

Abstract
Small doses of glucagon given subcutaneously in the research setting by an automated system prevent most cases of hypoglycemia in persons with diabetes. However, glucagon is very unstable and cannot be kept in a portable pump. Glucagon rapidly forms amyloid fibrils, even within the first day after reconstitution. Aggregation eventually leads to insoluble gels, which occlude pump catheters. Fibrillation occurs rapidly at acid pH, but is absent or minimal at alkaline pH values of ~10.
Glucagon also degrades over time; this problem is greater at alkaline pH. Several studies suggest that its primary degradative pathway is deamidation, which results in a conversion of asparagine to aspartic acid.
A cell-based assay for glucagon bioactivity that assesses glucagon receptor (GluR) activation can screen promising glucagon formulations. However, mammalian hepatocytes are usually problematic as they can lose GluR expression during culture. Assays for cyclic AMP (cAMP) or its downstream effector, protein kinase A (PKA), in engineered cell systems, are more reliable and suitable for inexpensive, high-throughput assessment of bioactivity.
Keywords: glucagon, cytotoxicity, amyloid, diabetes, assay
Introduction and Background
Glucagon, a 29-amino acid peptide, was discovered in 1922 by Kimball and Murlin during their efforts to purify insulin from pancreatic extracts. The discovery was accidental; they observed that after injection of insulin-containing extracts into animals, one of the preparations caused a rapid rise in blood glucose level before it caused the expected fall. Fortunately, they pursued this unexpected finding, verified it, and published the results in 1923 [1]. It was not until the 1950’s that glucagon was purified and crystallized. In this era, glucagon was also found to exhibit unusual biochemical behavior; shortly after incubation in aqueous solution, many slender fibrillar structures were noted [2]. As the glucagon solutions were incubated over time, the concentration of these fibrils increased. The solutions eventually formed firm gels that contained high concentrations of packed fibrils.
These fibrils are now known to exhibit a beta-pleated sheet amyloid protein configuration. Many subtypes of fibrils and protofibrils have been discovered; for excellent reviews, the reader is referred to the papers of Pedersen et al. [3, 4]. Though some compounds, including cyclodextrins, have been reported to reduce the tendency to form amyloid fibrils, there is no well-accepted additive or method that markedly prevents their formation.
In 2004, a very interesting and widely cited report regarding the effect of fibrillated solutions of glucagon on mammalian cells was published [5]. The authors observed that, under certain conditions, fibrillated glucagon was toxic to mammalian cells in culture. They emphasized the potentially dangerous nature of glucagon fibrils and pointed out that there are several deadly diseases characterized by the formation of amyloid fibrils, including Alzheimer’s Disease, Parkinson’s Disease, and prion diseases. More recently, amyloid formation of lipoproteins has been reported to be associated with the pathogenesis of atherosclerosis [6].
It should also be noted, however, that a recent comprehensive analysis of the conformation of secreted hormones in storage vesicles in vivo 7] demonstrated that most hormones are packed into amyloid fibrils prior to release, with dissociation to the monomeric form occurring after appearance in the circulation.
Shortly after the report of potentially toxic effects of glucagon in vitro, several reports appeared which showed the benefit of closed-loop administration of glucagon in animals [8, 9] and in humans [10, 11].
These human reports emphasized the benefit of administering glucagon (with insulin) in the closed-loop setting. In our study, subjects with type 1 diabetes received insulin plus placebo on one occasion and insulin plus glucagon (as commanded by an automated algorithm) on another occasion. In terms of effectiveness, the duration of time in the hypoglycemic range for the bihormonal approach was less than half of the duration for the insulin-only experiments using doses of glucagon (given subcutaneously) that are generally quite small, and range from 60-200 μg over 5-10 min [11]. For these studies, commercially available lyophilized glucagon indwelled within a pump was frequently reconstituted with diluent in order to minimize the effect of fibrillation.
These favorable clinical results suggest a definite need for a preparation of glucagon (or a suitable bioactive analog) that is sufficiently stable so that it can be indwelled in a portable pump for at least three days, the currently allowed maximum for portable pump use. Currently, the FDA-approved instructions for commercially available glucagon allow only for immediate usage of glucagon to treat severe hypoglycemia, after which the unused portion must be discarded.
