

Abstract
Acute nicotine enhances hippocampus-dependent learning through nicotine binding to β2-containing nicotinic acetylcholine receptors (nAChRs), but it is unclear if nicotine is targeting processes involved in short-term memory (STM) leading to a strong long-term memory (LTM) or directly targeting LTM. In addition, the molecular mechanisms involved in the effects of nicotine on learning are unknown. Previous research indicates that protein kinase A (PKA), extracellular regulated signaling kinase 1/2 (ERK1/2), and protein synthesis are crucial for LTM. Therefore, the present study examined the effects of nicotine on STM and LTM and the involvement of PKA, ERK1/2, and protein synthesis in the nicotine-induced enhancement of hippocampus-dependent contextual learning in C57BL/6J mice. The protein synthesis inhibitor anisomycin impaired contextual conditioning assessed at 4 hours but not 2 hours post-training, delineating time points for STM (2 hours) and LTM (4 hours and beyond). Nicotine enhanced contextual conditioning at 4, 8, and 24 hours but not 2 hours post-training, indicating nicotine specifically enhances LTM but not STM. Furthermore, nicotine did not rescue deficits in contextual conditioning produced by anisomycin, suggesting that the nicotine enhancement of contextual conditioning occurs through a protein synthesis-dependent mechanism. In addition, inhibition of dorsal hippocampal PKA activity blocked the effect of acute nicotine on learning and nicotine shifted the timing of learning-related PKA and ERK1/2 activity in the dorsal and ventral hippocampus. Thus, the present results suggest that nicotine specifically enhances LTM through altering the timing of PKA and ERK1/2 signaling in the hippocampus, and suggests that the timing of PKA and ERK1/2 activity could contribute to the strength of memories.
Keywords: learning, plasticity, protein synthesis, addiction, acetylcholine
1.0 Introduction
Nicotine alters hippocampal synaptic plasticity and learning (Kenney and Gould, 2008; Gould and Leach, 2013). Increasing evidence suggests that hippocampus-dependent learning, such as contextual fear conditioning and spatial object recognition (Parkinson et al., 1988; Phillips and LeDoux, 1992; Kim et al., 1993; Logue et al., 1997), are more sensitive to the effects of nicotine on learning than hippocampus-independent tasks (Gould and Wehner, 1999; Gould and Higgins, 2003; Davis et al., 2006; Kenney et al., 2011). Direct drug infusion studies confirmed that acute nicotine is acting in the dorsal hippocampus to enhance learning (Kenney et al., 2012; Raybuck and Gould, 2010) and further studies demonstrated that β2-containing nicotinic acetylcholine receptors (nAChRs), likely α4β2* (* indicates potential other subunits), are both necessary and sufficient for enhancement of hippocampus-dependent learning by acute nicotine (Davis and Gould, 2006; Davis et al., 2007; Kenney et al., 2010a). While it is known that nicotine enhances long-term memory (LTM) (Gould and Higgins, 2003), it is unknown if nicotine is enhancing short-term memory (STM) processes leading to stronger LTM or if nicotine is directly altering the cell signaling cascades that underlie LTM.
Multiple studies suggest that the signaling cascade involving cyclic adenosine monophosphate (cAMP), protein kinase A (PKA), and extracellular signal-regulated kinase 1/2 (ERK1/2) is involved in plasticity-related changes underlying LTM formation (for review, see Abel and Nguyen, 2008; Sweatt, 2004). Inhibiting PKA in the dorsal hippocampus impaired long-term contextual memory (Wallenstein et al., 2002; Ahi et al., 2004; Ma et al., 2009). Furthermore, Abel et al. (1997) reported that transgenic mice with reduced hippocampal PKA activity showed impaired contextual fear conditioning 24 hours after training but intact conditioning 1 hour after training. It has been shown that PKA can activate ERK1/2 (Roberson et al., 1999; Winder et al., 1999) and that ERK1/2 has similarly been shown to be critically involved in LTM (Sweatt, 2004). Contextual fear conditioning was associated with increased hippocampal ERK1/2 phosphorylation (Atkins et al., 1998; Triffilieff et al., 2006) and inhibition of ERK1/2, whether through pharmacological or genetic knockdown techniques, disrupted LTM for contextual fear conditioning (Triffilieff et al., 2006; Satoh et al., 2007). In addition, ERK1/2 is important for activation of the gene transcription factor cAMP response element-binding protein (CREB) (Triffilieff et al., 2006), which is critically involved in LTM (Bourtchuladze et al., 1994). Thus, activation of PKA and ERK1/2 may alter the gene transcription that undelies LTM. Furthermore, a recent model of LTM in Aplysia suggests that the strength of LTM formation may depend on the timing of activation of PKA and ERK1/2 (Zhang et al., 2012; for commentary see Abbott and Kandel, 2012).
Support for the contention that nicotine enhances learning through specifically acting on cell signaling cascades of LTM comes from multiple studies. For example, α4β2* nAChRs are abundantly expressed in the central nervous system and gate Ca2+ (Karadsheh et al., 2004; Tapia et al., 2007), which can lead to activation of the cAMP/PKA and ERK1/2 signaling pathways. Furthermore, nicotine facilitated the induction of long-term potentiation (LTP) and directly induced hippocampal LTP (Fujii et al., 1999; Matsuyama et al., 2000). The nicotine enhancement of LTP involved PKA (Welsby et al., 2009). Also, in hippocampal neuronal culture, nicotine activated ERK1/2 through a calcium (Ca2+) and PKA-dependent mechanism (Dajas-Bailador et al., 2002). In an initial study, we found that systemic inhibition of ERK1/2 blocked nicotine enhancement of learning (Raybuck and Gould, 2007). Thus, because LTM is distinguished from STM by the involvement of PKA, ERK1/2, and protein synthesis (Abel et al., 1997; Atkins et al., 1998; Bourtchouladze et al., 1998; Davis and Squire, 1984; Koh et al., 2002; Triffilieff et al., 2006; Satoh et al., 2007; Michel et al., 2011), the present experiments specifically examined whether nicotine enhances STM and/or LTM, whether PKA is involved in the enhancement of contextual fear conditioning by nicotine, and whether nicotine altered the timing of activation of learning-related PKA and ERK1/2 activity; dorsal and ventral hippocampus were compared because prior work suggests that these areas are functionally distinct (see Fanselow and Dong, 2010 for review) and that nicotine has different effects on learning in these areas (Kenney et al., 2010b; Kenney et al., 2012; Raybuck and Gould, 2010).
2.0 Materials and Methods
2.1 Subjects
Male C57BL/6J mice (8–12 wks old, Jackson Laboratories, Bar Harbor, ME) were housed 1–4 per cage and maintained on a 12 hour light-dark cycle (lights on at 7:00 AM) with ad libitum access to food and water. Cannulated mice were singly housed. All behavioral procedures were conducted between the hours of 9:00 AM – 5:00 PM. All behavioral and surgical procedures were approved by the Temple University Institutional Animal Care and Use Committee.
