

Abstract
Mitochondrial dysfunction is thought to play a role in the pathogenesis of a variety of disease states, including sepsis. An acquired defect in oxidative phosphorylation potentially causes sepsis-induced organ dysfunction. Cytochrome oxidase (CcOX), the terminal oxidase of the respiratory chain, is competitively inhibited early in sepsis and progresses, becoming noncompetitive during the late phase. We have previously demonstrated that exogenous cytochrome c can overcome myocardial CcOX competitive inhibition and improve cardiac function during murine sepsis at the 24-h point. Here, we evaluate the effect of exogenous cytochrome c on CcOX activity and survival in mice at the later time points. Exogenous cytochrome c (800 μg) or saline was intravenously injected 24 h after cecal ligation and puncture (CLP) or sham operation. Steady-state mitochondrial cytochrome c levels and heme c content increased significantly 48 h post-CLP and remained elevated at 72 h in cytochrome c-injected mice compared with saline injection. Cecal ligation and puncture inhibited CcOX at 48 h in saline-injected mice. However, cytochrome c injection abrogated this inhibition and restored CcOX kinetic activity to sham values at 48 h. Survival after CLP to 96 h after cytochrome c injection approached 50% compared with only 15% after saline injection. Thus, a single injection of exogenous cytochrome c 24 h post-CLP repletes mitochondrial substrate levels for up to 72 h, restores myocardial COX activity, and significantly improves survival.
Keywords: Cytochrome c, cytochrome oxidase, oxidative phosphorylation, mitochondria, sepsis, heart
INTRODUCTION
Mitochondrial dysfunction is thought to play a role in the pathogenesis of a variety of disease states, including sepsis (1-4). An acquired defect in oxidative phosphorylation potentially causes sepsis-induced organ dysfunction (5). A number of electron transport chain abnormalities have been demonstrated in a variety of tissues using sepsis and sepsisrelated models (6-13). Diminished function of any of the electron transport complexes can impair oxidative phosphorylation, limit aerobic adenosine triphosphate (ATP) synthesis, and lead to bioenergetic failure (5, 14).
Cytochrome oxidase (CcOX), the terminal oxidase of the respiratory chain, uses electrons donated by cytochrome c to reduce oxygen to water (15). Coupled with the reduction of oxygen, CcOX pumps hydrogen ions across the mitochondrial inner membrane to the intermembrane space to maintain the hydrogen ion gradient (15). This proton motive force is crucial for ATP synthesis. Myocardial CcOX is inhibited during murine sepsis (8). This inhibition is competitive and reversible early after cecal ligation and puncture (CLP) and progresses, becoming noncompetitive and irreversible during the late phase of sepsis (8). In addition, mitochondrial levels of the enzyme's substrate, cytochrome c, begin to decline 24 h post-CLP just before the onset of noncompetitive inhibition (8, 16). Cytochrome oxidase inhibition coupled with substrate limitation possibly impairs oxidative phosphorylation in the septic heart and is a potential cause of sepsis-associated myocardial depression.
The onset of the hypodynamic phase of sepsis coincides with maximal competitive CcOX inhibition and the initial decline in mitochondrial cytochrome c (8, 16). Increasing substrate availability during competitive enzyme inhibition can overcome inhibition and restore enzyme velocity (17). In prior work, we demonstrated that exogenous cytochrome c gains access to cardiomyocyte mitochondria, repletes mitochondrial levels of cytochrome c, restores myocardial CcOX activity, and improves cardiac function 24 h post-CLP (16). It is unknown if this effect is sustained over time. Here, we evaluate the effect of exogenous cytochrome c on myocardial CcOx activity and survival in the later time points of sepsis after a single injection at the 24-h time point.
MATERIALS AND METHODS
Induction of Sepsis
The care of the animals in this study was in accordance with National Institutes of Health and Institutional Animal Care and Use Committee guidelines. Under isoflurane general anesthesia (up to 2%), 6- to 8-week-old male C57Bl/6 mice (Charles River, Boston, Mass) underwent cecal ligation and double puncture with a 23-gauge needle or sham operation as previously described (8). All animals were administered 50 mL/kg saline (s.c.) immediately postprocedure and every 24 h. Once awake, animals were given access to food and water ad libitum. Mice were randomly killed at either 48 or 72 h postprocedure. Five animals per group were studied per experiment. All animals were euthanized with 150 mg/kg of pentobarbital (i.p.).