The effect of pH and other conditions on glucagon stability
In a recent study [12], we investigated whether the pH values during aging would affect the degree of fibrillation and cytotoxicity. We were particularly interested in studying the alkaline range in addition to the more commonly utilized acidic range (the latter of which is used in both of the preparations that are commercially available in the US). Because of its low solubility based on an isoelectric point of approximately 7, the neutral range is generally not suitable for evaluation of glucagon.
We were also interested in understanding the specific effects of pH and osmolarity on cytotoxicity, especially because we knew that some of the earlier studies did not report this information. The preparations were aged for various durations up to 7 days at 37° C.
A pH study was carried out to assess the direct effect of pH on cytotoxicity in NIH-3T3 cells. Glucagon aggregation and fibrillation were measured by the degree of reaction with the Congo red reagent.
In the pH study, we found that only those media pH values of 7.0-9.5 avoided cytotoxicity in NIH 3T3 cells. Similarly, we found that only the media osmolality range of 250-400 mOsm/l avoided cytotoxicity. Such findings are useful in interpreting studies in which compounds are tested for cytotoxicity. To the degree that the drug itself causes the cell culture media to fall out of these ranges, the appropriate interpretation would be that a toxic effect was at least partially attributable to the pH itself, quite apart from the compound being tested. For the commercially available glucagon, the pH of the cell culture media was approximately 6.5. Not surprisingly, even without aging, a high concentration of this acid preparation (5 mg/ml) caused immediate cytotoxicity, an effect likely due to pH or excipients.
For glucagon prepared at a pH of 8.5 or 10 without aging, there was no significant cytotoxicity at any concentration. However, after aging for 5 days, there was some evidence of toxicity at pH 8.5 at a concentration of 2.5 mg/ml. Importantly, after aging for 5 days at a pH of 10 in glycine buffer, there was no evidence of cytotoxicity [12].
Conditions that favor cytotoxicity also favor amyloid fibril formation and suggest that large amounts of fibrillar amyloid lead to cytotoxicity. Congo red amyloid intensity was greatest in glucagon prepared and aged at pH 3 in citrate buffer, indicating substantial amyloid fibril formation. At a mildly alkaline pH of 8.5, there was also substantial Congo red intensity after aging. In contrast, at a pH of 10, there was no Congo red staining for native glucagon, even after aging for 28 days. The results for size-exclusion chromatography (SEC) confirmed the Congo red findings. Using SEC, at pH values of 3, there was rapid loss of the monomeric glucagon peak over 1-3 days. At a pH of 8, the loss occurred over 1-3 weeks. However, at a pH of 10, there was remarkable preservation of the monomeric glucagon peak, with little if any loss even after 3-4 weeks [12]. Very recently, in unpublished studies, we sought to better understand the effect of differences in pH on glucagon fibrillation kinetics. Using very sensitive Thioflavin T and tryptophan fluorescence assays, we observed very rapid fibrillation between pH 8.6 and 9.6, starting within 24 hours of aging. However, with these sensitive tools, at higher pH values of 9.8-10, glucagon showed no evidence of amyloid fibril formation after one week [13].
These experiments have clarified several key concepts regarding the stability of glucagon.
In native glucagon, amyloid fibrils form very quickly (with substantial formation within 24 hours).
Amyloid formation occurs much more rapidly at acid pH than at alkaline pH. At pH 10, formation of amyloid is almost undetectable.
In studies in which compounds are investigated for potential cytotoxicity, experiments must be carried out within pH and osmolality ranges that are not inherently cytotoxic. (The cells commonly used for such assays, NIH-3T3 cells, require a pH of 7.0-9.5 and an osmolality of 250-400 mOsm/l).
Even at high glucagon concentrations, there is no evidence of cytotoxicity when aged at a pH of 10, though there is some cytotoxicity at pH 8.5.
The glycine buffer for pH 10 solutions need not be present in high concentration; 20 mM is sufficient to hold glucagon at 1 mg/ml at a pH of 10. In fact, we believe such a low concentration to be desirable. A low concentration of buffer would allow faster equilibration down to normal neutral body pH after subcutaneous injection in humans.