2.2 Drugs and Administration
Nicotine hydrogen tartrate salt (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% physiological saline and administered intraperitoneally (i.p.) at 0.09 mg/kg freebase 4–5 min prior to training and/or testing of contextual conditioning, as was conducted in other studies (Gould and Higgins, 2003; Davis et al., 2006). Anisomycin (Sigma-Aldrich, St. Louis, MO) was dissolved in hydrochloric acid and saline (pH adjusted to 7.0 with sodium hydroxide) and administered subcutaneously (s.c.) at 150 mg/kg 30 min prior to training of contextual conditioning, as was used in prior studies and yields >90% protein synthesis inhibition in the brain within two hours after administration (Abel et al., 1997; Flood et al., 1997; Lattal and Abel, 2004; Lewis and Gould, 2004). The PKA inhibitor PKI 14–22 amide, myristoylated (PKI; BMLP210-0500; Enzo Life Sciences, Plymouth Meeting, PA) was dissolved in 0.9% physiological saline and administered by microinfusion into the dorsal hippocampus 5 min before training. Microinfusions of PKI occurred bilaterally in dorsal hippocampus (2, 4, or 8 μg/mouse) at a rate of 0.50 μl/min with an injection volume of 0.50 μl/hemisphere, as based on previous work (Ma et al., 2009). Infusion cannulae were attached to polyethylene tubing (PE50; Plastics One, Roanoke, VA) connected to a 10 μl Hamilton syringe (Model 701; Hamilton, Reno, NV). Drug infusions were controlled by a microinfusion pump (KDS 100; KD Scientific, New Hope, PA). Cannulae were left in place for 1 min after infusion to allow for the drug to diffuse away from the cannulae.
2.3 Surgeries
Mice were anesthetized with isoflurane gas (5% induction, 2–3% maintenance) then placed into a stereotaxic device (David Kopf Instruments, Tujunga, CA). Bilateral, stainless–steel guide cannulae (C232G, 22 Gauge; Plastics One, Roanoke, VA) were implanted into the dorsal hippocampus (−1.7 mm posterior; ±1.5 mm mediolateral; −2.3 mm ventral from bregma) and fixed to the skull with dental cement. Coordinates were based upon a mouse brain atlas (Paxinos and Franklin, 2001) and previous research using these coordinates (Davis et al., 2007; Kenney et al., 2010b; André et al., 2011). Dummy cannulae (C232DC; Plastics One, Roanoke, VA) were inserted into the guide cannulae to prevent clogging. Following surgery, ketoprofen (2 mg/kg) was administered s.c. for post-operative pain. Animals were allowed at least 5 days to recover before behavioral procedures began. Cannulae placements were verified by histology and any animal with a placement outside of the dorsal hippocampus was excluded from data analysis.
2.4 Apparatus
Training and testing of contextual conditioning took place in four identical clear Plexiglas chambers (26.5 × 20.4 × 20.8 cm) housed in sound attenuating boxes (Med-Associates, St. Albans, VT). The floor of each chamber was made of metal bars connected to a shock generator and scrambler (Model ENV-414; Med-Associates, St. Albans, VT). Ventilation fans were mounted on the sides of each box to provide background noise. Illumination was provided by a 4 W light mounted above each box. Shock administration was controlled by a PC running LabView software (National Instruments, Austin, TX).
2.5 Behavioral Procedure
The behavioral procedure was performed as described previously (Davis et al, 2006). Freezing, defined as the complete absence of movement besides respiration, was sampled for 1 s every 10 s. On training day, mice were placed into the chambers and baseline freezing was scored for 120 s. At 148 s a 2 s 0.57 mA footshock unconditional stimulus (US) was administered. This footshock was followed by another footshock 148 s after the first. Mice remained in the chambers for an additional 30 s after the second footshock before being returned to their home cages. At 4 time points post-training (2, 4, 8, or 24 h), mice were returned to the original training chambers and freezing to the context was scored for 5 min to assess contextual fear memory.
2.6 PKA Kinase Activity Assay
Animals received an acute injection of either nicotine or saline and were either trained in contextual conditioning or returned to their home cages. At 4 different post-training time points or at equivalent post-training times (1, 2, 3, or 6 hours), mice were euthanized and hippocampi were dissected into dorsal and ventral sections (in a 1:1 ratio) on ice. Freshly dissected dorsal and ventral hippocampi were homogenized by an electronic sonicator in lysis buffer containing 20 mM MOPS, 5 mM EGTA, 2 mM EDTA, 1% v/v NP-40, 1 mM DTT, 1 mM Benzamidine and HALT Protease and Phosphatase Inhibitor Cocktail (PI-78445; Thermo Scientific, Rockford, IL). The homogenates were centrifuged at 14,000 rpm for 30 min at 4°C. The total protein concentration of the resulting supernatants was determined by DC™ Protein Assay (500–0112; Bio-Rad, Hercules, CA). Prior to the PKA activity assay, homogenates were diluted according to their total protein concentration and 20 μg of total protein was used for the assay. The activity of PKA was measured using an enzyme-linked immunosorbent assay-based kit (ADI-EKS-390A; Enzo Life Sciences, Plymouth Meeting, PA). Microtiter plate wells, pre-coated with the proprietary substrate of PKA, were soaked in 50 μl of kinase assay dilution buffer for 10 min and buffer was then carefully aspirated from the wells. Diluted homogenates (30 μl/sample) and standards (serial dilutions of active PKA) were then added to the wells of the microplate in triplicate. The kinase reaction was initiated by adding 10 μl of 1 mg/ml ATP and was carried out for 60 min at 30°C. After terminating the kinase reaction, 40 μl of phospho-specific substrate antibody was added to each well and the plate was incubated at room temperature for 60 min, followed by 4 washes with wash buffer. Anti-rabbit IgG:HRP-conjugated secondary antibody (40 μL) was then added to each well, and the plate incubated for 30 min at room temperature. The wells were again washed 4 times. The color, proportional to the kinase activity of PKA, was developed by incubation with 60 μl of stabilized tetramethlybenzidine substrate for 60 min. The color development reaction was terminated by 20 μl of acidic stop solution. Absorbance was measured at 450 nm in a microplate reader. The relative kinase activity for each sample was then interpolated from the standard curve.