Cytochrome c reduction
Cytochrome c from bovine heart (Sigma-Aldrich Corp., St. Louis, Mo) was reduced to ferrocytochrome c as previously described (18). Cytochrome c (50 mg) was dissolved in 10 mL of 0.01 M sodium phosphate buffer (pH 7.0). The solution was reduced with 2 mg of ascorbic acid. Excess ascorbate was dialyzed off against 0.01 M sodium phosphate buffer (pH 7.0) for 18 to 24 h at 4°C. This technique consistently yielded a 0.5% solution.
Exogenous ferrocytochrome c injection
For each experiment, 800 μg (150 μL) of reduced cytochrome c (40 mg/kg) was injected via tail vein under isoflurane anesthesia (up to 2%) 24 h after CLP or sham operation. Sham-injected cohorts received 150 μL of saline via tail vein.
The 800 μg dose was chosen based on previous data and preliminary work (16) (data not shown; 400 μg minimally increased mitochondrial cytochrome c in septic myocardium, whereas 800 μg increased levels by almost 2-fold).
Mitochondrial isolation
As previously described, cardiac ventricles were harvested and homogenized in ice-cold H medium (70 mM sucrose, 220 mM mannitol, 2.5 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, and 2 mM EDTA) (8, 19). The homogenate was spun at 1,500g for 10 min at 4°C. Supernatants were removed and centrifuged at 10,000g for 10 min at 4°C. Pellets were resuspended in H medium and centrifuged again at 10,000g for 10 min at 4°C. Pellets were again resuspended in H medium, and mitochondrial protein concentration was determined using the method of Lowry (8, 19, 20).
Steady-state levels of cardiac mitochondrial cytochrome c
Samples (10 μg) of mitochondrial protein were subjected to sodium dodecyl sulfate-acrylamide gel electrophoresis and immunoblotting as previously described (8, 21). Blots were labeled with a primary polyclonal antibody to bovine cytochrome c (Molecular Probes, Eugene, Ore) and secondarily exposed to donkey antirabbit immunoglobulin G (Santa Cruz Biotechnology Inc., Santa Cruz, Calif). The signal was detected with enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ), and density was measured using scanning densitometry. Five animals per group per experiment were evaluated.
Heme c determination
Mitochondrial heme c content was calculated from the difference in spectra (dithionate/ascorbate reduced minus air-oxidized) of mitochondria (0.5 - 1 mg) solubilized in 10% lauryl maltoside using an absorption coefficient of 20.5 mM-1 cm-1 at 550 to 535 nm (22, 23). Five animals per group were evaluated.
CcOX steady-state kinetics
Cytochrome oxidase kinetics were assayed by the method of Smith, in which the rate of oxidation of ferrocytochrome c was measured by following the decrease in absorbance at 550 nm (8, 19, 20). Assays were executed in a 1-mL reaction volume containing 50 mM PO4-2 (pH 7.0), 2% lauryl maltoside, and 1 μg of mitochondrial protein. Ferrocytochrome c was added at a concentration of 40 mM to initiate the reaction. Specific activity was calculated from mean values of three to four measurements using 21.1 mM-1 cm-1 as the extinction coefficient of ferrocytochrome c at 550 nm.
Survival
We followed survival in a separate cohort of animals in this two-treatment parallel-design study to 96 h. Each animal was injected via tail vein with either 800 μg (150 μL) of reduced cytochrome c (40 mg/kg) or equal volume of saline at 24 h after CLP or sham operation. Twenty animals per group were evaluated based on the probability being 80% that the study will detect a treatment difference at a two-sided 5.0% significance level if the true hazard ratio is 3 (24).
Statistics
Data are presented as mean ± SD. Statistical significance was assessed using ANOVA and post hoc Tukey test with P < 0.05. Statistical significance in the survival study was assessed using Kaplan-Meier survival curves and Mantel-Cox log-rank test.
