

Abstract
Heparan sulfate (HS) glucosaminyl 3-O-sulfotranferases sulfate the C3-hydroxyl group of certain glucosamine residues on heparan sulfate. Six different 3-OST isoforms exist, each of which can sulfate very distinct glucosamine residues within the HS chain. Among these isoforms, 3-OST1 has been shown to play a role in generating ATIII-binding HS anticoagulants whereas 3-OST2, 3-OST3, 3-OST4 and 3OST-6 have been shown to play a vital role in generating gD-binding HS chains that permit the entry of herpes simplex virus type 1 into cells. 3-OST5 has been found to generate both ATIII- and gD-binding HS motifs. Previous studies have examined the substrate specificities of all the 3-OST isoforms using HS polysaccharides. However, very few studies have examined the contribution of the epimer configuration of neighboring uronic acid residues next to the target site to 3-OST action. In this study, we utilized a well-defined synthetic oligosaccharide library to examine the substrate specificity of 3-OST3a and compared it to 3-OST1. We found that both 3-OST1 and 3-OST3a preferentially sulfate the 6-O-sulfated, N-sulfoglucosamine when an adjacent iduronyl residue is located to its reducing side. On the other hand, 2-O-sulfation of this uronyl residue can inhibit the action of 3-OST3a on the target residue. The results reveal novel substrate sites for the enzyme actions of 3-OST3a. It is also evident that both these enzymes have promiscuous and overlapping actions that are differentially regulated by iduronyl 2-O-sulfation.
Introduction
Heparan sulfate (HS) is a highly sulfated polysaccharide that is located on cell surfaces as well as in the extracellular matrix (ECM). The nascent HS chain consists of repeating units of N-acetyl glucosamine (GlcNAc) and glucuronic acid (GlcA). This backbone subsequently undergoes a series of modifications by various HS modifying enzymes located in the Golgi. GlcNAc residues can be N-deacetylated and N-sulfated by N-deacetylase-N-sulfotransferase (NDST). GlcA can be epimerized to iduronic acid (IdoA) by C5-epimerase. Additionally, a number of O-sulfotransferases (OST) can add sulfate groups to the C6 (by 6-OST) and C3 (by 3-OST) carbons of glucosamine (GlcN) residues and to the C2 carbon (by 2-OST) of IdoA or GlcA residues.1 These modifications create the enormous diversity that confers a wide array of biological functions. Among these modifications, 3-O-sulfation has been shown to play a vital role in creating HS chains that function as anticoagulants and as entry receptors for herpes simplex virus type 1 (HSV-1).2–5 It has also been shown that 3-O-sulfation has been associated with both cancer and embryonic development.6–9 Six different isoforms of 3-OST have been identified [3-OST1, 2, 3 (3a and 3b splice variants), 4, 5, and 6] and shown to generate distinct HS structures. 10–18 3-OST3a has a broad expression profile and is present in many different tissues including heart, placenta, lungs, liver and kidneys.11 One of its pathological functions is to create binding sites for viral gD glycoprotein and initiate the entry of HSV-1 into cells.5 3-OST1 is expressed in heart, brain, lungs and kidneys.11 3-OST1 has been shown to play a vital role in generating ATIII-binding HS anticoagulants.19 However, it has been also shown that mice deficient in 3-OST1 still have normal hemostasis.20,21 Therefore, it is important to carefully examine the contribution of other 3-OST isoforms to this crucial function of HS.
