

Abstract
Non-phenolic oxidative coupling: The first total synthesis of the C-2 symmetric indole alkaloid P-(+)-dispegatrine (1) is reported. A late-stage thallium(III)acetate mediated intermolecular oxidative coupling was employed to construct the C(9)-C(9′) bond with complete regio- and stereocontrol. The exclusive formation of a single atropodiastereomer 12 in this critical step arises due to internal asymmetric induction, as planned. In addition, the first total synthesis of four other monomeric sarpagine indole alkaloids is described.
Keywords: atropodiastereomer, biaryls, indole alkaloids, oxidative coupling, thallium
The Sarpagine-Macroline group of indole alkaloids consists of more than 150 members and is an important class of natural products with diverse biological activity.[1] A biogenetic link between the Sarpagine alkaloids and the Ajmaline-related alkaloids has been confirmed by Stöckigt et al.[2] Common to these three classes of alkaloids, is the core cycloocta[b]-indole framework I (Figure 1A), which has recently been a subject of biology-oriented synthesis (BIOS) by Waldmann et al.,[3] the analogues of which are promising targets for developing a novel class of potent and selective Mycobacterium protein tyrosine phosphatase B (MptpB) inhibitors against Mycobacterium tuberculosis.[4] The ‘S’ configuration at C(6) and C(10) and the β-ketoester moiety were key to the inhibitory activity.
Figure 1.

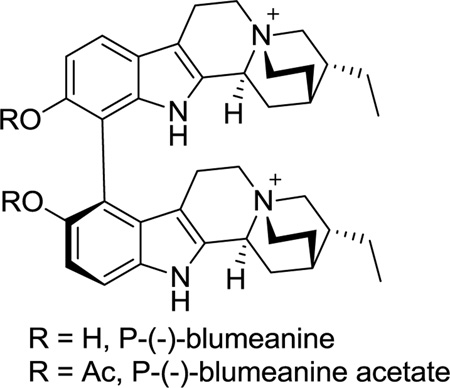
A) The cycloocta[b]-indole core structure with promising activity against M. tuberculosis.[4] B) Ring-A oxygenated sarpagine indole alkaloids 1–6
Bisphenolic, bisquaternary indole alkaloids are very rare.[5] Of the 300 or so dimeric indole alkaloids isolated to date, the sarpagine alkaloid dispegatrine 1[6] (Figure 1B) and blumeanine[7] are the only two dimeric indoles belonging to this class. (+)-Dispegatrine (1) and the monomer (+)-spegatrine (2) were isolated from the roots of Rauwolfia verticillata (Lour.) Baill var. hainanensis Tsiang by Yu et al.[6] Both 1 and 2 are known to exhibit antihypertensive activity with the affinity of the dimer 1 on both the α1- and α2-adrenoreceptors about an order of magnitude greater than that of the monomer 2.[8] While the structure of dispegatrine (1) was assigned during isolation, the apparent axial chirality at the C(9)-C(9′) biaryl axis was not determined. Although the authors attempted a semisynthesis of 1 via an oxidative phenolic coupling of 2 (Scheme 1A), the yield of the process was very low (0.25%).[6] A number of synthetically related monomeric alkaloids such as (+)-lochneram (3), (+)-10-methoxyvellosimine (4), (+)-lochnerine (5) and (+)-sarpagine (6) have also been reported (Figure 1B).[1c] The complex architecture combined with the promising bioactivity of 1,[8] 2,[8] and 5[9] rendered them very attractive targets. Herein is described the first total synthesis of the sarpagine alkaloids 1, 2 and 4–6 in a stereospecific manner.
Scheme 1.

A) Semisynthesis of 1[6]. B) Doubly Convergent Retrosynthetic Analysis of 1.