RESULTS
Exogenous cytochrome c increases mitochondrial levels in the septic heart
To determine the effect of injected cytochrome c on mitochondrial levels, we isolated ventricular mitochondria 48 or 72 h postprocedure and performed immunoblot analysis for steady-state levels of cytochrome c. In prior work, we demonstrated that mitochondrial cytochrome c levels decline to 60% of baseline in the septic heart 24 h post-CLP and remain persistently decreased (16). Although mitochondrial cytochrome c levels transiently decrease after sham operation, they return to basal levels by 72 h. Therefore, we assigned 72-h saline-injected sham-operated controls (Sham72sal) as surrogates for baseline values and arbitrarily set their band densities to 1.
Consistent with prior findings, steady-state levels of mitochondrial cytochrome c significantly decreased 48 h post-CLP in saline-injected mice (CLP48sal) (Fig. 1). However, after a single injection of exogenous cytochrome c at the 24-h point, mitochondrial levels increased by almost 2-fold at 48 h in septic mice (CLP48cyt) compared with CLP48sal. These levels remained significantly elevated at 72 h (CLP72cyt). Although relatively increased compared with CLP48sal, CLP72cyt steady-state levels were not statistically different from Sham72sal levels. This indicates that “supranormal” increases in mitochondrial substrate secondary to exogenous cytochrome c administration are evident for only 24 h postinjection.
FIG. 1. Exogenous cytochrome c repletes cardiac mitochondria with excess substrate up to 48 h after CLP.
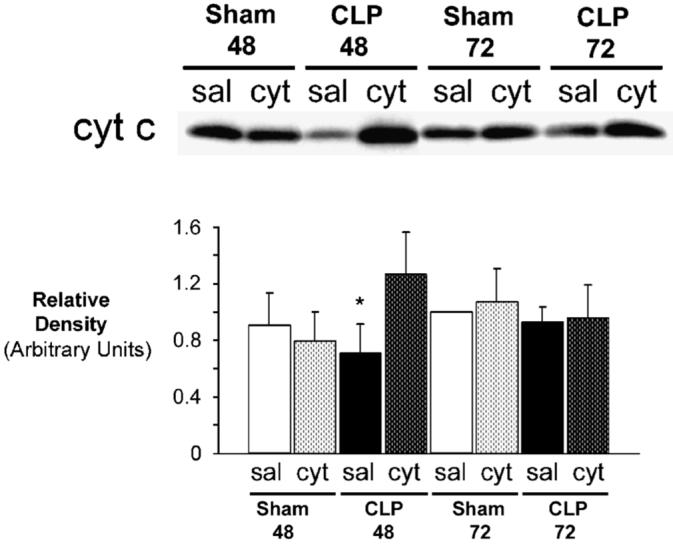
Septic mice and sham-operated controls underwent injection of cytochrome c or saline 24 h postprocedure. A, A representative Western blot of mitochondrial cytochrome c (cyt c)is shown. Sham48 and Sham72 represent sham-operated mice evaluated 48 and 72 h postprocedure, respectively. CLP48 and CLP72 represent septic mice evaluated 48 and 72 h post-CLP, respectively. Within each group, saline-injected cohorts (sal) and cytochrome c injected cohorts (cyt) are depicted. B, Graphical representation of relative densities. Values are expressed as mean T ± SD. Sham72sal values were set arbitrarily to 1 (n = 5 per group). *P < 0.05 vs. CLP48cyt, Sham72sal, Sham72cyt, CLP72sal, CLP72cyt.
There was no significant effect of exogenous cytochrome c on steady-state levels of any sham animal. Of note, cytochrome c levels in CLP72sal mice (and CLP72cyt for that matter) were essentially unchanged from Sham72sal levels. This may be a consequence of selection for survivors at this point. This is because mortality after CLP is 90% at 72 h (21). Therefore, those animals that survive to 72 h (a small minority) may do so because of adaptive responses to sepsis. Thus, an increase in CcOX substrate, cytochrome c, may represent one advantageous difference that promotes survival.
Exogenous cytochrome c restores myocardial heme c in sepsis
Cytochrome c contains a heme c center conjugated to the peptide backbone. Therefore, we measured myocardial heme c content in isolated mitochondria using spectrophotometry.