2.7 ERK1/2 Western Blotting
Dorsal and ventral hippocampi were dissected as described in the PKA Kinase Activity Assay section. After being dissected, tissue was flash frozen on dry ice and homogenized in 1X RIPA buffer solution (9806; Cell Signaling, Danvers, MA) containing HALT Protease and Phosphatase Inhibitor Cocktail (PI-78445; Thermo Scientific, Rockford, IL) diluted to 1X. Homogenates were spun at 14,000 rpm for 30 min at 4°C, and supernatants were collected. Protein concentration of each sample supernatant was determined as described previously. Each sample was diluted with 1X RIPA buffer to obtain 20μg of total protein, then was diluted 1:1 with Laemmeli sample buffer (161–0737; Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol. Samples were then loaded on TGX 4–20% gradient gels (456–1099; Bio-Rad, Hercules, CA). Electrophoresis was conducted for 80 min at 100V constant voltage in 1X Tris-Glycine-SDS running buffer (161–0732; Bio-Rad, Hercules, CA). Proteins were then transferred onto nitrocellulose membranes (162–0214; Bio-Rad, Hercules, CA) for 2 hours at 400mA constant current in ice-cold 1X Tris-Glycine buffer (161–0734; Bio-Rad, Hercules, CA) containing 20% methanol. Nitrocellulose membranes were then washed 3 times for 5 min with 1X Tris Buffered Saline (TBS) and blocked with 5% bovine serum albumin (BSA; A9418; Sigma-Aldrich, St. Louis, MO) in 1X TBS for 1 hour at room temperature. Incubation with primary antibody (anti-phospho-ERK1/2 rabbit mAb: Cell Signaling, Danvers, MA, 1/2000 in 5% BSA; anti-ERK1/2 rabbit mAb: Cell Signaling, Danvers, MA, 1/2000 in 5% BSA; monoclonal mouse anti-β-Actin: Sigma-Aldrich, St. Louis, MO, 1/5000 in 5% BSA) took place overnight in 4°C, except for anti-β-Actin incubations which took place at room temperature for 1 hour. Blots were washed 3 times for 5 min with 1X TBS with 0.1% Tween 20 prior to secondary antibody incubations. For phospho-ERK1/2 and ERK1/2, the secondary antibody was an HRP-conjugated anti-rabbit (Vector Laboratories, Burlingame, CA, 1/2000 in 5% BSA) and incubated for 1 hour at room temperature. For β-Actin, an HRP-conjugated anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA, 1/10000 in 5% BSA) was used and incubated for 1 hour at room temperature. Enhanced chemiluminescence reactions proceeded for 5 min (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific, Rockford, IL), and blots were imaged using a single 60 s exposure on a Kodak camera (Gel Logic 1500 Imaging System). Bands were quantified using ImageJ software (NIH, Bethesda, MD). Protein levels were determined using optical density values of target bands. Levels of phospho-ERK1/2 (i.e., ERK1/2 activity) were first normalized to ERK1/2 protein levels and then to β-actin loading control protein levels for final quantification.
2.8 Data Analyses
For behavioral measures, planned comparisons using independent samples t-tests were used to compare nicotine and saline at each post-training time point. The effects of anisomycin on the nicotine-induced enhancement of contextual conditioning were analyzed using one-way analysis of variance (ANOVA). Planned contrasts were used to determine specific effects against respective saline-treated control groups. Infusion data were analyzed using one-way or two-way (2 infusion × 2 acute) ANOVAs. Significant effects were followed by Tukey's post-hoc tests or Games-Howell post-hoc tests when the homogeneity of variance assumption was not met. Activity of PKA and ERK1/2 were analyzed using one-way ANOVAs followed by Bonferroni-corrected post-hoc simple contrasts against the saline home cage group and represented as fold-change relative to the respective saline home cage group. For all experiments, familywise α = 0.05.
3.0 Results
3.1 Determination of STM and LTM time points
Studies that have examined memory have generally used 1 hour as the STM time point and 24 hours as the LTM time point (Abel et al., 1997; Bourtchouladze et al., 1998; Nie and Abel, 2001). However, evidence indicates that LTM, as defined by the recruitment of protein synthesis, begins as early as 3 hours post-training in contextual conditioning (Bourtchouladze et al., 1998). Thus, we chose 2 hours to represent STM and 4 hours to represent the beginning of LTM formation. LTM, unlike STM, requires the synthesis of new proteins (Davis and Squire, 1984; Abel et al., 1997; Bourtchouladze et al., 1998). Therefore, to determine if 2 hours and 4 hours accurately reflected STM and LTM, separate groups of mice were trained in contextual conditioning after injections of saline or the protein synthesis inhibitor anisomycin then tested 2 or 4 hours later (Figure 1). When animals were tested 2 hours after training, there were no significant differences between saline- and anisomycin-treated animals [p > 0.05]. However, anisomycin produced deficits in contextual conditioning when tested 4 hours after training [t(14)=4.007, p < 0.05]. These results provide evidence that the 2 hour data reflect STM and that at 4 hours, protein-synthesis–mediated processes that support LTM are engaged.
Figure 1.
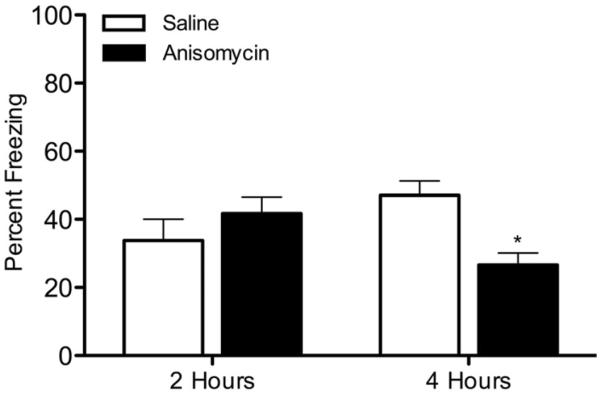
The effects of anisomycin on contextual conditioning. Mice were injected with saline or anisomycin (150 mg/kg), trained in contextual conditioning, then tested 2 or 4 hours later (n=8 for each group). * p < 0.05 for comparisons between anisomycin and saline groups within each time point. Data are presented as mean ± SEM.
3.2 The effects of nicotine on STM and LTM
Previous research has shown that nicotine enhances contextual conditioning when assessed at least 24 hours after training (Gould and Wehner, 1999; Gould and Higgins, 2003), which would reflect LTM. It is unknown if enhancement occurs at earlier time points, including periods that would reflect STM. To determine if nicotine enhances STM in addition to LTM, saline or nicotine was injected prior to training and testing of contextual conditioning with testing occurring either 2, 4, 8, or 24 hours after training (Figure 2). Nicotine had no effect when animals were tested 2 hours post-training, but nicotine enhanced contextual conditioning when animals were tested 4 hours post-training [t(20)=2.626, p < 0.05]. Animals tested 8 hours [t(20)=2.211, p < 0.05] and 24 hours [t(20)=2.604, p < 0.05] after training also showed nicotine enhancement of contextual conditioning.
Figure 2.