Direct biaryl bond formation can be classified into two types: (a) the reductive metal-catalyzed coupling reaction (e.g Ullmann, Suzuki, Stille, etc.) and (b) the oxidative (phenolic/non-phenolic) coupling reaction.[10] Although the regioselectivity of the reductive coupling processes is predetermined by the use of activated coupling partners (which may have to be synthesized separately), this could mean additional steps to the synthetic route. The direct oxidative (phenolic/non-phenolic) coupling of non-activated substrates on the other hand, is the most powerful and economical method for the synthesis of biaryls.[10,11] The lack of preactivation, however, could affect the regioselectivity of the process, especially in the intermolecular mode. For example, the presence of more than one reactive site in the phenolic substrate could lead to a mixture of products (via ortho- and para- coupling) and subsequent overoxidation to quinones either from the coupled product or the original substrate could lower the yield of the process. Atropselectivity in both cases is most commonly achieved by employing chiral catalysts[10–12] or in a few cases by taking advantage of the existing chiral centers in the substrate[13] and/or by double diastereodifferentiation [14]
The formation of the biaryl bond in a large number of bisphenolic biaryl natural products is assumed and in some cases proven to be a result of an oxidative phenolic coupling.[15] The existence of 1 as a single atropodiastereomer both in nature and during semisynthesis, suggests the simultaneous formation of the biaryl bond with internal asymmetric induction by the sarpagine framework of the monomer 2. We based the strategy on these reports. The retrosynthetic analysis of 1 is illustrated in Scheme 1B. A potentially biomimetic intermolecular biaryl coupling could be employed to construct the C(9)-C(9′) bond in 1. Since oxidative phenolic coupling of 2,[6] yielded only a trace amount of 1, a non-phenolic Scholl-type oxidative coupling[16] of the sarpagine alkaloid (+)-lochnerine (5) could be attempted to this end. We have been interested in the total synthesis of sarpagine indole alkaloids and a general route for the synthesis of related alkaloids via the asymmetric Pictet-Spengler reaction has been developed. Our strategy toward the synthesis of the monomers 2–6 and in turn 1 relied on this approach.[1a]
Analogous to the previous work of Zhao et al., in the Na-Me series,[17] the 5-methoxy-Na-H-D(+)-tryptophan ethyl ester 7 was converted to the key tetracyclic core 8 in six steps via the asymmetric Pictet-Spengler approach. Attempts to effect the key enolate mediated palladium catalyzed cyclization of 8 under the modified conditions [Pd2(dba)3/DPEphos/t-BuONa][18] furnished the pentacyclic ketone 9 in lower yields (50%), accompanied by acetylene byproduct (not shown). This problem was circumvented by subjecting the vinyl iodide 8 to the much milder conditions of Bonjoch et al.[19] which furnished the pentacyclic ketone 9 in 73% yield (Scheme 2). Wittig reaction of 9, followed by a hydrolysisepimerization sequence provided the more stable α-aldehyde in (+)-10-methoxyvellosimine (4). Reduction of the aldehyde 4 with NaBH4/EtOH furnished (+)-lochnerine (5) in 90% yield. Demethylation of 5 with BBr3/CH2Cl2 furnished the phenolic monomer (+)-sarpagine (6) in 80% yield. Subsequent quaternization of the Nb-nitrogen function in 6 at room temperature with excess methyl iodide provided the Nb-methiodide salt, which on stirring with silver chloride in ethanol[20] furnished (+)-spegatrine chloride (2). The spectral data of synthetic 2 and 4–6 were in excellent agreement to that reported for the natural products and resulted in the first total synthesis of the alkaloids.[1c]
Scheme 2.

General approach to the total synthesis of the sarpagine indole alkaloids 2, 4–6. THF = tetrahydrofuran.
Intermolecular non-phenolic oxidative dimerizations of complex aryl substrates are very rare.[21] A majority of the documented examples constitute simpler aromatic systems or substrates with no competing reactive sites.[10d, 22] In order to test the feasibility of such an intermolecular oxidative coupling on the electron-rich indole alkaloid (+)-lochnerine (5) [with two possible reactive sites, C(9) and C(11)], it was decided to first carry out a model reaction on a more robust substrate. The Na-methyl beta-carboline 10, synthesized earlier[17] was employed for this purpose. In the absence of a chiral catalyst, such a model reaction could also provide an insight into the possible substrate-dependent atropselectivity of the process.