Myocardial heme c content significantly decreased 48 h after CLP and sham operation in saline-injected mice (Fig. 2). Exogenous cytochrome c injection at the 24-h point significantly increased myocardial heme c content of CLP48cyt. This increase approached Sham72sal levels. There was no significant effect of exogenous cytochrome c on the heme c content in the other groups. As with steady-state levels of cytochrome c protein, heme c content of both CLP72sal and CLP72cyt was unchanged from that of Sham72sal controls. Consistent with the immunoblot data, this suggests selection for survivors and a potentially important adaptive difference.
FIG. 2. Myocardial heme c content is restored by exogenous cytochrome c 48 h after CLP.
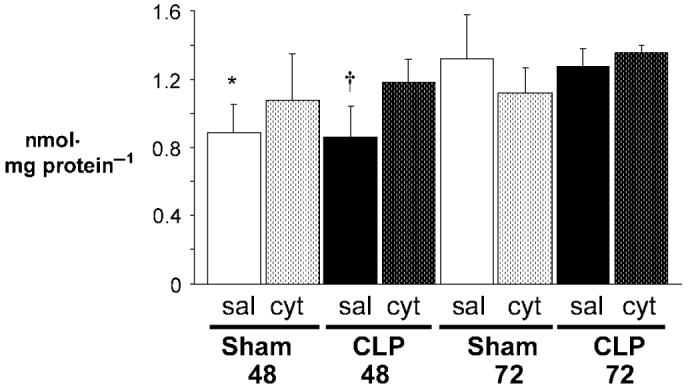
Heme c content was determined from the difference in spectra (reduced minus oxidized). Sham48 and Sham72 represent sham-operated mice evaluated 48 and 72 h postprocedure, respectively. CLP48 and CLP72 represent septic mice evaluated 48 and 72 h post-CLP, respectively. Saline-injected cohorts (sal) and cytochrome c-injected cohorts (cyt) are depicted (n = 5 per group). Values are mean ± SD. *P < 0.05 vs. Sham72sal, CLP72sal, CLP72cyt; †P < 0.05 vs. CLP48cyt, Sham72sal, CLP72sal, CLP72cyt.
Exogenous cytochrome c abrogates myocardial CcOX inhibition 48 h post-CLP
Prior work demonstrated that myocardial CcOX is competitively inhibited 24 h post-CLP and noncompetitively inhibited 48 h post-CLP (8). Furthermore, exogenous cytochrome c administered to septic mice restored mitochondria with excess cytochrome c and overcame CcOX inhibition at the 24-h point (16). It is unknown if this effect is sustained over time during the late phase of sepsis when CcOX inhibition progresses and becomes noncompetitive. Therefore, we measured CcOX activity at 48 and 72 h post-CLP after a single injection of cytochrome c at the 24-h point.
Consistent with prior findings, CLP inhibited myocardial CcOX at 48 h in CLP48sal as evidenced by significantly decreased CcOX activity (Fig. 3). Injection of exogenous cytochrome c 24 h post-CLP restored CcOX activity at 48 h to sham values and overcame the inhibition. Exogenous cytochrome c had no significant effect on CcOX activity within sham and CLP groups at the other time points. Cytochrome oxidase activity was also not significantly different between all sham cohorts. Although CcOX activity was significantly improved at 72 h in both cytochrome c-injected and salineinjected CLP mice compared with CLP48sal, enzyme velocity was significantly slower than Sham48cyt, Sham72cyt, and CLP48cyt mice. Importantly, decreased CcOX activity in both 72-h CLP groups occurred despite “baseline” levels of mitochondrial cytochrome c and heme c content. However, the increase in activity compared with C48s is consistent with the notion of selection for survivors and an adaptive response in these animals. Changes in CcOX activity in the 48- and 72-h septic mice suggests CcOX inhibition can only be abrogated by excess or “supranormal” availability of CcOX substrate, cytochrome c.
FIG. 3. Exogenous cytochrome c restores CcOX activity 48 h after CLP.