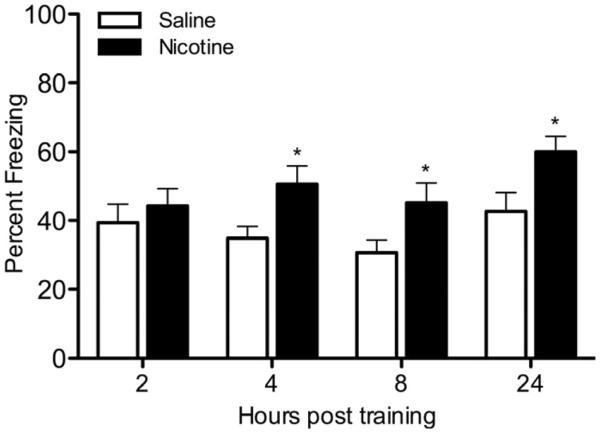
The effects of nicotine on contextual conditioning at various times post-training. Mice were injected with saline or nicotine (0.09 mg/kg), trained in contextual conditioning, then tested with saline or nicotine 2, 4, 8, or 24 hours post-training (n=11 for each group). * p < 0.05 for comparisons between saline and nicotine groups within each time point. Data are presented as mean ± SEM.
3.3 The effects of protein synthesis inhibition on the nicotine enhancement of contextual conditioning
LTM is dependent upon protein synthesis (Davis and Squire, 1984; Abel et al., 1997; Bourtchouladze et al., 1998). To determine if nicotine enhances contextual conditioning in a protein synthesis-dependent pathway, mice received injections of saline or anisomycin prior to training and saline or nicotine prior to training and testing of contextual conditioning (Figure 3). A one-way ANOVA revealed a significant effect of drug treatment on contextual conditioning [F(3, 60)=46.900, p < 0.05]. Planned contrasts revealed that mice receiving nicotine and saline froze significantly more to the context than mice receiving only saline [p < 0.05]. In addition, mice that received saline and anisomycin or nicotine and anisomycin froze significantly less to the context than mice that received only saline [p < 0.05], and, concordantly, there was no effect of nicotine on learning in the anisomycin group [p > 0.05].
Figure 3.
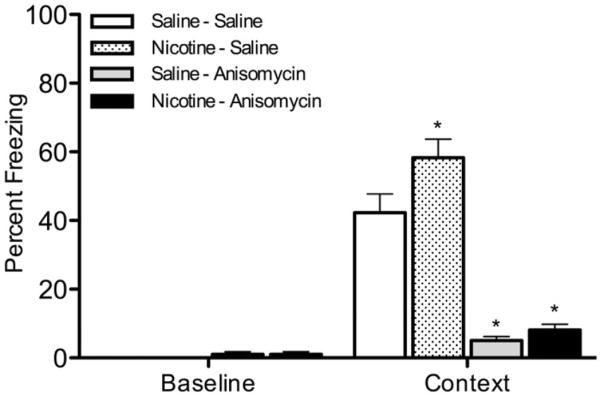
The effects of anisomycin on nicotine enhancement of contextual conditioning. Mice received injections of saline or anisomycin (150 mg/kg) prior to training and saline or nicotine (0.09 mg/kg) prior to training and testing of contextual conditioning (n=16 for each group). * p < 0.05 for comparisons with saline-saline controls. Data are presented as mean ± SEM.
3.4 The involvement of PKA in nicotine enhancement of contextual conditioning
Protein kinase A is involved in LTM (Abel et al., 1997; Bourtchouladze et al., 1998; Wallenstein et al., 2002; Ahi et al., 2004; Ma et al., 2009). Therefore, we examined the involvement of PKA in the nicotine enhancement of contextual conditioning. First, a dose subthreshold for disrupting fear conditioning (2 μg) of PKI, an inhibitor of PKA, was determined (Figure 4). Mice received bilateral dorsal hippocampal microinfusions of saline, 2, 4, or 8 μg of PKI prior to training of contextual conditioning. A one-way ANOVA revealed a significant effect of PKI on contextual freezing [F(3, 27)=5.608, p < 0.05]. Post-hoc tests revealed that animals receiving 4 and 8 μg of PKI froze significantly less to the context than all other groups [p < 0.05]. This finding replicates previous work demonstrating that 4 μg PKI disrupts contextual conditioning (Ma et al., 2009).
Figure 4.
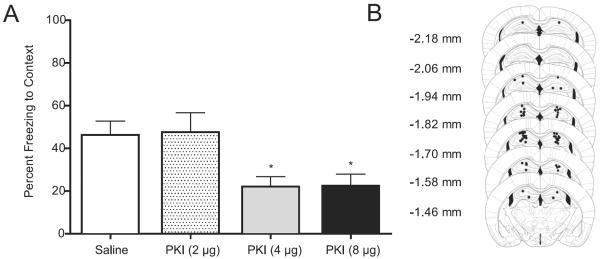
The effects of PKI bilaterally infused into the dorsal hippocampus on contextual conditioning. (A) Mice received infusions of saline or PKI (2, 4, or 8 μg; n=7–8 for each dose) prior to training of contextual conditioning. (B) Representative pictures of the infusion sites in dorsal hippocampus; figures modified from Paxinos and Franklin (2001). * p < 0.05 for comparisons with saline-treated animals. Data are presented as mean ± SEM.
Next, to determine the involvement of PKA in the nicotine enhancement of contextual conditioning, mice received either saline or 2 μg of PKI bilaterally into the dorsal hippocampus prior to injections of systemic saline or nicotine (Figure 5). A two-way ANOVA revealed a significant nicotine X PKI interaction [F(1, 48)=7.694, p < 0.05]. Post-hoc tests revealed that mice infused with saline then injected with nicotine froze significantly more to the context than all other groups [p < 0.05]. In addition, mice infused with PKI, whether receiving systemic saline or nicotine, were not different from saline controls [p > 0.05].
Figure 5.

The effects of PKI bilaterally infused into the dorsal hippocampus on nicotine-enhanced contextual conditioning. (A) Mice received infusions of saline or PKI (2 μg) prior to training of contextual conditioning, as well as injections of saline or nicotine (0.09 mg/kg) prior to training and testing (n=12–14 for each group). (B) Representative pictures of the infusion sites in dorsal hippocampus; figures modified from Paxinos and Franklin (2001). * p < 0.05 for comparisons between animals treated with saline-saline. # p < 0.05 for comparisons between animals treated with nicotine-saline. Data are presented as mean ± SEM.