Of the various combinations documented,[10d] the hypervalent iodine(III) mediated coupling conditions developed by Kita et al.[22b, 23] and the thallium(III) mediated conditions of Taylor et al.[22c, 24] seemed best suited to the substrates under study here. As illustrated in Scheme 3, β-carboline 10 when subjected to iodine(III) or thallium(III) mediated oxidative coupling afforded a mixture of the atropodiastereomers 11a and 11b with complete regioselectivity. In general, the hypervalent iodine mediated oxidations [phenyliodine(III)bis(trifluoroacetate) (PIFA) or phenyliodine diacetate (PIDA) at −78 °C to 0 °C) generated a number of highly colored baseline impurities. At best a combined yield of 30% (11a : 11b = 4 : 1) was obtained with a combination of PIFA (0.8 equiv), BF3·Et2O (3 equiv) at −40 °C. However, with the thallium(III) salt [Tl(OCOCF3)3 or Tl(OCOCH3)3] as the oxidant, the reaction was much cleaner with very little baseline material. Upon optimization, the milder thallium(III) acetate (0.7 equiv), BF3·Et2O (3 equiv) at −40 °C provided a combined yield of 67% (b.r.s.m.), in favor of 11b. Upon chromatographic separation, the lower Rf atropodiastereomer 11b was recrystallized from EtOH. X-ray analysis determined the axial chirality of 11b as P(S).[25] Based on the results of the model reaction, oxidative dehydrodimerization of 5 was next attempted.
Scheme 3.

Oxidative non-phenolic coupling of beta-carboline 10. Recovered 10 = 14%. b.r.s.m. = based on recovered starting material.
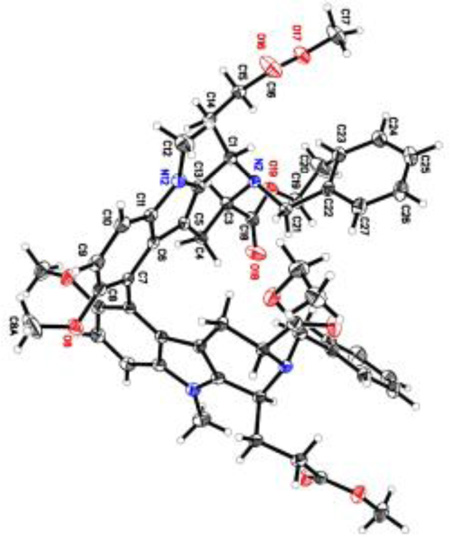
As illustrated in Scheme 4, a combination of thallium(III)acetate (0.65 equiv) and BF3·Et2O (3.0 equiv) in acetonitrile at −40 °C afforded the key C(9)-C(9′) biaryl 12 as the sole atropodiastereomer in 60% yield (b.r.s.m) with complete regioselectivity in agreememt with the retrosynthetic analysis. Formation of a small amount of the aryl-thallium byproduct A (8%) at C(9), is in accordance with the mechanistic studies by Kochi et al.[26] The free indole Na-H and the primary hydroxyl function at C(17) remained unaffected. Attempts to achieve complete conversion of the starting material 5 by increasing the equivalents of Tl(OCOCH3)3 led to increased formation of the arylthallium adduct (A) and baseline impurities. Column chromatographic purification of the dimer 12, followed by recrystallization from methanol provided white crystals for X-ray analysis, which established the axial configuration as P(S).[27] As anticipated, the rigid sarpagine framework and the existing stereogenic centers in 5 exerted complete atropselection in the key biaryl coupling step, thereby forming a single atropodiastereomer 12. This would be in agreement with a potential biomimetic coupling in the plant since the other atropodiastereomer of 1 was not reported there or observed by us.