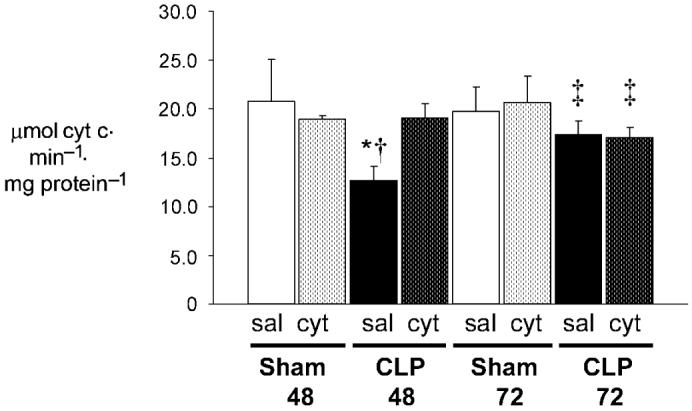
Steady-state CcOX kinetic activity was determined by measuring the oxidation of ferrocytochrome c at 550 nm. Sham48 and Sham72 represent sham-operated mice evaluated 48 and 72 h postprocedure, respectively. CLP48 and CLP72 represent septic mice evaluated 48 and 72 h post-CLP, respectively. Saline-injected cohorts (sal) and cytochrome c-injected cohorts (cyt) are depicted (n = 5 per group). Values are mean ± SD. *P < 0.01 vs. all; †P < 0.001 vs. Sham72. ‡P < 0.05 vs. Sham48cyt, CLP48cyt, Sham72cyt.
Exogenous cytochrome c improves survival in the late phase of sepsis
Changes in mitochondrial substrate levels and CcOX activity are only physiologically significant if there is an impact on survival. Therefore, we followed survival to 96 h in a separate cohort of animals after injection at 24 h of either cytochrome c or saline.
A single injection of exogenous cytochrome c significantly improved survival after CLP compared with saline injection (Fig. 4). Only 15% of animals survived to 96 h after CLP in the saline-injected group, whereas 50% survived to 96 h after CLP and cytochrome c injection. As expected, there was no mortality in either sham group.
FIG. 4. Survival rate post-CLP improves after injection of exogenous cytochrome c.
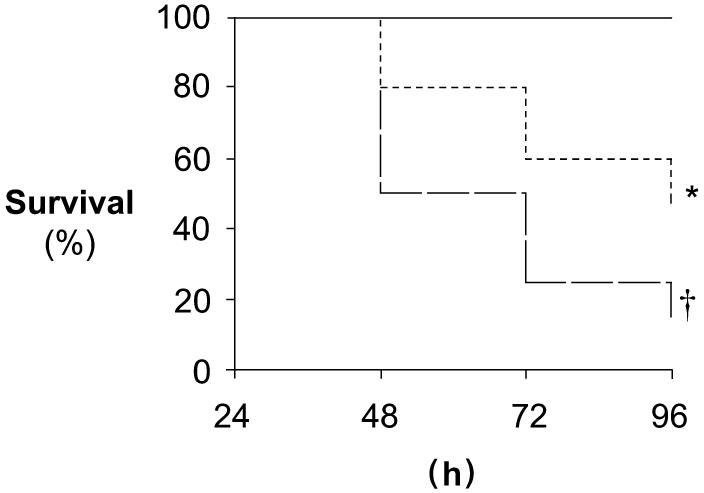
Survival to 96 h was followed in a separate cohort of animals after injection of either cytochrome c or saline post-CLP and sham operation. Kaplan-Meier survival plot is depicted. Solid line represents both saline-injected and cytochrome c-injected sham animals. Short dashed line represents cytochrome c-injected CLP animals. Long dashed line represents saline-injected CLP animals (n = 20 per group). *P < 0.05 vs. all; †P < 0.05 vs. all.
DISCUSSION
Impaired oxidative phosphorylation and bioenergetic failure have been proposed to underlie sepsis-associated organ dysfunction and may lead to death (5). Historically, this has been difficult to demonstrate because impairments in mitochondrial function have not been readily reversible. Cytochrome oxidase, the terminal oxidase of the electron transport chain, is crucial to aerobic ATP production. Myocardial CcOX is progressively inhibited in sepsis, and availability of its substrate cytochrome c becomes limited during the late phase of sepsis (8, 16). Enzyme inhibition together with substrate limitation during sepsis is a potential cause of myocardial depression and mortality during the hypodynamic phase.