Though the substrate specificity of 3-OST1 has been rigorously studied, the substrate specificity of other isoforms including 3-OST3a has not been examined extensively. The Rosenberg group has shown that 3-OST3a preferentially acts on GlcNH2, GlcNH26S and GlcNS residues that have an IdoA2S residue at their non-reducing side.10,15,22 Furthermore, earlier studies reported the crystal structures of 3-OST 1, 3, 5, and identified two amino acid residues that are responsible for the specific activity of 3-OST3a on the IdoA2S-GlcNH2/NS ± 6S.23 However, it is still unknown whether the epimer configuration of the neighboring uronic acid residue, i.e. GlcA or IdoA, located at the reducing side of the target site, influences the action of 3-OST3a. This structural information could not be obtained previously because commonly used fragmentation approaches such as heparitinase digestion or nitrous acid treatment cleaved the chains at the reducing side of the target GlcN residue. In this study, we utilized a library of chemically synthesized, well-defined oligosaccharides to study the effect of precursor structures, in particular the influence of epimer configuration of adjacent uronyl residues, on the action of 3-OST3a. We found that 3-OST3a preferentially sulfates the GlcNS6S residue when an IdoA residue is located to its reducing side. On the other hand, 2-O-sulfation of this IdoA inhibits the action of 3-OST3a on the target residue.
Results
It has previously been shown that 3-OST3a can sulfate GlcNH2, GlcNH26S and GlcNS that are located to the reducing side of IdoA2S.10,15 However, all previous substrate specificity studies relied on the analysis of 3-O-sulfotransferase modified heparan sulfate polysaccharides that were fragmented using nitrous acid or heparitinases. The heterogeneity of heparan sulfate makes it difficult to thoroughly analyze all the modified products. Moreover, enzymatic digestion leads to a loss of epimer configuration and chemically-induced chain scission occasionally causes isomerisation, thus complicating the study of the effect of neighboring uronic acid residues located next to the target glucosamine residues. In this study, we utilized a library of well-defined tetra- and hexa-saccharides to study the effect of precursor structures on the enzymatic action of 3-OST3a (Table 1). We also compared the enzymatic action of 3-OST3a to that of 3-OST1. This library of oligosaccharides was previously synthesized and extensively studied by the Boons group.24 By utilizing HPLC coupled to a radiometry detector and capillary LC coupled to electrospray ionization-time-of-flight MS (ESI-TOF-LC-MS), we could detect and distinguish all possible modified products.
Table 1.
Structures of oligosaccharides examined as potential substrates for 3-OST3a and 3-OST1. Potential 3-OST substrate sites are marked with asterisks
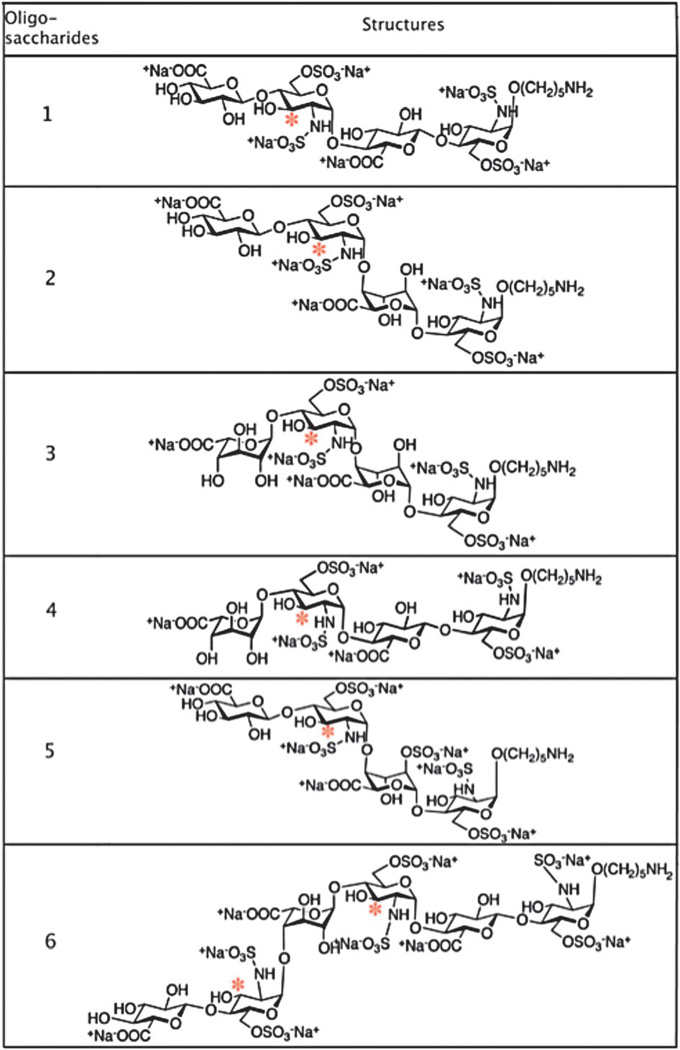 |
