

Summary
In growing tissues, cell fitness disparities provoke interactions that promote stronger cells at the expense of the weaker in a process called cell competition. The mechanistic definition of cell fitness remains unclear, as does how differences are recognized. In Drosophila cells with extra Myc activity acquire “super-competitor” status upon confrontation with wild-type (WT) cells, prompting the latters’ elimination via apoptosis. Confrontation enhances Myc cell fitness by increasing glycolytic flux and promoting expansion of the population. p53 is induced in these cells and promotes their enhanced metabolism. Whereas p53 loss in noncompeting Myc cells is inconsequential, it impairs metabolism, reduces viability and prevents the killing activity of Myc super-competitor cells. We propose that p53 acts as a general sensor of competitive confrontation to enhance the fitness of “winner” cells. Our findings suggest that the initial confrontation between pre-cancerous and WT cells could enhance cancer cell fitness and promote tumor progression.
Introduction
In growing epithelia, information about growth, metabolic status or genetic identity is shared locally among cells to establish themselves as relatively weaker or stronger. The sensing of differences in fitness results in competition for tissue occupancy and improves the proliferation potential of the more robust “winner” cells at the expense of the relatively less robust “loser” cells. This conserved homeostatic process, called cell competition, facilitates the health of growing tissues and aids in tissue size regulation (reviewed in (Baker, 2011; Johnston, 2009). The best characterized examples of cell competition occur between wild-type (WT) Drosophila cells and cells mutant for one of a number of ribosomal proteins (collectively called Minute, or M mutants), or between WT cells and cells expressing higher or lower amounts of Drosophila Myc (hereafter called Myc), the sole homolog of the c-Myc transcriptional regulator and oncoprotein. Indeed, primordial wing cells that differ less than 2-fold in Myc expression compete vigorously for occupancy of the adult wing (de la Cova et al., 2004; Johnston et al., 1999; Moreno and Basler, 2004). Evidence indicates that intercellular signaling mediates competitive behavior. Winner cells transmit a killing signal to loser cells, which die by apoptosis, and loser cell participation promotes expansion of the winner cells (de la Cova et al., 2004; Rhiner et al., 2010; Senoo-Matsuda and Johnston, 2007). Cell competition is thought to be an evolutionarily conserved mechanism of ensuring optimal organ fitness, via recognition and elimination of cells deemed dangerous to the animal (Johnston, 2013). Recent reports suggest that a Myc-based cell fitness surveillance system operates at early mouse embryonic stages to optimize development (Claveria et al., 2013; Sancho et al., 2013).
How cell fitness is mechanistically defined and how fitness differences are recognized remain unclear. Studies have identified genes expressed in loser cells (de la Cova et al., 2004; Portela et al., 2010; Rhiner et al., 2010), but what defines winner cells has received little attention. Broadly, cell fitness is its capacity to reproduce and populate a tissue. However, cell competition relies on relative differences in cell fitness, making winner fitness difficult to define: WT cells are winners when growing next to M/+ cells (Morata and Ripoll, 1975) or cells mutant for diminutive (dm), encoding Drosophila Myc (Johnston et al., 1999; Wu and Johnston, 2010), or c-Myc (Claveria et al., 2013), but are losers when next to cells with more Myc (Claveria et al., 2013; de la Cova et al., 2004; Moreno and Basler, 2004; Sancho et al., 2013), more Yki, the transducer of the Hippo tumor suppressor pathway (Neto-Silva et al., 2010; Tyler et al.; Ziosi et al.), or more Wnt/Wingless (Vincent et al., 2011) or JAK/STAT activity (Rodrigues et al., 2012); or with less p53 activity (Bondar and Medzhitov, 2010; Dejosez et al., 2013; Marusyk et al., 2010). Cell fitness is thus under constant surveillance in growing tissues and mechanisms exist to recognize disparities when they arise.