In prior work, we demonstrated that injection of exogenous cytochrome c 24 h post-CLP repletes cardiac mitochondria with supranormal levels of cytochrome c (16). This increase in substrate overcomes myocardial CcOX inhibition and improves cardiac function 30 min after injection. Here, we evaluated the effect of a single injection of cytochrome c at the 24-h point on myocardial CcOX function and survival during the later time points. After cytochrome c injection, mitochondrial levels of cytochrome c remained persistently elevated 48 h post-CLP compared with saline injection and sham controls. This increase, however, was only sustained for 24 h because mitochondrial levels of cytochrome c returned to sham values by 72 h post-CLP. Consequently, only supranormal levels of mitochondrial cytochrome c completely overcame myocardial CcOX inhibition as evidenced by mildly depressed CcOX activity in both 72-h septic cohorts. This indicates ongoing CcOX inhibition during the late phase of sepsis despite baseline levels of mitochondrial cytochrome c even in selected survivors. Therefore, it is safe to conclude that the effective half-life of exogenous cytochrome c is less than 48 h when injected at 24 h post-CLP. The survival data also support this conclusion.
Although overall survival post-CLP improved after injection of cytochrome c, the greatest effect was on survival to the 48-h point. Taken together, these data indicate that increasing the frequency of injection of exogenous cytochrome c to every 24 h may be more beneficial in sepsis to maintain elevated mitochondrial substrate levels and overcome ongoing CcOX inhibition during the later time points.
The 72-h CLP cohorts deserve some special mention here. Prior evaluation of myocardial CcOX has consistently demonstrated a progressive deterioration in steady-state levels of mRNA and protein of its key subunits and reduced activity in the late phase of sepsis (8). In this study, we found maintained steady-state levels and content of mitochondrial cytochrome c and heme c in both septic groups at 72 h compared with sham controls. Furthermore, although CcOX was mildly inhibited in these animals compared with sham controls, CcOx activity was significantly increased compared with CLP48sal. The combination of relatively increased cytochrome c levels, heme c content, and CcOX activity 72 h post-CLP compared with CLP48sal mice supports the notion of selection for survivors. Furthermore, finding increased cytochrome c and heme c in the setting of ongoing CcOX inhibition at 72 h represent a potentially adaptive response to sepsis and support our hypothesis and findings that increased substrate availability can overcome CcOX inhibition and promote survival.
Limitations
Although we have previously evaluated cardiac function immediately after cytochrome c injection, we did not measure myocardial function as a part of this study. In previous work, we demonstrated that exogenous cytochrome c improves cardiac contractility and relaxation 30 min after injection 24 h post-CLP (16). However, although injection of cytochrome c enhanced survival into the later time points of sepsis, we cannot assume that cardiac function is restored in the late phase as well. In addition, we cannot assume that survival benefits after cytochrome c injection during sepsis are solely due to primary cardiac effects. It is possible that exogenous cytochrome c has systemic effects and improves extracardiac organ function and may impart a beneficial secondary effect on cardiac function during sepsis. Such possibilities require exploration.
Although exogenous cytochrome c gained access to cardiomyocyte mitochondria, and abrogated sepsis induced CcOx inhibition, we have not evaluated how exogenous cytochrome c traverses the plasma membrane and gets imported into mitochondria. It is apparent from this and prior studies that exogenous cytochrome c readily gains access to cardiomyocyte mitochondria during sepsis but, to a lesser degree, in sham-operated and healthy controls. This may be due to increased membrane permeability during sepsis. Membrane integrity, an ATP-dependent process, is known to be impaired in sepsis (25-28). Although this is one possibility, a host of other mechanisms may exist, and clearly, these need to be explored.
Another important consideration is apoptosis. Cytochrome c release from mitochondria is known to initiate the intrinsic apoptosis pathway (29). We have not evaluated if exogenous cytochrome c causes apoptosis. Thus, the effect of exogenous cytochrome c on the mitochondrial apoptosis pathway needs to be evaluated.
Finally, exogenous cytochrome c may have an effect on reactive oxygen species (ROS). Stimulated electron transport may result in ROS production (30). Alternatively, cytochrome c may scavenge ROS. Both effects can impact on cell and organ function. Therefore, ROS levels and production will need to be assessed in sepsis with regard to exogenous cytochrome c.