We also sought to address the activity of fresh and amyloid-rich, aged glucagon solutions after subcutaneous injection into non-diabetic pigs, since earlier data from our group and the Boston group suggested that aged solutions might have greater bioactivity than one might expect. The Yorkshire pigs were treated with octreotide to suppress their endogenous production of glucagon, in order to isolate the effect to exogenous glucagon. To understand the pharmacokinetic effect of glucagon, we also collected periodic serum samples for measurement of glucagon levels during these studies. We found that the pharmacodynamic (PD) hyperglycemic effect of fibrillated glucagon aged for one week was substantially similar to the PD effect of fresh glucagon. When the glucagon plasma levels were measured, we observed a tendency for the rise in glucagon to be slightly delayed (approximately 10-15 minutes) in the aged vs freshly prepared formulation. This finding suggests that fibrillated glucagon, in which the glucagon monomers are held together by weak bonds, almost certainly dissociates over time to monomeric glucagon, which can be absorbed into the bloodstream [12]. Nonetheless, for safety and regulatory reasons, fibrillated glucagon is unsuitable for human drug use.
Degradation of glucagon
Many proteins degrade over time, and deamidation of asparagine and glutamine residues are a known degradation mechanism in mammals. Robinson and Robinson have suggested that this process is a normal mechanism that appropriately regulates protein activity lifetime in animals [14]. Deamidation is typically pH-dependent, with the lowest rates occurring around pH 4-6, with marked rate increases occurring at both high and low pH extremes [14]. Asparagine deamidation is more common than glutamine deamidation, and is better understood. At acidic pH, direct hydrolysis is the major mechanism for deamidation. At neutral and alkaline pH, succinimide ring formation contributes to deamidation, which is inhibited by steric hindrance from neighboring amino acid side chains. Glutamine deamidation is less well understood, but kinetic evidence points to direct hydrolysis [15]. The intermediate formations in asparagine involve a five-membered ring, while glutamine intermediates involve a less-stable six-membered ring [16].
Experimentally, low pH glucagon has demonstrated deamidation at glutamine and asparagine residues and peptide cleavage at aspartate residues [16]. Glucagon has three glutamine residues and one asparagine, making four of its 29 amino acids susceptible to deamidation. Our preliminary work, presented in abstract form [13], showed a pH-dependent degradation of native glucagon, which increased with rising pH from pH 8.6 to 10. The degradation products were analyzed by liquid chromatography-mass spectrometry (LCMS), which showed deamidation as the major degradative pathway, accompanied by some oxidation and chain cleavage. Deamidated glucagon appears on LC-MS analysis as a separate, well-defined peak with a retention time 2-3 minutes shorter than that of native glucagon. Its mass is increased by 1 Dalton over native glucagon. We observed that, by day 3 of aging at 370C at pH 10, 35% of the glucagon at pH 10 has been deamidated, compared to only 20% at pH 9 at the same time point [13].
These data confirm that both the aggregation and degradation of glucagon are strongly pH-dependent. Because there is no one pH value that avoids both processes, it is likely that stabilizing agents will be needed to optimize glucagon formulations.
There are other approaches that show promise in achieving a stable, liquid glucagon formulation. First, a team at Biodel, Inc., found a way to stabilize glucagon at neutral pH by the use of a surfactant accompanied by a sugar. Like the alkaline preparation discussed above and the other approaches discussed below, the formulation has not been approved by the FDA. The Biodel formulation was found to be reasonably stable by reverse-phase HPLC over several weeks [17]. For more details regarding the Biodel approach, the reader is referred to a published patent application [18].
Another approach is to create a mutant form of glucagon by substituting non-native amino acids into the peptide chain. Chabenne and DiMarchi et al. reported that such a substitution near the C-terminal end of the glucagon molecule achieved a substantial degree of biostability [19]. For a summary of glucagon analogs proposed by this group, whose technology is now owned by Roche, see patent application [20]. The group from Xeris Pharmaceuticals presented preliminary evidence at the 2012 ADA Scientific Sessions (http://xerispharma.com/ADA_Poster_FINAL_5_30.pdf) showing that native glucagon can be stabilized in liquid form with the use of a non-aqueous solvent (dimethyl sulfoxide). In their presentation, they reported that this formulation largely avoids glucagon degradation and fibrillation. For their patent application, see [21]. Other approaches designed to stabilize glucagon include the method of Arecor, Inc., which uses excipients that exchange protons with proteins [22].