Six different oligosaccharides containing GlcA or IdoA at different locations were chosen for this study (Table 1). The first four oligosaccharides are tetrasaccharides containing either a GlcA or IdoA adjacent to the expected modification site (marked with an asterisk, Table 1). The sequences of these tetrasaccharides are GlcA/IdoA-GlcNS6S-GlcA/IdoA-GlcNS6S. Oligosaccharide 5 contains a 2-O-sulfated iduronic acid residue in its sequence: GlcA-GlcNS6S-IdoA2S-GlcNS6S. Oligosaccharide 6 is a hexasaccharide with the following sequence: GlcA-GlcNS6SIdoA-GlcNS6S-GlcA-GlcNS6S.
These six oligosaccharides were modified with either 3-OST3a or 3-OST1 in the presence of [35S]PAPS as the sulfate donor. The 3-O-sulfated products could then be detected due to the addition of radiolabeled sulfate ([35S]SO42−). HPLC analysis showed that only oligosaccharides 2, 3 and 6 were radiolabeled by 3-OST3a (Fig. 1 and Fig. S1A, ESI†). Thus, it is clear that 3-OST3a can only act on three substrates: GlcA-GlcNS6S-IdoA-GlcNS6S (2), IdoA-GlcNS6S-IdoA-GlcNS6S24 and GlcA-GlcNS6S-IdoA-GlcNS6S-GlcA-GlcNS6S (6). On the other hand, 3-OST1 modification was detected on oligosaccharides 1, 2, 3, 5, and 6 (Fig. 2 and Fig. S1B, ESI†). Thus, oligosaccharides 2, 3 and 6 were modified by both isoforms whereas oligosaccharides 1 and 5 were exclusively modified by 3-OST1. Our finding that oligosaccharide 5 is the substrate for 3-OST1 but not for 3-OST3a is consistent with earlier findings that 3-OST1 modifies GlcNS6S residues that are flanked by GlcA at their non-reducing sides and IdoA2S residues at their reducing side.19,25 Furthermore, it is interesting to note that the reducing end GlcNS6S residue in oligosaccharide 5 is not modified by 3-OST3a even though it is located adjacent to IdoA2S. It has previously been observed that 3-OST1 modification preferentially occurs on the internal GlcNS residues whereas reducing end terminal GlcNS fails to undergo modification.25 Liu et al. have shown that 3-OST1 can act on HS structures that lack IdoA residues.26 This is consistent with our observation that oligosaccharide 1, which lacks IdoA, is a substrate for 3-OST1. On the other hand, oligosaccharide 3 is also a substrate for 3-OST1, albeit with much less [35S] incorporation (Fig. S1, ESI†). This suggests that 3-OST1 prefers GlcNS6S residues that contain GlcA at the non-reducing side. It has been reported in several studies that 3OST1 and 3OST3a act on distinct sites within HS chains. Surprisingly, earlier studies have not found that 3-OST1 and 3-OST3 can act on the same oligosaccharide—as in the case of oligosaccharides 2, 3, and 6. Based on these interesting observations, we next focused our efforts to locate the 3-OST3a modification site within these oligosaccharides. Oligosaccharides 2 and 3 have one potential modification site whereas oligosaccharide 6 has two potential modification sites. However, the radiolabeled results could not pinpoint which GlcNS6S residues on the oligosaccharides were modified upon 3-O-sulfation by 3OST3a.