In Drosophila tissues ectopic Myc expression drives cellular growth but developmental constraints prevent acceleration of cell division, thus tissue mass is promoted by increasing cell size, not cell number (Johnston et al., 1999). In cell culture, however, it stimulates both growth and division, leading to a faster proliferation rate (Senoo-Matsuda and Johnston, 2007). In mosaic wing imaginal discs or in mixed cell populations in culture, interactions between WT and Myc-expressing cells cause Myc cells to acquire “super-competitor” behavior that increases their reproductive fitness and enables them to overtake the tissue by killing off their WT neighbors. This behavior is analogous to cancer and suggests that cancer cells and super-competitor cells may use similar mechanisms to surpass normal controls on tissue growth (Baker and Li, 2008; Johnston, 2013; Moreno, 2008). Many of c-Myc’s target genes regulate glucose metabolism and its increased expression promotes aerobic glycolysis, known as the Warburg effect (Dang, 1999). This metabolic switch is common in tumor cells and may facilitate their rapid expansion (Assaily and Benchimol, 2006; Young and Anderson, 2008), and in combination with its potent growth-promoting properties makes Myc activity powerfully oncogenic. The discovery of Myc’s super-competitor capacity makes its role in tumor promotion potentially even more destructive.
We undertook these studies to probe the molecular basis of the fitness of Myc super-competitor cells. Using a combination of in vivo and cell culture approaches, we confirm that simple increases in Myc expression reprogram metabolism in Drosophila, and demonstrate that the tumor suppressor p53 is induced adaptively to regulate mitochondrial respiration. Strikingly, in competitive mosaics, p53 promotes considerable enhancement of metabolic flux in Myc cells in a distinct role. Here, p53 endows and protects super-competitor behavior of Myc cells: in its absence, Myc super-competitors no longer kill WT cells, cannot expand as a population, have increased genomic instability, and undergo catastrophic induction of apoptosis. Myc-expressing cells only require the protective role of p53 when in the presence of WT neighbor cells. We propose that p53 detects confrontation between Myc and WT cells and mediates a genetic program that elevates Myc cells to super-competitor status. Our experiments demonstrate the determinative nature of cellular context and suggest a mechanism by which confrontation between emerging cancer cells and nearby WT cells may promote mammalian tumor formation by further boosting cell metabolism and fitness.
Results
Myc expression stimulates glycolysis but impairs oxidative phosphorylation
Virtually nothing is known about the basis of the fitness advantage of Myc super-competitor cells during cell competition, but Myc proteins have well-known roles in regulation of ribosome biogenesis and cellular metabolism. Stimulation of glucose uptake and conversion of pyruvate to lactate instead of acetyl Co-A for Krebs cycle utilization (Figure 1A) is a hallmark of cancer cells and thought to promote increased proliferation (Hanahan and Weinberg, 2011), thus we asked if metabolic changes underlay Myc super-competitive behavior. We found that in the absence of competition, a simple increase in Myc expression stimulates glycolysis by examining glucose metabolism in mono-cultures of Drosophila S2 cells stably transfected with a Cu-inducible Myc transgene (hereafter called Myc cells). While Cu induction on its own had little effect on metabolism (Supp. Figure 1), induction of Myc led to significant increases in expression of genes encoding glycolytic enzymes, including phosphoglucose isomerase (Pgi), phosphofructokinase (Pfk), triose phosphate isomerase (Tpi), the glucose transporter glut1, and impl3, encoding lactate dehydrogenase (Ldh) (Figure 1B). Glucose consumption was also increased in Cu-induced Myc cells, but not in uninduced Myc cells (not shown) or control Cu-induced S2 (WT) cells (Figure 1D), that was accompanied by an increase in intracellular lactate (Figure 1E). Stimulation of glycolytic gene expression and flux by Myc is thus a conserved regulatory process. We also noted up-regulation of genes encoding proteins functioning in mitochondria to promote oxidative respiration in Myc-expressing cells, including synthesis of cytochrome C oxidase (Scox), required for assembly of cytochrome c oxidase (COX, complex IV) (Yang et al., 2010), succinate dehydrogenase (Sdh), a component of the electron transport chain (ETC; complex II), and pyruvate dehydrogenase (Pdh), which catalyzes decarboxylation of pyruvate and generates acetyl CoA for the Krebs cycle (Figure 1C). Despite increased expression of these genes, steady-state ATP in Myc cells was 30% lower than in WT cells (Figure 1F). Myc expression thus reconfigures cellular metabolism so that oxidative phosphorylation is displaced as the predominant source of energy production by increased glycolysis and lactic acid fermentation, resembling the Warburg effect observed in cancer cells (Dang, 2010).