The foregoing methods mentioned here also have the potential to stabilize liquid glucagon for use as a rescue injection for persons with diabetes who suffer from marked symptomatic hypoglycemia in the setting of an emergency. Currently, it is necessary for the companions of such individuals to quickly mix a lyophilized powder with an aqueous diluent and then inject the solution subcutaneously or intramuscularly into the patient. Such a procedure is very difficult for a lay person to accomplish, especially in the heat of an emergency. If a stable liquid solution were available, the procedure would be simpler, safer, and less anxiety-provoking.
In order to avoid the need for manual mixing, Xeris is developing a glucagon pen that uses a non-aqueous liquid solvent that requires no mixing. In addition, Enject, Inc., has developed a pen device that, upon injection, automatically mixes the diluent with the lyophilized glucagon powder. Though not yet commercially available, this device is summarized in the following document (http://www.enject.com/uploads/Enject_article_BioCentury.pdf).
Tolerability of subcutaneously-administered of alkaline proteins in humans
There is a paucity of information about tolerability of alkaline drug solutions that are administered subcutaneously. In an off-label fashion, furosemide, a highly alkaline solution, is sometimes given by the subcutaneous route, but the parenteral formulation is approved only for intravenous use [23]. Because of the question of tolerability of glucagon at a high pH, we carried out a study in which we administered a common, well-tolerated protein (human albumin) to normal human subjects. In a double-blind fashion, on separate occasions, a physician administered albumin (1 mg/ml) buffered at pH 7.4 (with phosphate buffer) and albumin buffered at pH 10 (with glycine buffer). Each subject received 4 injections: albumin at each pH given fast (over 10 seconds) or slow (over 60 seconds). Subjects rated their experience of discomfort on a 6-point scale, from 0-5, with 0 representing no discomfort. The results [24] demonstrated that:
Slow injections caused slightly more discomfort than fast injections.
Albumin at pH 10 caused slightly more discomfort than albumin at pH 7.4.
However, even at pH 10, the discomfort was rated as minimal or slight, approximately 1 on a scale of 0-5.
Using the Draize scale, there was no more visible inflammation at the subcutaneous site with the alkaline injection vs. the neutral injection.
Methods for assessment of glucagon bioactivity in vitro
As outlined above, many preparations of glucagon are chemically unstable and lose bioactivity over time. For this reason, it is important that glucagon analogs or formulations be demonstrated to be bioactive using in vitro assays before initiation of in vivo animal or clinical studies. In contrast to in vivo studies, cellular assays of glucagon action represent a high-throughput, inexpensive assay that can be used to quickly screen different glucagon formulations. Cell-based assays are based on the fact that the major biological actions of glucagon, such as inhibition of hepatic glucose production, result from activation of the cell-surface glucagon receptor (GluR) in hepatocytes. The GluR is a classical 7-transmembrane G protein-coupled receptor that is linked to adenylate cyclase, which generates the second messenger cAMP after ligand binding [25]. The increased intracellular cAMP then binds to the regulatory subunit of PKA, resulting in the release of the PKA catalytic subunit, which then is able to phosphorylate downstream effectors of the GluR signaling cascade [26]. The unactivated PKA complex is localized in a punctate pattern due to association with cytoskeletal cellular structures, while activated PKA catalytic subunits become dispersed in the cytoplasm as they interact with target substrates. Thus, GluR activation can be assessed by determination of glucagon-stimulated dispersion of PKA.
Glucagon activity in vitro can be assessed in hepatocytes by directly measuring rises in glucose levels in the cell media or reduction in intracellular glycogen stores. Such direct measurements require the use of hepatocytes that are able to store glycogen and retain responsiveness to glucagon. However, it is important to note that both the storage of glycogen and the expression of the GluR are differentiation-dependent processes that may be lost in transformed hepatocyte-derived cell lines. Thus, cell line choice is critical. HepG2 cells, a commonly used hepatoma line that retains many hepatocyte-specific characteristics, did not respond to glucagon in our hands or in the experience of others [27]. Though some investigators have successfully used primary rat or human hepatocytes to assay the glucagon effect [28, 29], primary hepatocytes are expensive, inconvenient, and their performance, in our experience, is inconsistent.