Fig. 1.

Radiochromatograms of oligosaccharide products generated by modification with 3-OST3a and [35S]PAPS. Radiochromatograms indicated that oligosaccharides 2, 3 and 6 are substrates for 3-OST3a whereas other oligosaccharides are not substrates.
Fig. 2.

Radiochromatograms of oligosaccharide products generated by modification of oligosaccharides 1, 2, 3, 5, and 6 by 3-OST1 and [35S]PAPS.
To determine which residue was modified, the oligosaccharides were treated with 3-OST3a again in the presence of non-radioactive [32S]PAPS. The modified oligosaccharide products were then digested with heparitinase I, II and III to disaccharides and analyzed by LC-MS. Heparitinases catalyze the eliminative cleavage of heparin and heparan sulfate at the α(1,4)-glycosidic linkage between the glucosamine and the uronic acid.27,28 Since all oligosaccharides were terminated with the aminopentyl linker, cleavage with heparitinase created specific disaccharides species: a disaccharide containing no unsaturated bond, a disaccharide containing 4,5-unsaturated uronic acid (DUA), and a disaccharide containing both the aminopentyl linker and the 4,5-unsaturated uronic acid. For clarification, the potential modifications of oligosaccharide 2 and its disaccharides upon heparitinase digestion are presented in Fig. S2 (ESI†). Due to the mass differences among these cleavage products, we could distinguish all possible modified disaccharides from unmodified disaccharides and were able to pinpoint the 3-O-sulfation site (Table 2). MS data of 3-O-sulfated disaccharides revealed the precise location of 3-O-sulfation sites within the oligosaccharides 2, 3 and 6 (Fig. 3). MS spectra of unmodified disaccharides as well as intact 3-O-sulfated oligosaccharide products are shown in Fig. S3 and S4 (ESI†). In the case of oligosaccharides 2 and 3, one 3-O-sulfated disaccharide with m/z = 593.96 was found, indicating that 3-OST3a sulfated the penultimate GlcNS6S residue from the non-reducing end of the oligosaccharide (Fig. 3). For oligosaccharide 6, two 3-O-sulfated disaccharides with m/z = 593.96 and 575.97 were found, indicating that 3-OST3a sulfated both the penultimate residue from the non-reducing end and the internal GlcNS6S residue. Since the only difference between these two disaccharides is the hydroxyl group at the C4 position of the uronic acid residue, we expect that the ionization to be similar or close to similar. The fact that the relative intensity of the peak with m/z = 593.96 is far higher than that of the peak with m/z = 575.97 suggests that the penultimate GlcNS6S residue is preferred over the internal GlcNS6S residue by 3-OST3a (Fig. 3). However, one may have to use isotope enriched disaccharides as internal standards to estimate quantitatively differential preference. It is interesting to note that the penultimate GlcNS6S residue has IdoA at its reducing side whereas the less preferred internal GlcNS6S residue, the third residue from the aminopentyl linker, has GlcA at its reducing side.
Table 2.
Potential 3-O-sulfated disaccharides resulting from 3-OST3a modified oligosaccharides with their expected mass
| Oligosaccharide | Structure | Possible modified disaccharide product | Exact mass |
|---|---|---|---|
| 2 | GlcA-GlcNS6S-IdoA-GlcNS6S-(CH2)5NH2 | GlcA-GlcNS6S3S | 594.98 |
| ΔUA-GlcNS6S3S-(CH2)5NH2 | 662.06 | ||
| 3 | IdoA-GlcNS6S-IdoA-GlcNS6S-(CH2)5NH2 | IdoA-GlcNS6S3S | 594.98 |
| ΔUA-GlcNS6S3S-(CH2)5NH2 | 662.06 | ||
| 6 | GlcA-GlcNS6S-IdoA-GlcNS6S-GlcA-GlcNS6S-(CH2)5NH2 | GlcA-GlcNS6S3S | 594.98 |
| ΔUA-GlcNS6S3S | 576.97 | ||
| ΔUA-GlcNS6S3S-(CH2)5NH2 | 662.06 |
Fig. 3.