3.5 The effects of nicotine and learning on the temporal activation of hippocampal PKA
Because the timing of PKA activity may be important for LTM (Zhang et al., 2012), PKA activity in the dorsal and ventral hippocampus was examined to determine if nicotine altered the timing of PKA activity. At 1 hour post-training in the dorsal hippocampus, a one-way ANOVA revealed a significant effect of group on PKA activity [F(3, 15)=8.375, p < 0.01] (Figure 6A). Post-hoc tests revealed that PKA activity was significantly higher in the Saline + Training (ST) group relative to the Saline + Home Cage (SH) group [p < 0.001]. No other significant differences were observed against the SH group [p > 0.05]. At 1 hour post-training in the ventral hippocampus, a one-way ANOVA revealed a significant effect of group on PKA activity [F(3, 12)=5.362, p < 0.05] (Figure 6A). Post-hoc tests revealed that PKA activity in the ST group was significantly higher than the SH group [p < 0.05]. No other significant differences against SH were observed [p > 0.05].
Figure 6.

Activity levels of PKA at 1, 2, 3, or 6 hours post-training or injection (n=4 for each group). Mice either received acute injections of saline (SH) or nicotine (0.09 mg/kg; NH) then were left in their home cages or acute injections of saline (ST) or nicotine (0.09 mg/kg; NT) then trained in contextual conditioning. Bilateral dorsal and ventral hippocampi were then removed (A) 1 hour post-training, (B) 2 hours post-training, (C) 3 hours post-training, or (D) 6 hours post-training and then subjected to the PKA activity assay. Data are presented as fold change in kinase activity relative to the respective SH group. * p < 0.05, ** p < 0.01, *** p < 0.001 compared to the SH group only. Data are presented as mean ± SEM.
At 2 hours post-training in the dorsal hippocampus, a one-way ANOVA revealed a significant effect of group on PKA activity [F(3, 12)=12.340, p < 0.01] (Figure 6B). Post-hoc tests revealed that the NT group had the largest increase in PKA activity compared to the SH group [p < 0.001]. PKA activity levels in the NH and ST groups were also significantly greater than the SH group [p < 0.05]. At 2 hours in the ventral hippocampus, the one-way ANOVA approached significance but there was not an overall significant effect on PKA activity levels [F(3, 12)=3.133, p = 0.07]. Because the ANOVA approached significance, an exploratory post hoc analysis was conducted, which revealed that the NT group had significantly reduced PKA activity relative to the SH group [p < 0.05].
Finally, at 3 (Figure 6C) and 6 hours post-training (Figure 6D), one-way ANOVAs did not reveal any significant effect of group on PKA activity levels in either the dorsal [3 hour: F(3, 12)=0.726, p > 0.05; 6 hour: F(3, 12)=0.645, p > 0.05] or ventral [3 hour: F(3, 12)=1.172, p > 0.05; 6 hour: F(3, 12)=0.450, p > 0.05] hippocampus, indicating a return to baseline values (i.e., SH group). These data suggest a time-dependent involvement of PKA in the dorsal and ventral hippocampus during contextual conditioning with nicotine shifting the time course of the learning-related PKA activity in the dorsal hippocampus and suppressing PKA activity in the ventral hippocampus.
3.6 The effects of nicotine and learning on the temporal activation of hippocampal ERK1/2
ERK1/2 is involved in LTM (Sweatt, 2004). This experiment examined if nicotine altered ERK1/2 activity across multiple time points. At 1 hour post-training in the dorsal hippocampus (Figure 7A), a one-way ANOVA revealed a significant effect of group on ERK1/2 activity [F(3, 20)=5.84, p < 0.01]. However, post-hoc contrasts found no significant differences for any group compared to controls (SH) [p > 0.05]. In the ventral hippocampus, a significant group effect was also seen [F(3, 20)=3.382, p < 0.05] but as before, comparisons found no significant differences for any group compared to SH [p > 0.05].
Figure 7.

Activity levels of ERK1/2 at 1, 2, 3, or 6 hours post-training or injection (n=4 for each group). Mice either received acute injections of saline (SH) or nicotine (0.09 mg/kg; NH) then were left in their home cages or acute injections of saline (ST) or nicotine (0.09 mg/kg; NT) then trained in contextual conditioning. Bilateral dorsal and ventral hippocampi were then removed (A) 1 hour post-training, (B) 2 hours post-training, (C) 3 hours post-training, or (D) 6 hours post-training and then subjected to the ERK1/2 western blotting procedure. Data are presented as fold change in kinase activity relative to the respective SH group. * p < 0.05, ** p < 0.01, *** p < 0.001 compared to the SH group only. Data are presented as mean ± SEM.
At 2 hours post-training in the dorsal hippocampus (Figure 7B), a one-way ANOVA revealed a significant effect of group on ERK1/2 activity [F(3, 20)=4.087, p < 0.05]. Post-hoc contrasts found that the only significant difference was increased ERK1/2 activity in the ST group compared to the SH group [p < 0.05]. No significant difference was seen in the ventral hippocampus at this time point [F(3, 19)=1.091, p > 0.05].
At 3 hours post-training in the dorsal hippocampus (Figure 7C), a one-way ANOVA revealed a significant effect of group on ERK1/2 activity [F(3, 20)=11.066, p < 0.001]. Post-hoc contrasts revealed that the NH, ST, and NT groups had significantly greater ERK1/2 activity compared to the SH group [p < 0.01]. In the ventral hippocampus, a significant group effect was also found [F(3, 20)=10.726, p < 0.001]. Post-hoc contrasts revealed that the NH and ST groups had greater ERK1/2 activity than the SH group [p < 0.01].
At 6 hours post-training (Figure 7D), no significant difference across groups was found in the dorsal hippocampus [F(3, 19)=2.800, p > 0.05]. A one-way ANOVA revealed a significant effect of group in the ventral hippocampus [F(3, 17)=4.203, p < 0.05], but post-hoc contrasts revealed no significant differences compared to the SH group [p > 0.05]. Similar to what was seen with PKA, these data also suggest a time-dependent involvement of ERK1/2 activity in the dorsal and ventral hippocampus during contextual conditioning with nicotine again shifting the timing of ERK1/2 activity in the hippocampus.
4.0 Discussion
The primary issues addressed in this study were whether acute nicotine modified STM, LTM, or both types of memory and what molecular processes might underlie nicotine-enhanced learning. It has become clear that STM occurs within the first 3 hours after training and is protein synthesis-independent whereas LTM occurs at later time points and is protein synthesis-dependent as demonstrated here and in prior studies (Davis and Squire, 1984; Abel et al., 1997; Bourtchouladze et al., 1998). One of the striking findings from this study was that nicotine selectively enhanced LTM; no memory enhancement was observed 2 hours after training, yet nicotine enhanced memory 4 hours after training and later. This transition from STM to LTM may involve activation of a molecular switch that engages cell signaling cascades involved in the protein synthesis necessary for LTM formation.