The alternative is to generate GluR-expressing lines by transient [27, 30, 31] or stable [32] expression of a human or rodent GluR cDNA. Stable expression generally produces lower effects of GluR than transient expression. The lower levels may be adequate for binding studies but are not sufficient for generation of sufficiently detectable levels of cAMP; thus, it can be difficult to select clones that express the appropriate level of GluR without resulting in ligand-induced desensitization [33].
An alternative approach, favored by the authors of this review, involves the use of a Chinese hamster ovary cell line available from Thermo-Scientific in Lafayette, CO (http://www.thermoscientific.com/ecomm/servlet/productsdetail_11152 11961931-1) that, although not expressing endogenous GluR, has been stably transfected with a cDNA encoding the human GluR as well as a cDNA encoding a fluorescent version of PKA comprised of a fusion protein of the catalytic subunit of PKA and green fluorescent protein (GFP). GluR activation is assessed by measurement of the disappearance of highly fluorescent PKA aggregates after ligand treatment with the use of fluorescence microscopy. This approach is suitable for relatively high-throughput screening in a multi-well format and allows time-course and dose-response studies using different ligands to activate PKA.
A figure of this glucagon assay using PKA-GFP activation is shown as Figure 1. It can be seen that fluorescence (expressed as normalized activity on the Y-axis) declines with increasing glucagon effect. Note that the region over which fluorescence declines corresponds to the physiological range in mammals (30-300 pg/ml) over which glucagon exerts its biologic effects.
Figure 1.
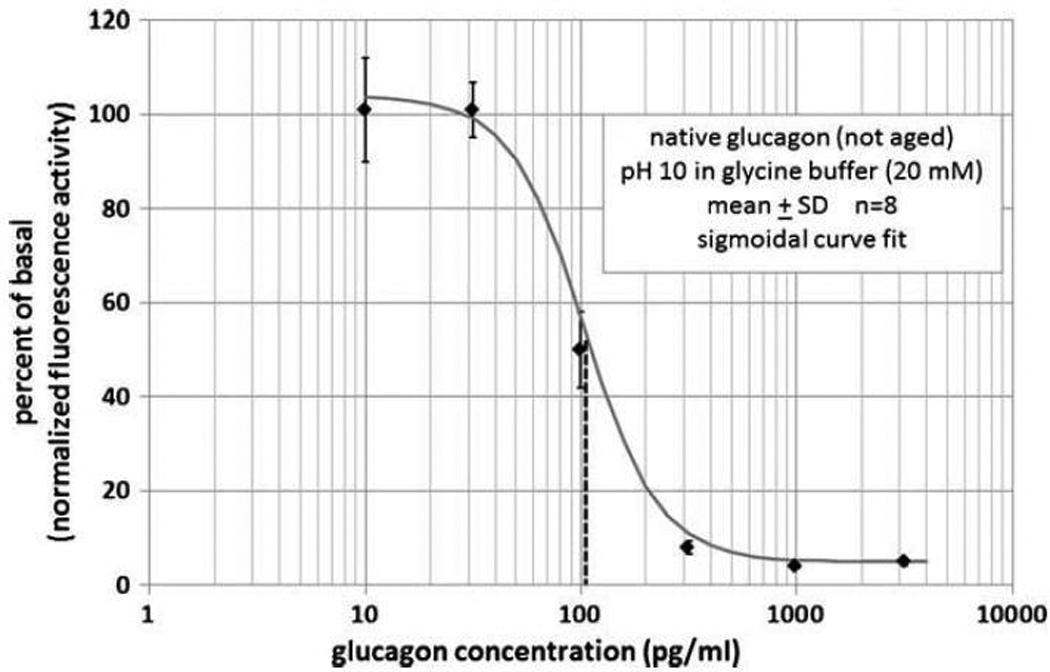
Conclusions
In summary, automated delivery of low-dose glucagon is proving useful in preventing hypoglycemia, a feared complication of insulin-treated diabetes. However, glucagon is a highly unstable peptide prone to spontaneous polymerization to an amyloid form (which can be minimized at alkaline pH) and spontaneous degradation. A cell-based PKA-based fluorescent bioassay is a convenient way of assessing glucagon formulations in vitro.