CONCLUSIONS
Mitochondrial dysfunction may be a cause of sepsis-associated myocardial depression. Cytochrome oxidase inhibition coupled with decreased cytochrome c availability is one such defect that can impair oxidative phosphorylation in sepsis. Overcoming CcOX inhibition with excess substrate is one strategy to restore mitochondrial function in the septic heart. Twenty four hours post-CLP, at the onset of the late, hypo-dynamic phase of sepsis, CcOX is competitively inhibited, and substrate begins to decrease. Exogenous cytochrome c injected at the 24-h point readily gains access to cardiomyocyte mitochondria, repletes mitochondria with supranormal levels of substrate, and overcomes CcOX inhibition during sepsis. Here, we demonstrate that after the 24-h injection of cyto-chrome c, supranormal levels of substrate persist until 48 h post-CLP and overcome sepsis-induced CcOX inhibition. Although mitochondrial levels of cytochrome c return to sham levels by 72 h post-CLP, CcOX activity is relatively improved. Importantly, a single injection of exogenous cyto-chrome c improves survival in the late phase of sepsis.
Acknowledgments
This study was supported by National Institutes of Health/National Institutes of General Medical Science (grant nos. 1K08GM074117 to R.J.L. and 5R01GM059930 to C.S.D.) and a Maria Fareri Children's Hospital Foundation Grant (to R.J.L.).
Footnotes
The authors have no financial interests to disclose.
REFERENCES
- 1.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep. 2003;5:365–371. doi: 10.1007/s11908-003-0015-9. [DOI] [PubMed] [Google Scholar]
- 4.Singer M, Brealey D. Mitochondrial dysfunction in sepsis. Biochem Soc Symp. 1999;66:149–166. doi: 10.1042/bss0660149. [DOI] [PubMed] [Google Scholar]
- 5.Fink MP. Bench-to-bedside review: cytopathic hypoxia. Critical Care. 2002;6:491–499. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callahan LA, Supinski GS. Sepsis induces diaphragm electron transport chain dysfunction and protein depletion. Am J Respir Crit Care Med. 2005;172:861–868. doi: 10.1164/rccm.200410-1344OC. [DOI] [PubMed] [Google Scholar]
- 7.Chen HW, Hsu C, Lu TS, Wang SJ, Yang RC. Heat shock pretreatment prevents cardiac mitochondrial dysfunction during sepsis. Shock. 2003;20:274–279. doi: 10.1097/00024382-200309000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Levy RJ, Vijayasarathy C, Raj NR, Avadhani NG, Deutschman CS. Competitive and noncompetitive inhibition of myocardial cytochrome c oxidase in sepsis. Shock. 2004;21:110–114. doi: 10.1097/01.shk.0000108400.56565.ab. [DOI] [PubMed] [Google Scholar]
- 9.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286:R491–R497. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 10.Trumbeckaite S, Opalka JR, Neuhof C, Zierz S, Gellerich FN. Different sensitivity of rabbit heart and skeletal muscle to endotoxin-induced impairment of mitochondrial function. Eur J Biochem. 2001;268:1422–1429. doi: 10.1046/j.1432-1327.2001.02012.x. [DOI] [PubMed] [Google Scholar]
- 11.Gellerich FN, Trumbeckaite S, Opalka JR, Gellerich JF, Chen Y, Neuhof C, Redl H, Werdan K, Zierz S. Mitochondrial dysfunction in sepsis: evidence from bacteraemic baboons and endotoxaemic rabbits. Biosci Rep. 2002;22:99–113. doi: 10.1023/a:1016017224003. [DOI] [PubMed] [Google Scholar]
- 12.Gellerich FN, Trumbeckaite S, Hertel K, Zierz S, Muller-Werdan U, Werdan K, Redl H, Schlag G. Impaired energy metabolism in hearts of septic baboons: diminished activities of complex I and complex II of the mitochondrial respiratory chain. Shock. 1999;11:336–341. [PubMed] [Google Scholar]