Scheme 4.

Completion of the total synthesis of the P-atropodiastereomer of (+)-dispegatrine (1). Recovered 5 = 14%, A = arylthallium adduct, C24H29N2O6Tl (8%), b.r.s.m = based on recovered starting material.
With the axial chirality established in the key intermediate 12, the total synthesis of dispegatrine (1) was completed in two steps as illustrated in Scheme 4. The C(10)-C(10′) methoxy groups were demethylated with 11 equiv of BBr3/CH2Cl2 at −78 °C to furnish the bisphenol. Nb-quaternization of the highly polar bisphenol required heating of the reaction mixture in a sealed tube at 40 °C with a large excess of MeI to provide the methiodide salt. Further treatment with AgCl/MeOH at room temperature completed the total synthesis of P-dispegatrine (1). Spectral analysis (1H NMR and HRMS) of the synthetic (+)-dispegatrine (1) were in good agreement with the literature values,[6] except the proton chemical shifts for H-5,5′. In order to obtain better spectroscopic data, the bismethyl ether 13 was synthesized by carrying out the the Nb-quaternization of 12 with MeI/MeOH at rt or 40 °C. Analogous to blumeanine (isolated as its diacetate),[7] chromatographic purification and isolation of the bisquaternary salt 13 proved to be much easier in comparison to (+)- dispegatrine (1). The 2D NMR correlation experiments (see Supporting Information) on the dimer 13 established the position of the H-3,3′ and H-5,5′ protons. In the absence of an authentic sample[28] [for circular dichroism (CD) analysis or thin layer chromatography (TLC) comparison], it is impossible to unequivocally report that synthetic 1 is identical to the natural product even though the 1H NMR is in good agreement.[29] However the fact that the biomimetic coupling by Yu et al.[6] gave only the natural isomer and our oxidative coupling gave the P-atropodiastereomer from similar scaffolds strongly suggests that they presumably are the same.
In summary, a general approach to the ring-A oxgenated Na-H sarpagine indole alkaloids has led to the first asymmetric total synthesis of the dimeric indole alkaloid P-(+)-dispegatrine (1) as well as the monomers 2, 4–6. The synthesis is notable especially for execution of the direct oxidative dimerization of (+)-lochnerine (5), in the presence of the free indole Na-H, highly basic Nb-nitrogen atom and the C(17) hydroxyl group. To the best of our knowledge, this is the first report of a direct intermolecular oxidative nonphenolic coupling of highly functionalized and sensitive substrates (5 and 10) and has expanded the scope of the thallium(III)-mediated oxidative coupling reaction in heteroaromatic biaryl synthesis. Additionally an indirect alternative to the less predictable phenolic coupling, especially in complex aryl substrates is presented. Further work to understand the mechanism of the key biaryl coupling reaction is in progress and will be reported in due course.
Supplementary Material
Footnotes
We wish to acknowledge the NIMH (in part) and the Lynde and Harry Bradley Foundation for support of this work. X-ray crystallographic studies were carried out at the Naval Research Laboratory and supported by NIDA-NRL Interagency Agreement Number Y1-DA1101.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Chitra R. Edwankar, Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, 3210, N. Cramer Street, Milwaukee, WI-53201 (USA)
Rahul V. Edwankar, Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, 3210, N. Cramer Street, Milwaukee, WI-53201 (USA)
Jeffrey R. Deschamps, Center for Biomolecular Science and Engineering, Naval Research Laboratory, Code 6930, Washington, D. C. 20375 (USA)
James M. Cook, Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, 3210, N. Cramer Street, Milwaukee, WI-53201 (USA).
References
- 1.For reviews on Sarpagine-Macroline indole alkaloids see: Edwankar CR, Edwankar RV, Rallapalli SK, Cook JM. Nat. Prod. Commun. 2008;3:1839–1870. Lewis SE. Tetrahedron. 2006;62:8655–8681. Lounasmaa M, Hanhinen P, Westersund M. In: The Alkaloids. Cordell GA, editor. Vol.52. San Diego, CA: Academic Press; 1999. pp. 103–195. and references therein.