Cited References
- 1.Kimball CP, Murlin JR. Aqueous extracts of pancreas: some precipitation reactions of insulin. J. Biol. Chem. 1923;66:337. [Google Scholar]
- 2.Staub A, Behrens OK. The glucagon content of crystalline insulin preparations. J. Clin. Invest. 1954;33:1629–1633. doi: 10.1172/JCI103043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pedersen JS. The nature of amyloid-like glucagon fibrils. J Diabetes Sci Technol. 2010;4:1357–1367. doi: 10.1177/193229681000400609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pedersen JS, Dikov D, Flink JL, Hjuler HA, Christiansen G, Otzen DE. The changing face of glucagon fibrillation: structural polymorphism and conformational imprinting. J Mol Biol. 2006;355:501–523. doi: 10.1016/j.jmb.2005.09.100. [DOI] [PubMed] [Google Scholar]
- 5.Onoue S, Ohshima K, Debari K, Koh K, Shioda S, Iwasa S, Kashimoto K, Yajima T. Mishandling of the therapeutic peptide glucagon generates cytotoxic amyloidogenic fibrils. Pharm Res. 2004;21:1274–1283. doi: 10.1023/b:pham.0000033016.36825.2c. [DOI] [PubMed] [Google Scholar]
- 6.Teoh CL, Griffin MD, Howlett GJ. Apolipoproteins and amyloid fibril formation in atherosclerosis. Protein Cell. 2011;2:116–127. doi: 10.1007/s13238-011-1013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, Eisenberg D, Rivier J, Sawchenko P, Vale W, Riek R. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Khatib FH, Jiang J, Damiano ER. A feasibility study of bihormonal closed-loop blood glucose control using dual subcutaneous infusion of insulin and glucagon in ambulatory diabetic swine. J Diabetes Sci Technol. 2009;3:789–803. doi: 10.1177/193229680900300428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward W, Engle J, Duman H, Bergstrom C, Kim S, Federiuk I. The Benefit of Subcutaneous Glucagon During Closed-Loop Glycemic Control in Rats With Type 1 Diabetes. IEEE, Sensors Journal. 2008;8:88–96. [Google Scholar]
- 10.El-Khatib FH, Russell SJ, Nathan DM, Sutherlin RG, Damiano ER. A bihormonal closed-loop artificial pancreas for type 1 diabetes. Sci Transl Med. 2010;2:27ra27. doi: 10.1126/scitranslmed.3000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castle JR, Engle JM, El Youssef J, Massoud RG, Yuen KC, Kagan R, Ward WK. Novel use of glucagon in a closed-loop system for prevention of hypoglycemia in type 1 diabetes. Diabetes Care. 2010;33:1282–1287. doi: 10.2337/dc09-2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward WK, Massoud RG, Szybala CJ, Engle JM, El Youssef J, Carroll JM, Roberts CT, Jr, DiMarchi RD. In vitro and in vivo evaluation of native glucagon and glucagon analog (MAR-D28) during aging: lack of cytotoxicity and preservation of hyperglycemic effect. J Diabetes Sci Technol. 2010;4:1311–1321. doi: 10.1177/193229681000400604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson M, Stonex T, Caputo N, El Youssef J, Branigan D, Castle JR, David L, Ward WK. Development of a Stable Formulation of Liquid Glucagon for Use in a Bihormonal Pump; Presented at 2012 Scientific Session, American Diabetes Association, Philadelphia; 2012. [Google Scholar]
- 14.Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci U S A. 2001;98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joshi AB, Sawai M, Kearney WR, Kirsch LE. Studies on the mechanism of aspartic acid cleavage and glutamine deamidation in the acidic degradation of glucagon. J Pharm Sci. 2005;94:1912–1927. doi: 10.1002/jps.20405. [DOI] [PubMed] [Google Scholar]
- 16.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of protein pharmaceuticals: an update. Pharm Res. 2010;27:544–575. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 17.Steiner SS, Li M, Hauser R, Pohl R. Stabilized glucagon formulation for bihormonal pump use. J Diabetes Sci Technol. 4:1332–1337. doi: 10.1177/193229681000400606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steiner SS, Hauser R, Li M, Feldstein R, Pohl R. Stabilized glucagon solutions. US Patent Application # 12/891,240