- 13.Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med. 2002;30:276–284. doi: 10.1097/00003246-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Fink MP. Cytopathic hypoxia. Is oxygen use impaired in sepsis as a result of an acquired intrinsic derangement in cellular respiration? Crit Care Clin. 2002;18:165–175. doi: 10.1016/s0749-0704(03)00071-x. [DOI] [PubMed] [Google Scholar]
- 15.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 16.Piel DA, Gruber PJ, Weinheimer CJ, Courtois MR, Robertson CM, Coopersmith CM, Deutschman CS, Levy RJ. Mitochondrial resuscitation with exogenous cytochrome c in the septic heart. Crit Care Med. 2007;35:2120–2127. doi: 10.1097/01.ccm.0000278914.85340.fe. [DOI] [PubMed] [Google Scholar]
- 17.Streyer L. Introduction to Enzymes. W.H. Freeman and Company; New York: 1988. pp. 177–200. [Google Scholar]
- 18.Wharton DC, Tzagoloff A. Cytochrome Oxidase From Beef Heart Mitochondria. Vol X. Academic Press; New York: 1967. pp. 245–250. [Google Scholar]
- 19.Piel DA, Khan AR, Waibel R, Birbach M, Spray TL, Deutschman CS, Gaynor JW, Levy RJ. Chronic hypoxemia increases myocardial cytochrome oxidase. J Thorac Cardiovasc Surg. 2005;130:1101–1106. doi: 10.1016/j.jtcvs.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 20.Vijayasarathy C, Biunno I, Lenka N, Yang M, Basu A, Hall IP, Avadhani NG. Variations in the subunit content and catalytic activity of the cytochrome c oxidase complex from different tissues and different cardiac compartments. Biochim Biophys Acta. 1998;1371:71–82. doi: 10.1016/s0005-2736(97)00278-2. [DOI] [PubMed] [Google Scholar]
- 21.Weiss YG, Bouwman A, Gehan B, Schears G, Raj N, Deutschman CS. Cecal ligation and double puncture impairs heat shock protein 70 (HSP-70) expression in the lungs of rats. Shock. 2000;13:19–23. doi: 10.1097/00024382-200013010-00004. [DOI] [PubMed] [Google Scholar]
- 22.Ozawa T, Tanaka M, Shimomura Y. Crystallization of the middle part of the mitochondrial electron transfer chain: Cytochrome bc1-cytochrome c complex. Proc Natl Acad Sci U S A. 1980;77:5084–5086. doi: 10.1073/pnas.77.9.5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka M, Ogawa N, Ihara K, Sugiyama Y, Mukohata Y. Cytochrome aa(3) in Haloferax volcanii. J Bacteriol. 2002;184:840–845. doi: 10.1128/JB.184.3.840-845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richardson DB. Power calculations for survival analyses via Monte Carlo estimation. Am J Ind Med. 2003;44:532–539. doi: 10.1002/ajim.10310. [DOI] [PubMed] [Google Scholar]
- 25.Sawada N, Murata M, Kikuchi K, Osanai M, Tobioka H, Kojima T, Chiba H. Tight junctions and human diseases. Med Electron Microsc. 2003;36:147–156. doi: 10.1007/s00795-003-0219-y. [DOI] [PubMed] [Google Scholar]
- 26.Ye J, Tsukamoto T, Sun A, Nigam SK. A role for intracellular calcium in tight junction reassembly after ATP depletion-repletion. Am J Physiol. 1999;277:F524–F532. doi: 10.1152/ajprenal.1999.277.4.F524. [DOI] [PubMed] [Google Scholar]
- 27.Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit Care Clin. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 28.McConnell KW, Coopersmith CM. Epithelial cells. Crit Care Med. 2005;33:S520–S522. doi: 10.1097/01.ccm.0000187004.09189.1b. [DOI] [PubMed] [Google Scholar]
- 29.Walford GA, Moussignac RL, Scribner AW, Loscalzo J, Leopold JA. Hypoxia potentiates nitric oxide-mediated apoptosis in endothelial cells via peroxynitrite-induced activation of mitochondria-dependent and -independent pathways. J Biol Chem. 2004;279:4425–4432. doi: 10.1074/jbc.M310582200. [DOI] [PubMed] [Google Scholar]
- 30.Nohl H, Gille L, Staniek K. Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol. 2005;69:719–723. doi: 10.1016/j.bcp.2004.12.002. [DOI] [PubMed] [Google Scholar]