveals the mechanisms through which BCL-2 can repress autophagy in addition to its anti-apoptotic function. [DOI] [PubMed] [Google Scholar]
- 41.Schwarten M, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 42.Luo S, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zalckvar E, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012;19:788–797. doi: 10.1038/cdd.2011.149. References 43 and 45 unravel the mechanisms through which DAPK stimulate autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gandesiri M, et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis. 2012;17:1300–1315. doi: 10.1007/s10495-012-0757-7. [DOI] [PubMed] [Google Scholar]
- 47.Guenebeaud C, et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell. 2010;40:863–876. doi: 10.1016/j.molcel.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 48.Widau RC, Jin Y, Dixon SA, Wadzinski BE, Gallagher PJ. Protein phosphatase 2A (PP2A) holoenzymes regulate death-associated protein kinase (DAPK) in ceramide-induced anoikis. J Biol Chem. 2010;285:13827–13838. doi: 10.1074/jbc.M109.085076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He C, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 51.Wang RC, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3 -kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 53.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Iaccarino I, Hancock D, Evan G, Downward J. c-Myc induces cytochrome c release in Rat1 fibroblasts by increasing outer mitochondrial membrane permeability in a Bid-dependent manner. Cell Death Differ. 2003;10:599–608. doi: 10.1038/sj.cdd.4401211. [DOI] [PubMed] [Google Scholar]
- 55.Hart LS, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–4634. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lock R, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci USA. 2012;109:13325–13330. doi: 10.1073/pnas.1120193109. References 56 to 58 illustrate the importance of autophagy to survive stress occurring in cells expressing oncogenic RAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmukler E, et al. Ras inhibition enhances autophagy, which partially protects cells from death. Oncotarget. 2013;4:142–152. doi: 10.18632/oncotarget.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14:1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grasso D, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1–USP9x–p62, prevents pancreatic cell death. J Biol Chem. 2011;286:8308–8324. doi: 10.1074/jbc.M110.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fortunato F, et al. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–360. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 64.Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amir M, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20:878–887. doi: 10.1038/cdd.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandilands E, Serrels B, Wilkinson S, Frame MC. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2012;13:733–740. doi: 10.1038/embor.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mathew R, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. Details the molecular and cellular mechanisms through which autophagy inhibition triggers carcinogenesis in the liver. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oral O, et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis. 2012;17:810–820. doi: 10.1007/s10495-012-0735-0. [DOI] [PubMed] [Google Scholar]
- 70.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wirawan E, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1:e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pagliarini V, et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012;19:1495–1504. doi: 10.1038/cdd.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fujita N, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122:2554–2566. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yousefi S, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nature Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. Illustrates how limited proteolysis of an autophagy-inducing gene product generates a protein with a new pro-apoptotic function. [DOI] [PubMed] [Google Scholar]
- 76.Lamy L, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell. 2013;23:435–449. doi: 10.1016/j.ccr.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nature Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8:1–3. doi: 10.4161/auto.8.1.16618. [DOI] [PubMed] [Google Scholar]
- 79.Galluzzi L, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH. Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development. 2013;140:1321–1329. doi: 10.1242/dev.089490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manjithaya R, Subramani S. Autophagy: a broad role in unconventional protein secretion? Trends Cell Biol. 2011;21:67–73. doi: 10.1016/j.tcb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Young MM, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nezis IP, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–531. doi: 10.1083/jcb.201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu L, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saftig P, Beertsen W, Eskelinen EL. LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy. 2008;4:510–512. doi: 10.4161/auto.5724. [DOI] [PubMed] [Google Scholar]