MS spectra of 3-O-sulfated disaccharide products. The expected molecular weights of possible disaccharide products are presented in Table 2.
Current work demonstrates that 3-OST3a preferentially sulfates GlcA/IdoA-GlcNS6S-IdoA (Fig. 4) and that the presence of an IdoA residue at the reducing side of the target GlcNS6S promotes 3-OST3a activity. 3-OST3a could sulfate the sequence, GlcA-GlcNS6S-IdoA, in the oligosaccharide 2, but not the sequence, GlcA-GlcNS6S-IdoA2S, in the oligosaccharide 5, which suggests that 2-O-sulfation of the IdoA, at the reducing side of the target site, is inhibitory for 3-OST3a. It is also interesting to note that 3-OST3a could sulfate IdoA-GlcNS6S-GlcA (6), albeit less preferentially, only if the target site was located downstream of a GlcA-GlcNS6S-IdoA sequence. It is possible that there could be a conformational change, after 3-OST3a sulfates the first glucosamine residue, in the oligosaccharide structure that may allow the enzyme to slide along the oligosaccharide sequence and continue to sulfate the next glucosamine residue on the reducing side in the oligosaccharide 6.
Fig. 4.

List of oligosaccharides tested as substrates for 3-OST3a and their modified products.
Discussion
The substrate specificities of the 3OST isoforms have been extensively studied before. However, in these studies, heparitinase treatment resulted in the loss of stereochemical information of the proximal uronyl residues next to the target residue. Furthermore, the employment of a nitrous acid degradation technique in earlier studies resulted in the removal of the uronyl residue at the reducing side of the target residue. Therefore, the influence of the epimer configuration of adjacent uronyl residues could not be elucidated until now. In addition, the heterogeneous nature of HS precursors used in those earlier studies exacerbated the difficulties involved in elucidating the influence of epimer configuration. Therefore, it is essential to use defined oligosaccharides to elucidate the substrate specificity of the 3-OST isoforms in order to lay a foundation for uncovering their physiological functions. In the present study, we systematically investigated the substrate specificity of 3-OST3a and compared its specificity to that of 3-OST1 using well-defined oligosaccharide precursors that had different epimer configurations.
In this study, we expressed and purified 3-OST3a and 3-OST1 recombinant enzymes using a baculovirus system. We then radiolabeled a library of synthetic oligosaccharides with these enzymes in the presence of [35S]PAPS. The reaction mixture was then analyzed using HPLC. It was found that oligosaccharide structures with the following sequence were preferentially modified by 3-OST3a: GlcA/IdoA-GlcNS6SIdoA and less preferentially IdoA-GlcNS6S-GlcA. It was also found that 3-OST3a failed to modify the oligosaccharides that consisted of the following sequences: GlcA-GlcNS6S-IdoA2S and GlcA-GlcNS6S-GlcA. These sequence requirements have not been reported previously.
After the identification of novel sequence requirements for 3-OST3a, we focused our efforts to compare the differential substrate preferences of 3-OST3a and 3-OST1. It is surprising to note that both 3-OST1 and 3-OST3a modified oligosaccharides 2, 3 and 6. Thus, both of these isoforms preferred an iduronyl residue at the reducing side of the target residue. Furthermore, oligosaccharide 4, carrying IdoA-GlcNS6S-GlcA, was not a substrate for either of these isoforms. This suggests that the presence of GlcA at the reducing side of the target residue is unfavorable for 3-OST enzyme action. Nevertheless, oligosaccharide 1 was moderately modified by 3-OST1 but not by 3-OST3a. This suggests that there are subtle differences in the substrate specificity between these enzyme isoforms. It is also interesting that both oligosaccharides 2 and 5 were substrates for 3-OST1 whereas only the oligosaccharide 2 was the substrate for 3-OST3a. The only difference between these two oligosaccharides is the presence of the 2-O-sulfate group on the IdoA residue, located at the reducing side of the target residue. Additionally, even though oligosaccharide 4, carrying IdoA-GlcNS6S-GlcA sequence, was not at all modified by 3-OST3a, the same sequence in oligosaccharide 6 was modified, albeit less efficiently. Based on the results presented here, we propose that both 3-OST1 and 3-OST3a have a significant overlap in their substrate specificity and that their activity is differentially regulated by 2-O-sulfation (Fig. 5).