Multiple lines of evidence suggest both PKA and ERK1/2 are involved in LTM but not STM. Long-term potentiation (LTP) reflects changes in synaptic plasticity that may model processes involved in STM and LTM formation. Similar to stages of memory formation, LTP has an early phase (E-LTP) that lasts 1 to 3 hours and does not require protein synthesis or PKA activity and a late phase (L-LTP) that lasts for at least 8 hours and is blocked by inhibitors of PKA and protein synthesis (Frey et al., 1993; Huang and Kandel, 1994; Huang, 1998). Studies with transgenic mice deficient in hippocampal PKA activity showed that these mice had intact E-LTP and STM but deficits in L-LTP and LTM that began to emerge as early as 3 hours post-training (Abel et al., 1997; Bourtchouladze et al., 1998). Likewise, ERK1/2 is involved in LTP (English and Sweatt, 1997) and mice with disrupted ERK1/2 signaling due to genetic modification of MEK1 have normal E-LTP but disrupted L-LTP (Kelleher et al., 2004). Furthermore, inhibiting PKA activity impaired contextual conditioning at 24 hours (LTM) but not 1 hour (STM) post-training (Schafe et al., 1999), impaired conditioned taste aversion at 24 hours (LTM) but not 3 hours (STM) post-training (Koh et al., 2002), and impaired operant learning in Aplysia at 24 hours (LTM) but not 30 minutes (STM) post-training (Michel et al., 2011). Similarly, inhibiting ERK1/2 disrupted LTM but not STM for contextual fear conditioning (Trifilieff et al., 2006) and ERK2 knockdown mice also had deficits in LTM but not STM for contextual fear conditioning (Satoh et al., 2007). These studies strongly suggest that PKA and ERK1/2 are molecular switches important for the formation of LTM.
In line with our observation that nicotine selectively enhances LTM and prior work demonstrating that PKA is involved in LTM memory formation (Abel et al., 1997; Bourtchouladze et al., 1998; Schafe et al., 1999; Koh et al., 2002; Michel et al., 2011), PKA was critically involved in the nicotine enhancement of contextual fear conditioning. Specifically, direct dorsal hippocampal infusion of the PKA inhibitor PKI, at a dose that did not alter learning, blocked the enhancement of learning by acute nicotine, suggesting increased PKA signaling contributed to acute nicotine enhancement of learning. Because PKA can activate ERK1/2 (Roberson et al., 1999) and ERK1/2 is involved in synaptic plasticity and LTM (Atkins et al., 1998; Sweatt, 2004; Winder et al., 1999; Trifilieff et al., 2006), it follows that nicotine enhancement of learning might also involve elavated ERK1/2 signaling. In support, a prior study found that a systemic administration of an ERK1/2 inhibitor subthreshold for disrupting learning blocked the enhancement of contextual fear conditioning by acute nicotine (Raybuck and Gould, 2007). Finally, the present study found that no enhancement of learning by nicotine was observed when the protein synthesis inhibitor anisomycin was administered, suggesting that nicotine enhances learning through a PKA/ERK1/2/protein synthesis-dependent process.
A question that remains is how nicotine-related alterations in PKA and ERK1/2 signaling could lead to changes in cell signaling that support more efficient learning. Recently, it has been proposed that the timing of PKA and ERK1/2 activation may alter synaptic plasticity. A computational model of enhanced learning and memory, which relies on the dynamic responses of PKA and ERK1/2 to external stimulation, suggests that shifting the rapid and transient increase in PKA activity to coincide with a gradual increase in ERK1/2 activity results in enhanced learning (Zhang et al., 2012; for commentary see Abbott and Kandel, 2012). Predictions made with the model were confirmed experimentally by examining long-term sensitization in Aplysia. The findings from Zhang and colleagues (2012) that shifting the timing of PKA activation may lead to enhanced learning are particularly interesting because the present study found that nicotine shifted the timing of PKA and ERK1/2 activation in the hippocampus. Saline-treated mice trained in contextual conditioning showed an increase in hippocampal PKA activity 1 hour post-training whereas nicotine-treated mice showed an increase in PKA activity 2 hours post-training. A similar shift in ERK1/2 activation was seen; for the saline-treated training group, ERK1/2 activation was significantly increased at 2 hours post training whereas a significant increase in ERK1/2 activity was not seen in the nicotine-treated mice until 3 hours post training. Prior work has shown that nicotine enhances contextual learning and not specifically the context-shock association (Kenney and Gould, 2008). Thus, the shifting of PKA and ERK activity by nicotine could enhance processing and remembering contextual information; this could be beneficial for learning about potential dangers such as during fear conditioning but could be maladaptive by facilitating context-drug associations such as those that occur during conditioned place preference for nicotine (Portugal and Gould, 2009). It is of note that the nicotine control group and the nicotine fear conditioned group showed similar patterns of PKA and ERK activation. This could suggest that nicotine alone is sufficient to change the temporal pattern of PKA and ERK activation and that this then facilitates learning. It is also possible that the shift in PKA and ERK activation in the nicotine control group is related to learning that was not measured; that is learning about the context of the nicotine injections. While the current results suggest shifts in PKA and ERK1/2 activation may contribute to the enhanced learning, further work is required to understand how this could enhance learning.
The findings from the present study and prior work allow us to propose a cell signaling model of how nicotine may enhance hippocampus-dependent LTM. Evidence suggests that NMDA receptors, which gate Ca2+ and are critically involved in synaptic plasticity (for review, see Malenka and Nicoll 1993), and nAChRs may mediate similar learning-related processes (André et al., 2011). Like NMDA receptors, nAChRs gate Ca2+ (Karadsheh et al., 2004; Tapia et al., 2007), which can activate a number of intracellular signaling cascades including the cAMP/PKA pathway. Moreover, PKA can then activate ERK1/2 (Roberson et al., 1999). It is possible that increased Ca2+ influx due to nicotine alters the timing and level of PKA and ERK1/2 activation. This change in PKA and ERK1/2 activity might recruit additional cell signaling cascades that could strengthen synaptic plasticity. In support, prior work found that the nicotine enhancement of learning was critically dependent on c-jun N terminal kinase (JNK1 or MAPK8) activation in the dorsal hippocampus during the memory consolidation phase (Kenney et al., 2010b). The activation of JNK1 is involved in development and neurite outgrowth and can phosphorylate microtubule-associated protein 2 (MAP2) (Chang et al., 2003; Barnat et al., 2010; Hirai et al., 2011); these types of structural changes can support synaptic plasticity and contextual learning (Woolf et al., 1999; Muller et al., 2002; Khuchua et al., 2003). Thus, the shifting of PKA and ERK1/2 associated with nicotine treatment prior to training could facilitate activation of JNK1 or facilitate coordinated activation of an unidentified target by PKA, ERK, and JNK1. These possibilities require testing.