- 2.Ruppert M, Ma X, Stöckigt J. Curr. Org. Chem. 2005;9:1431–1444. [Google Scholar]
- 3.For a review on biology-oriented synthesis (BIOS) see: Wetzel S, Bon RS, Kumar K, Waldmann H. Angew. Chem. Int. Ed. 2011;50:10800–10826. doi: 10.1002/anie.201007004.
- 4.a) Nören-Müller A, Reis-Corrêa JI, Prinz H, Rosenbaum C, Saxena K, Schwalbe H, Vestweber D, Schunk S, Schwarz O, Schiewe H, Waldmann H. Proc. Natl. Acad. Sci. USA. 2006;103:10606–10611. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nören-Müller A, Wilk W, Saxena K, Schwalbe H, Kaiser M, Waldmann H. Angew. Chem. Int. Ed. 2008;47:5973–5977. doi: 10.1002/anie.200801566. [DOI] [PubMed] [Google Scholar]
- 5.a) Cordell GA, Saxton JE. In: The Alkaloids. Manske RHF, Rodrigo RGA, editors. Vol.20. New York: Academic Press; 1981. pp. 3–295. [Google Scholar]; b) Kam T-S, Choo Y-M. In: The Alkaloids. Cordell GA, editor. Vol.63. San Diego: Academic Press; 2006. pp. 182–345. [Google Scholar]
- 6.Lin M, Yang B, Yu D-Q. Acta Pharm. Sinica. 1986;21:114–118. [PubMed] [Google Scholar]
-
7.For isolation of (−)-blumeanine see:
Arbain D, Firmansyah, D D, Sargent MV, Skelton BW, White AH. J. Chem. Soc., Perkin Trans. 1. 1998:2537–2540.
- 8.Feng Y, Gao H, Zeng G. Acta Pharm. Sinica. 1986;21:1–6. [PubMed] [Google Scholar]
- 9.a) Kazumasa Z, Ikumi K, Nobuhide I, Takahiro H, Yusuke H, Toshio K, Safinar II, Lajis NH, Morita Hiroshi M. J. Nat. Med. 2012 PubMed ID:22350216. [Google Scholar]; b) Svoboda GH, Blake DA. In: The Catharanthus Alkaloids. Taylor WI, Farnsworth NR, editors. New York: Marcel Dekker; 1974. p. 45. and references therein. [Google Scholar]
- 10.For reviews on the synthesis of biaryl natural poducts see Bringmann G, Gulder T, Gulder TAM, Breuning M. Chem. Rev. 2011;111:563–639. doi: 10.1021/cr100155e. Bringmann G, Price Mortimer AJ, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem. Int. Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661. Wallace TM. Org. Biomol. Chem. 2006;4:3197–3210. doi: 10.1039/b608470m. Cepanec I. Synthesis of Biaryls. 1st ed. San Diego: Elsevier; 2004.