Fig. 5.

Schematic model of the action of 3-OST1 and 3-OST3a at the target sites (*) representing the influence of neighbouring epimer configuration and 2-O-sulfate group.
In summary, the action of 3-OST1 and 3-OST3a on the HS substrates depends on the epimer configuration and the 2-O-sulfation. It is important to note that even though polymers are the substrates for HS biosynthetic enzymes in vivo, polymers cannot be helpful in elucidating the influence of adjacent uronic acid configuration. Therefore, oligomeric substrates are utilized instead in this study to be able to pinpoint the influence of epimer configuration on the action of 3-OST enzymes. Future studies will focus on examining the substrate specificities of other 3OST isoforms and studying the influence of 6-O-sulfation with the aid of additional synthetic HS oligosaccharides.
Materials and methods
Materials
Oligosaccharides that were used in the current study were synthesized and characterized in an earlier report.24 Heparitinase (Hep) I, II, and III were cloned and expressed in E. coli BL21. The DEAE-sepharose gel was purchased from Amersham Biosciences. The analytical strong-anion exchange column and capillary reverse phase C18 column were purchased from various commercial vendors. [35S]Na2SO4 and Ultima-FloAP scintillation fluid were purchased from Perkin Elmer Life and Analytical Sciences. [35S]PAPS was prepared as reported earlier.29 [32S]PAPS was purchased from Sigma-Aldrich. HS disaccharide standards were purchased from Iduron and Sigma-Aldrich. Insect cell Sf-900 SFM medium was purchased from Invitrogen. Dibutylamine was used as an ion-pairing agent in LC-MS analysis.30 All other reagents and solvents were from Sigma-Aldrich unless otherwise stated.
Expression of 3-OST3a
The 3-OST3a recombinant enzyme was expressed using a baculovirus system. 20 ml of 3-OST3a viral stock was added to 1 × 109 Sf9 cells in 1 L Sf-900 SFM medium. Infected cells were shaken at 90 rpm in a humidified shaker-incubator maintained at 28 1C for 4 days. The cell suspension was then centrifuged at 1000 × g for 30 min to pellet cells. PIPES and phenylmethylsulfonyl fluoride were added to the supernatant to a final concentration of 10 mM and 1 mM respectively. The supernatant was then adjusted to pH 7.0, chilled on ice for 30 min, and subsequently centrifuged at 4000 × g for 30 min. The solution was diluted twice with water, filtered and loaded onto a 100-ml column of ToyoPearl AF-heparin 650M. The column was washed with 600 ml of PCG-50 (10 mM PIPES, pH 7.0, 1% glycerol, 0.2% CHAPS, 50 mM NaCl) and eluted with a 450 ml linear gradient of 50–1000 mM NaCl in PCG. The elution of the protein was monitored by measuring its absorbance at 280 nm. The protein containing fractions were pooled and concentrated using an Amicon YM-10 filter. 3-OST1 recombinant enzyme was also expressed and purified in a similar manner for comparative studies.