Finally, increasing evidence suggests that the dorsal and ventral hippocampi are distinct structures that mediate different behaviors (Fanselow and Dong, 2010). The dorsal hippocampus may be more involved in spatial and contextual learning (Moser et al., 1995) while the ventral hippocampus may be more involved in emotional behavior (Henke, 1990). The effects of nicotine on learning also segregate along the dorsal-ventral plane. For both trace and contextual fear conditioning, infusion of nicotine into the dorsal hippocampus enhanced learning whereas infusions into the ventral hippocampus disrupted learning (Raybuck and Gould, 2010; Kenney et al., 2012). Expression of nAChR subtypes also varies along the dorsal–ventral axis of the hippocampus (Mugnaini et al., 2002), which may contribute to the different effects of nicotine infusion into dorsal versus ventral hippocampus on learning. Further, systemic administration of nicotine prior to learning results in different activation of cell signaling cascades in the dorsal versus ventral hippocampus. Learning in the presence of nicotine increased in JNK1 phosphorylation in the dorsal but not the ventral hippocampus (Kenney et al., 2010b), and in the present study nicotine increased learning-related PKA and ERK1/2 activity in the dorsal but not the ventral hippocampus. If the dorsal and ventral hippocampi are mediating different processes, administration of nicotine during learning may favor dorsal hippocampal processes, which could facilitate the learning of context/spatial information.
5.0 Conclusion
In summary, acute nicotine enhances hippocampus-dependent learning (Kenney and Gould, 2008; Kenney et al., 2011; Melichercik et al., 2012) and the present findings indicate that nicotine specifically enhances LTM without affecting STM. The enhancement of contextual fear conditioning by nicotine is dependent upon PKA and ERK1/2 signaling. Interestingly, nicotine shifted the temporal activation of dorsal hippocampal PKA and ERK1/2 activity and differentially activated PKA and ERK1/2 in the dorsal versus ventral hippocampus during learning. This suggests that the timing of PKA and ERK1/2 activity may contribute to the strength of memory formation. In addition, the study demonstrated that nicotine acts directly on molecular substrates of LTM, suggesting that either the molecular substrates of STM and LTM are separate or that nicotine can increase PKA and ERK1/2 learning-related activity without modifying downstream cell signaling cascades involved in STM.
Highlights
-
1)
Acute nicotine enhances hippocampus-dependent LTM but not STM
-
2)
Increased PKA activation is necessary for enhancement of LTM by acute nicotine
-
3)
Nicotine and learning interact to shift activation of hippocampal PKA and ERK1/2
-
4)
Nicotine increases dorsal but not ventral hippocampus signaling during learning
Acknowledgements
We would like to thank Christopher de Solis and Bianca Case-Whiteside for their help with histology and Sheree Logue for reading an earlier version of this manuscript. We would like to acknowledge grant support from the National Institute on Drug Abuse (NIDA, DA024787, DA017949, TJG). DSW was supported by a NIDA diversity supplement (DA024787-01A1S1). PTL and SJS were supported by a NIDA training grant (T32DA007237, Ellen Unterwald).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no conflicts of interest.
References
- Abbott LF, Kandel ER. A computational approach enhances learning in Aplysia. Nature Neuroscience. 2012;15:178–179. doi: 10.1038/nn.3030. [DOI] [PubMed] [Google Scholar]
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Progeress in Brain Research. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behavioural Brain Research. 2004;149:17–31. doi: 10.1016/s0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- André JM, Leach PT, Gould TJ. Nicotine ameliorates NMDA receptor antagonist-induced deficits in contextual fear conditioning through high-affinity nicotinic acetylcholine receptors in the hippocampus. Neuropharmacology. 2011;60:617–625. doi: 10.1016/j.neuropharm.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nature Neuroscience. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F. Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. Journal of Neuroscience. 2010;30:7804–7816. doi: 10.1523/JNEUROSCI.0372-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learning & Memory. 1998;5:365–374. [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LSB, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Developmental Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. Journal of Neurochemistry. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychological Bulletin. 1984;96:518–559. [PubMed] [Google Scholar]
- Davis JA, Gould TJ. The effects of DHBE and MLA on nicotine-induced enhancement of contextual fear conditioning in C57BL/6 mice. Psychopharmacology (Berl) 2006;184:345–352. doi: 10.1007/s00213-005-0047-y. [DOI] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ. Hippocampal alpha4beta2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. Journal of Neuroscience. 2007;27:10870–10877. doi: 10.1523/JNEUROSCI.3242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Porter J, Gould TJ. Nicotine enhances both foreground and background contextual fear conditioning. Neuroscience Letters. 2006;394:202–205. doi: 10.1016/j.neulet.2005.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt DJ. A Requirement for the Mitogen-activated Protein Kinase Cascade in Hippocampal Long Term Potentation. Journal of Biological Chemistry. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood JF, Rosenzweig MR, Bennett EL, Orme AE. The influence of duration of protein synthesis inhibition on memory. Physiol Behav. 1973;10:555–562. doi: 10.1016/0031-9384(73)90221-7. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Research. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Higgins SJ. Nicotine enhances contextual fear conditioning in C57BL/6J mice at 1 and 7 days post-training. Neurobiology of Learning and Memory. 2003;80:147–157. doi: 10.1016/s1074-7427(03)00057-1. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Leach PT. Cellular, molecular, and genetic substrates underlying the impact of nicotine on learning. Neurobiology of Learning and Memory. 2013 doi: 10.1016/j.nlm.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TJ, Wehner JM. Nicotine enhancement of contextual fear conditioning. Behavioral Brain Research. 1999;102:31–39. doi: 10.1016/s0166-4328(98)00157-0. [DOI] [PubMed] [Google Scholar]
- Henke PG. Hippocampal pathway to the amygdala and stress ulcer development. Brain Research Bulletin. 1990;25:691–695. doi: 10.1016/0361-9230(90)90044-z. [DOI] [PubMed] [Google Scholar]
- Hirai S, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. Journal of Neuroscience. 2011;31:6468–6480. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EP. Synaptic plasticity: going through phases with LTP. Current Biology. 1998;8:R350–352. doi: 10.1016/s0960-9822(98)70219-2. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learning & Memory. 1994;1:74–82. [PubMed] [Google Scholar]