- 19.Chabenne JR, DiMarchi MA, Gelfanov VM, DiMarchi RD. Optimization of the native glucagon sequence for medicinal purposes. J Diabetes Sci Technol. 2010;4:1322–1331. doi: 10.1177/193229681000400605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiMarchi MA, Smiley DL, DiMarchi M, Chabenne JR, Day J. Glucagon analogs exhibiting enhanced solubility and stability. US Patent Application # 12/999,284
- 21.Prestrelski S, Fang WJ, Carpenter JF, Kinzell J. Glucagon formulations for the treatment of hypoglycemia. US Patent Application # 13/186,275
- 22.Jezek J. Stable aqueous systems comprising proteins. US Patent Application 11/994,408
- 23.Verma AK, da Silva JH, Kuhl DR. Diuretic effects of subcutaneous furosemide in human volunteers: a randomized pilot study. Ann Pharmacother. 2004;38:544–549. doi: 10.1345/aph.1D332. [DOI] [PubMed] [Google Scholar]
- 24.Ward WK, Castle JR, Branigan D, Massoud RG, El Youssef J. Discomfort from an Alkaline Formulation Delivered Subcutaneously in Humans: Albumin at pH 7 versus pH 10. Clinical Drug Investigation. 2012;32:433–438. doi: 10.2165/11632840-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brubaker PL, Drucker DJ. Structure-function of the glucagon receptor family of G protein-coupled receptors: the glucagon, GIP, GLP-1, and GLP-2 receptors. Receptors Channels. 2002;8:179–188. [PubMed] [Google Scholar]
- 26.Almholt K, Tullin S, Skyggebjerg O, Scudder K, Thastrup O, Terry R. Changes in intracellular cAMP reported by a Redistribution assay using a cAMP-dependent protein kinase-green fluorescent protein chimera. Cell Signal. 2004;16:907–920. doi: 10.1016/j.cellsig.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Y, Cypess AM, Muse ED, Wu CR, Unson CG, Merrifield RB, Sakmar TP. Glucagon receptor activates extracellular signal-regulated protein kinase 1/2 via cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2001;98:10102–10107. doi: 10.1073/pnas.131200398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pilar Lopez M, Gomez-Lechon MJ, Castell JV. Role of glucose, insulin, and glucagon in glycogen mobilization in human hepatocytes. Diabetes. 1991;40:263–268. doi: 10.2337/diabetes.40.2.263. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Lechon MJ, Ponsoda X, Castell JV. A microassay for measuring glycogen in 96-well-cultured cells. Anal Biochem. 1996;236:296–301. doi: 10.1006/abio.1996.0170. [DOI] [PubMed] [Google Scholar]
- 30.Cypess AM, Unson CG, Wu CR, Sakmar TP. Two cytoplasmic loops of the glucagon receptor are required to elevate cAMP or intracellular calcium. J Biol Chem. 1999;274:19455–19464. doi: 10.1074/jbc.274.27.19455. [DOI] [PubMed] [Google Scholar]
- 31.Cascieri MA, Koch GE, Ber E, Sadowski SJ, Louizides D, de Laszlo SE, Hacker C, Hagmann WK, MacCoss M, Chicchi GG, Vicario PP. Characterization of a novel, non-peptidyl antagonist of the human glucagon receptor. J Biol Chem. 1999;274:8694–8697. doi: 10.1074/jbc.274.13.8694. [DOI] [PubMed] [Google Scholar]
- 32.Parker JC, McPherson RK, Andrews KM, Levy CB, Dubins JS, Chin JE, Perry PV, Hulin B, Perry DA, Inagaki T, Dekker KA, Tachikawa K, Sugie Y, Treadway JL. Effects of skyrin, a receptor-selective glucagon antagonist, in rat and human hepatocytes. Diabetes. 2000;49:2079–2086. doi: 10.2337/diabetes.49.12.2079. [DOI] [PubMed] [Google Scholar]
- 33.Ikegami T, Cypess AM, Bouscarel B. Modulation of glucagon receptor expression and response in transfected human embryonic kidney cells. Am J Physiol Cell Physiol. 2001;281:C1396–C1402. doi: 10.1152/ajpcell.2001.281.4.C1396. [DOI] [PubMed] [Google Scholar]