Enzymatic modification of synthetic oligosaccharide substrates
All reactions were performed in a buffer consisting of 25 mM MES (pH 7.0), 0.02% Triton X-100, 2.5 mM MgCl2, 2.5 mM MnCl2, 1.25 mM CaCl2 and 0.75 mg ml−1 BSA. 5 mg of an oligosaccharide was incubated with 10 ml 3-OST3a or 3-OST1 (B20 mg ml−1) in the presence of [35S]PAPS (0.5 × 107 CPM) or [32S]PAPS (10 mg) in a 50 ml reaction. The radioactive samples were then analyzed using analytical SAX-HPLC coupled with an in-line radiometry detector. The oligosaccharides were eluted with a linear gradient of 0 to 1 M NaCl in 70 mM phosphate buffer (pH 3.0) for 60 min. HS disaccharide standards were co-injected and detected at 232 nm. The non-radioactive samples were digested with Hep I, II and III, and analyzed using liquid chromatography–mass spectrometry (LC-MS). Disaccharides were separated on a capillary C18 column (0.3 × 250 mm) using a linear gradient of acetonitrile at a flow rate of 5 ml min−1 over 70 min. 5 mM dibutylamine was used as an ion-pairing agent. C18-HPLC was coupled to an electrospray ionization time-of-flight MS (Bruker Daltonics, USA) and analyzed in the negative ion mode under the following conditions: cone gas 50 l h−1, nozzle temperature 130 1C, drying gas (N2) flow 450 l h−1, spray tip potential 2.3 kV, and nozzle potential 35 V.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants (GM075168 and HL107152), Mizutani Foundation for Glycoscience Award, and American Heart Association National Scientist Development Award (0830360N) to BK, TN also acknowledges support from a graduate fellowship from the Vietnam Education Foundation.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05221g
Notes and references
- 1.Habuchi H, Habuchi O, Kimata K. Glycoconjugate J. 2004;21:47–52. doi: 10.1023/B:GLYC.0000043747.87325.5e. [DOI] [PubMed] [Google Scholar]
- 2.Lam LH, Silbert JE, Rosenberg RD. Biochem. Biophys. Res. Commun. 1976;69:570–577. doi: 10.1016/0006-291x(76)90558-1. [DOI] [PubMed] [Google Scholar]
- 3.Hook M, Bjork I, Hopwood J, Lindahl U. FEBS Lett. 1976;66:90–93. doi: 10.1016/0014-5793(76)80592-3. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl U, Backstrom G, Thunberg L, Leder IG. Proc. Natl. Acad. Sci. U. S. A. 1980;77:6551–6555. doi: 10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 6.Cadwallader AB, Yost HJ. Dev. Dyn. 2006;235:3423–3431. doi: 10.1002/dvdy.20991. [DOI] [PubMed] [Google Scholar]
- 7.Kamimura K, Rhodes JM, Ueda R, McNeely M, Shukla D, Kimata K, Spear PG, Shworak NW, Nakato H. J. Cell Biol. 2004;166:1069–1079. doi: 10.1083/jcb.200403077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bui C, Ouzzine M, Talhaoui I, Sharp S, Prydz K, Coughtrie MW, Fournel-Gigleux S. FASEB J. 2010;24:436–450. doi: 10.1096/fj.09-136291. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto K, Asada K, Fukutomi T, Okochi E, Yagi Y, Hasegawa T, Asahara T, Sugimura T, Ushijima T. Oncogene. 2003;22:274–280. doi: 10.1038/sj.onc.1206146. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Shriver Z, Blaiklock P, Yoshida K, Sasisekharan R, Rosenberg RD. J. Biol. Chem. 1999;274:38155–38162. doi: 10.1074/jbc.274.53.38155. [DOI] [PubMed] [Google Scholar]