- 11.a) Ashenhurst JA. Chem. Soc. Rev. 2010;39:540–548. doi: 10.1039/b907809f. [DOI] [PubMed] [Google Scholar]; b) Kozlowski MC, Morgan BJ, Linton EC. Chem. Soc. Rev. 2009;38:3193–3207. doi: 10.1039/b821092f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Cammidge AN, Crépy KVL. Tetrahedron. 2004;60:4377–4386. [Google Scholar]; b) Yin J, Buchwald SL. J. Am. Chem. Soc. 2000;122:12051–12052. [Google Scholar]
- 13.Huang S, Petersen TB, Lipshutz BH. J. Am. Chem. Soc. 2010;132:14021–14023. doi: 10.1021/ja1065202. [DOI] [PubMed] [Google Scholar]
- 14.a) Park YS, Grove CI, Gonzalez-Lopez M, Urgaonkar S, Fettinger JC, Shaw JT. Angew. Chem. Int. Ed. 2011;50:3730–3733. doi: 10.1002/anie.201007298. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bringmann G, Hamm A, Schraut M. Org. Lett. 2003;5:2805–2808. doi: 10.1021/ol0347693. [DOI] [PubMed] [Google Scholar]
- 15.Keserú GM, Nógrádi M. In: Studies in Natural Products Chemistry. Atta-ur-Rahman, editor. Vol.20. Amsterdam: Elsevier; 1998. pp. 263–322. [Google Scholar]
- 16.a) Kovacic P, Jones MB. Chem. Rev. 1987;87:357–379. [Google Scholar]; b) Scholl R, Seer C. Justus Liebigs Ann. Chem. 1912;394:111–177. [Google Scholar]; c) Scholl R, Mansfeld J. Ber. Dtsch. Chem. Ges. 1910;43:1734–1746. [Google Scholar]
- 17.Zhao S, Liao X, Wang T, Flippen-Anderson J, Cook JM. J. Org. Chem. 2003;68:6279–6295. doi: 10.1021/jo030055u. [DOI] [PubMed] [Google Scholar]
- 18.Liao X, Zhou H, Yu J, Cook JM. J. Org. Chem. 2006;71:8884–8890. doi: 10.1021/jo061652u. [DOI] [PubMed] [Google Scholar]
- 19.Solé, D D, Urbaneja X, Bonjoch J. Adv. Synth. Catal. 2004;346:1646–1650. [Google Scholar]
- 20.Yamazaki N, Dokoshi W, Kibayashi C. Org. Lett. 2001;3:193–196. doi: 10.1021/ol0003228. [DOI] [PubMed] [Google Scholar]
- 21.Bringmann G, Tasler S. Tetrahedron. 2001;57:331–343. [Google Scholar]
- 22.a) Keller PA, Yepuri NR, Kelso MJ, Mariani M, Skelton BW, White AH. Tetrahedron. 2008;64:7787–7795. [Google Scholar]; b) Tohma H, Morioka H, Takizawa S, Arisawa M, Kita Y. Tetrahedron. 2001;57:345–352. [Google Scholar]; c) McKillop A, Turell AG, Young DW, Taylor EC. J. Am. Chem. Soc. 1980;102:6504–6512. [Google Scholar]
- 23.a) Dohi T, Ito M, Yamaoka N, Morimoto K, Fujioka H, Kita Y. Tetrahedron. 2009;65:10797–10815. [Google Scholar]; b) Takada T, Arisawa M, Gyoten M, Hamada R, Tohma H, Kita Y. J. Org. Chem. 1998;63:7698–7706. [Google Scholar]
- 24.McKillop A, Taylor EC. Recent Advances in Organothallium Chemistry. In: Stone FGA, West R, editors. Advances in Organometallic Chemistry. Vol.11. New York: Academic Press; 1973. pp. 147–206. [Google Scholar]
-
25.ORTEP view of the crystal structure of 11b. CCDC 893770 (11b) contains the supplementeray crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
- 26.Lau W, Kochi JK. J. Am. Chem. Soc. 1984;106:7100–7112. [Google Scholar]
- 27.CCDC 893771 (12) contains the supplementeray crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
- 28.A request was made for an authentic sample of (+)-dispegatrine
- 29.For examples of homo- and hetero-atropodiastereomeric natural products that have almost identical 1H NMR and 13C NMR spectra see: Hirasawa Y, Hara M, Nugroho AE, Sugai M, Zaima K, Kawahara N, Goda Y, Awang K, Hadi AHA, Litaudon M, H J. Org. Chem. 2010;75:4218–4223. doi: 10.1021/jo1006762. Tatsuta K, Yamazaki T, Mase T, Yoshimoto T. Tetrahedron Lett. 1998;39:1771–1772. Fukuyama Y, Asakawa Y. J. Chem. Soc., Perkin Trans. 1. 1991:2737–2741.


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.