- 86.Ma X, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/ reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 88.Kessel DH, Price M, Reiners JJ., Jr ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy. 2012;8:1333–1341. doi: 10.4161/auto.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boya P, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–1334. doi: 10.1084/jem.20021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 93.Huang C, et al. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32:1362–1376. doi: 10.1038/jcbfm.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. A seminal paper demonstrating the impact of pre-mortem autophagy on the clearance of apoptotic cells. [DOI] [PubMed] [Google Scholar]
- 96.Chekeni FB, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michaud M, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. Evidence that inhibition of autophagy in tumour cells may subvert anticancer immunosurveillance and hence reduce the efficacy of chemotherapies. [DOI] [PubMed] [Google Scholar]
- 98.Sukkurwala AQ, et al. Immunogenic calreticulin exposure occurs through a phylogenetically conserved stress pathway involving the chemokine CXCL8. Cell Death Differ. 2013 Jan; doi: 10.1038/cdd.2013.73. doi: http://dx.doi.org/10.1038/cdd.2013.73. [DOI] [PMC free article] [PubMed]
- 99.Bao JX, et al. Lysosome–membrane fusion mediated superoxide production in hyperglycaemia-induced endothelial dysfunction. PLoS ONE. 2012;7:e30387. doi: 10.1371/journal.pone.0030387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy. 2012;8:1741–1756. doi: 10.4161/auto.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Uhl M, et al. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009;16:991–1005. doi: 10.1038/cdd.2009.8. [DOI] [PubMed] [Google Scholar]
- 102.Ko A, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signaling. Cell Death Differ. 2013 Sep; doi: 10.1038/cdd.2013.124. doi: http://dx.doi.org/10.1038/cdd.2013.124. [DOI] [PMC free article] [PubMed]
- 103.Ma Y, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 104.Calabrese EJ, Iavicoli I, Calabrese V. Hormesis: its impact on medicine and health. Hum Exp Toxicol. 2013;32:120–152. doi: 10.1177/0960327112455069. [DOI] [PubMed] [Google Scholar]
- 105.Hetz C, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Rev Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rello-Varona S, et al. Autophagic removal of micronuclei. Cell Cycle. 2012;11:170–176. doi: 10.4161/cc.11.1.18564. [DOI] [PubMed] [Google Scholar]
- 108.Kuo TC, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nature Cell Biol. 2011;13:1214–1223. doi: 10.1038/ncb2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 111.Hamasaki M, et al. Autophagosomes form at ER–mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 112.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. An exhaustive review on pro-survival and lethal signalling pathways converging on mitochondria, as well as on the mechanisms that decide whether mitochondrial membranes undergo deadly permeabilization. [DOI] [PubMed] [Google Scholar]
- 114.Galonek HL, Hardwick JM. Upgrading the BCL-2 network. Nature Cell Biol. 2006;8:1317–1319. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 115.Kroemer G, Martin SJ. Caspase-independent cell death. Nature Med. 2005;11:725–730. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 116.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 117.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 118.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nature Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 119.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–112. doi: 10.1038/sj.cdd.4402231. An important paper describing the contribution of autophagy to pathological loss of neurons in nematodes. [DOI] [PubMed] [Google Scholar]
- 120.Erdelyi P, et al. Shared developmental roles and transcriptional control of autophagy and apoptosis in Caenorhabditis elegans. J Cell Sci. 2011;124:1510–1518. doi: 10.1242/jcs.080192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tracy K, Baehrecke EH. The role of autophagy in Drosophila metamorphosis. Curr Top Dev Biol. 2013;103:101–125. doi: 10.1016/B978-0-12-385979-2.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. First genetic evidence that autophagic cell death occurrs during the development of Drosophila. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Denton D, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chang T-K, et al. Uba1 functions in Atg7- and Atg3-independent autophagy. Nature Cell Biol. 2013;15:1067–1078. doi: 10.1038/ncb2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gozuacik D, et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- 126.Byun JY, et al. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis. 2009;30:1880–1888. doi: 10.1093/carcin/bgp235. [DOI] [PubMed] [Google Scholar]
- 127.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 128.Koike M, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. An example of how autophagy may mediate deleterious effects in a model of pressure overload affecting the heart muscle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sun Y, et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Science Signal. 2012;5:ra16. doi: 10.1126/scisignal.2001931. [DOI] [PubMed] [Google Scholar]
- 131.Sentelle RD, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nature Chem Biol. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.