- 11.Shworak NW, Liu J, Petros LM, Zhang L, Kobayashi M, Copeland NG, Jenkins NA, Rosenberg RD. J. Biol. Chem. 1999;274:5170–5184. doi: 10.1074/jbc.274.8.5170. [DOI] [PubMed] [Google Scholar]
- 12.Moon AF, Edavettal SC, Krahn JM, Munoz EM, Negishi M, Linhardt RJ, Liu J, Pedersen LC. J. Biol. Chem. 2004;279:45185–45193. doi: 10.1074/jbc.M405013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Liu J. Biochim. Biophys. Acta. 2005;1725:190–200. doi: 10.1016/j.bbagen.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Razi N, Lindahl U. J. Biol. Chem. 1995;270:11267–11275. doi: 10.1074/jbc.270.19.11267. [DOI] [PubMed] [Google Scholar]
- 15.Liu J, Shworak NW, Sinay P, Schwartz JJ, Zhang L, Fritze LM, Rosenberg RD. J. Biol. Chem. 1999;274:5185–5192. doi: 10.1074/jbc.274.8.5185. [DOI] [PubMed] [Google Scholar]
- 16.Lawrence R, Yabe T, Hajmohammadi S, Rhodes J, McNeely M, Liu J, Lamperti ED, Toselli PA, Lech M, Spear PG, Rosenberg RD, Shworak NW. Matrix Biol. 2007;26:442–455. doi: 10.1016/j.matbio.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence R, Kuberan B, Lech M, Beeler DL, Rosenberg RD. Glycobiology. 2004;14:467–479. doi: 10.1093/glycob/cwh057. [DOI] [PubMed] [Google Scholar]
- 18.Mochizuki H, Yoshida K, Gotoh M, Sugioka S, Kikuchi N, Kwon YD, Tawada A, Maeyama K, Inaba N, Hiruma T, Kimata K, Narimatsu H. J. Biol. Chem. 2003;278:26780–26787. doi: 10.1074/jbc.M301861200. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Shworak NW, Fritze LM, Edelberg JM, Rosenberg RD. J. Biol. Chem. 1996;271:27072–27082. doi: 10.1074/jbc.271.43.27072. [DOI] [PubMed] [Google Scholar]
- 20.Shworak NW, HajMohammadi S, de Agostini AI, Rosenberg RD. Glycoconjugate J. 2002;19:355–361. doi: 10.1023/A:1025377206600. [DOI] [PubMed] [Google Scholar]
- 21.HajMohammadi S, Enjyoji K, Princivalle M, Christi P, Lech M, Beeler D, Rayburn H, Schwartz JJ, Barzegar S, de Agostini AI, Post MJ, Rosenberg RD, Shworak NW. J. Clin. Invest. 2003;111:989–999. doi: 10.1172/JCI15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu ZL, Lech M, Beeler DL, Rosenberg RD. J. Biol. Chem. 2004;279:1861–1866. doi: 10.1074/jbc.M311398200. [DOI] [PubMed] [Google Scholar]
- 23.Xu D, Moon AF, Song D, Pedersen LC, Liu J. Nat. Chem. Biol. 2008;4:200–202. doi: 10.1038/nchembio.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arungundram S, Al-Mafraji K, Asong J, Leach FE, 3rd, Amster IJ, Venot A, Turnbull JE, Boons GJ. J. Am. Chem. Soc. 2009;131:17394–17405. doi: 10.1021/ja907358k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuberan B, Lech MZ, Beeler DL, Wu ZL, Rosenberg RD. Nat. Biotechnol. 2003;21:1343–1346. doi: 10.1038/nbt885. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Jones CL, Liu J. Chem. Biol. 2007;14:986–993. doi: 10.1016/j.chembiol.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korn ED, Payza AN. J. Biol. Chem. 1956;223:859–864. [PubMed] [Google Scholar]
- 28.Linker A, Hovingh P. J. Biol. Chem. 1965;240:3724–3728. [PubMed] [Google Scholar]
- 29.Lansdon EB, Fisher AJ, Segel IH. Biochemistry. 2004;43:4356–4365. doi: 10.1021/bi049827m. [DOI] [PubMed] [Google Scholar]
- 30.Kuberan B, Lech M, Zhang L, Wu ZL, Beeler DL, Rosenberg RD. J. Am. Chem. Soc. 2002;124:8707–8718. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.