- Karadsheh MS, Shah MS, Tang X, Macdonald RL, Stitzel JA. Functional characterization of mouse α4β2 nicotinic acetylcholine receptors stably expressed in HEK293T cells. Journal of Neurochemistry. 2004;91:1138–1150. doi: 10.1111/j.1471-4159.2004.02801.x. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, III, Govindarajan A, Jung H-Y, Kang H, Tonegawa S. Translational Control by MAPK Signaling in Long-Term Synaptic Plasticity and Memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kenney JW, Adoff MD, Wilkinson DS, Gould TJ. The effects of acute, chronic, and withdrawal from chronic nicotine on novel and spatial object recognition in male C57BL/6J mice. Psychopharmacology (Berl) 2011;217:353–365. doi: 10.1007/s00213-011-2283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Florian C, Portugal GS, Abel T, Gould TJ. Involvement of hippocampal jun-N terminal kinase pathway in the enhancement of learning and memory by nicotine. Neuropsychopharmacology. 2010b;35:483–492. doi: 10.1038/npp.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Molecular Neurobiology. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Nicotine enhances context learning but not context-shock associative learning. Behav Neurosci. 2008;122:1158–1165. doi: 10.1037/a0012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Raybuck JD, Gould TJ. Nicotinic receptors in the dorsal and ventral hippocampus differentially modulate contextual fear conditioning. Hippocampus. 2012;22:1681–1690. doi: 10.1002/hipo.22003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Wilkinson DS, Gould TJ. The enhancement of contextual fear conditioning by ABT-418. Behavioral Pharmacology. 2010a;21:246–249. doi: 10.1097/FBP.0b013e32833a5b9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuchua Z, Wozniak DF, Bardgett ME, Yue Z, McDonald M, Boero J, Hartman RE, Sims H, Strauss AW. Deletion of the N-terminus of murine map2 by gene targeting disrupts hippocampal ca1 neuron architecture and alters contextual memory. Neuroscience. 2003;119:101–111. doi: 10.1016/s0306-4522(03)00094-0. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Rison RA, Fanselow MS. Effects of amygdala, hippocampus, and periaqueductal gray lesions on short- and long-term contextual fear. Behavioral Neuroscience. 1993;107:1093–1098. doi: 10.1037//0735-7044.107.6.1093. [DOI] [PubMed] [Google Scholar]
- Koh MT, Thiele TE, Bernstein IL. Inhibition of protein kinase A activity interferes with long-term, but not short-term, memory of conditioned taste aversions. Behavioral Neuroscience. 2002;116:1070–1074. [PubMed] [Google Scholar]
- Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proceedings of the National Academy of Sciences. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MC, Gould TJ. Latent inhibition of cued fear conditioning: an NMDA receptor-dependent process that can be established in the presence of anisomycin. European Journal of Neuroscience. 2004;20:818–826. doi: 10.1111/j.1460-9568.2004.03531.x. [DOI] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behavioral Neuroscience. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- Ma N, Abel T, Hernandez PJ. Exchange protein activated by cAMP enhances long-term memory formation independent of protein kinase A. Learning & Memory. 2009;16:367–370. doi: 10.1101/lm.1231009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends in Neuroscience. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. European Journal of Neuroscience. 2000;12:3741–3747. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- Melichercik AM, Elliott KS, Bianchi C, Ernst SM, Winters BD. Nicotinic receptor activation in perirhinal cortex and hippocampus enhances object memory in rats. Neuropharmacology. 2012;62:2096–2105. doi: 10.1016/j.neuropharm.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Michel M, Green CL, Lyons LC. PKA and PKC are required for long-term but not short-term in vivo operant memory in Aplysia. Learning & Memory. 2011;18:19–23. doi: 10.1101/lm.2026311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MB, Moser EI, Forrest E, Andersen P, Morris RG. Spatial learning with a minislab in the dorsal hippocampus. Proceedings of the National Academy of Sciences. 1995;92:9697–9701. doi: 10.1073/pnas.92.21.9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaini M, Tessari M, Tarter G, Merlo Pich E, Chiamulera C, Bunnemann B. Upregulation of [3H]methyllycaconitine binding sites following infusion of nicotine, without changes of α7 or α6 subunit mRNA: an autoradiography and in situ hybridization study in rat brain. European Journal of Neuroscience. 2002;16:1633–1646. doi: 10.1046/j.1460-9568.2002.02220.x. [DOI] [PubMed] [Google Scholar]
- Muller D, Nikonenko I, Jourdain P, Alberi S. LTP, memory and structural plasticity. Current Molecular Medicine. 2002;2:605–611. doi: 10.2174/1566524023362041. [DOI] [PubMed] [Google Scholar]
- Nie T, Abel T. Fear conditioning in inbred mouse strains: an analysis of the time course of memory. Behavioral Neuroscience. 2001;115:951–956. doi: 10.1037//0735-7044.115.4.951. [DOI] [PubMed] [Google Scholar]
- Parkinson JK, Murray EA, Mishkin M. A Selective Mnemonic Role for the Hippocampus in Monkeys: Memory for the Location of Objects. Journal of Neuroscience. 1988;8:4159–4167. doi: 10.1523/JNEUROSCI.08-11-04159.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behavioral Neuroscience. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Portugal GS, Gould TJ. Nicotine withdrawal disrupts new contextual learning. Pharmacology Biochemistry and Behavior. 2009;92:117–123. doi: 10.1016/j.pbb.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck JD, Gould TJ. Extracellular signal-regulated kinase 1/2 involvement in the enhancement of contextual fear conditioning by nicotine. Behavioral Neuroscience. 2007;121:1119–1124. doi: 10.1037/0735-7044.121.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck JD, Gould TJ. The role of nicotinic acetylcholine receptors in the medial prefrontal cortex and hippocampus in trace fear conditioning. Neurobiology of Learning and Memory. 2010;94:353–363. doi: 10.1016/j.nlm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. Journal of Neuroscience. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular Signal-Regulated Kinase 2 (ERK2) Knockdown Mice Show Deficits in Long-Term Memory; ERK2 Has a Specific Function in Learning and Memory. Journal of Neuroscience. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learning & Memory. 1999;6:97–110. [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Current Opinion in Neurobiology. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Molecular Pharmacology. 2007;71:769–776. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Trifilieff P, Herry C, Vanhoutte P, Caboche J, Desmedt A, Riedel G, Mons N, Micheau J. Foreground contextual fear memory consolidation requires two independent phases of hippocampal ERK/CREB activation. Learning & Memory. 2006;13:349–358. doi: 10.1101/lm.80206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallenstein GV, Vago DR, Walberer AM. Time-dependent involvement of PKA/PKC in contextual memory consolidation. Behavioural Brain Research. 2002;133:159–164. doi: 10.1016/s0166-4328(01)00476-4. [DOI] [PubMed] [Google Scholar]
- Welsby PJ, Rowan MJ, Anwyl R. Intracellular mechanisms underlying the nicotinic enhancement of LTP in the rat dentate gyrus. European Journal of Neuroscience. 2009;29:65–75. doi: 10.1111/j.1460-9568.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Zinnerman MD, Johnson GV. Hippocampal microtubule-associated protein-2 alterations with contextual memory. Brain Research. 1999;821:241–249. doi: 10.1016/s0006-8993(99)01064-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu R-Y, Heberton GA, Smolen P, Baxter DA, Cleary LJ, Byrne JH. Computational design of enhanced learning protocols. Nature Neuroscience. 2012;15:294–297. doi: 10.1038/nn.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]