Figure 1. Myc induces glycolysis and reduces oxidative phosphorylation.
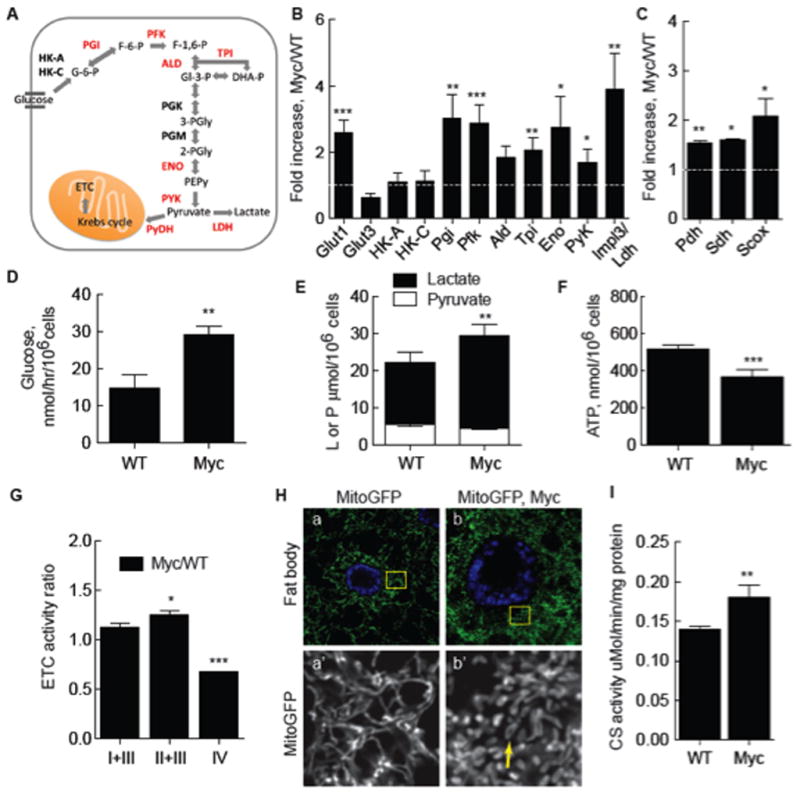
A. Schematic of glycolysis. Enzymes catalyzing reactions in glucose breakdown are bolded, those induced by Myc expression are red. See text for abbreviations.
B. Quantitative (q) RT-PCR of glycolytic gene expression in Myc cells from monocultures. mRNA expression is normalized to actin5C mRNA and shown as fold increase in Myc cells relative to WT cells. Significance is compared to expression in WT cells. All error bars in this figure are SD.
C. qRT-PCR of genes required for mitochondrial respiration, as described in B.
D. Glucose consumption in WT or Myc cell mono-cultures.
E. Intracellular lactate and pyruvate in mono-cultures of WT or Myc cells.
F. Steady-state ATP in WT or Myc cell mono-cultures.
G. ETC complex activity, normalized to CS activity, in Myc cells expressed as a ratio over activity in WT cells.
H. Mito-GFP-labeling of mitochondria in larval fat body cells shows mitochondrial morphology. a) WT cells. Area within each yellow square is magnified in single channel images below. b) WT Myc-expressing cells. Mitochondrial number and morphology are altered, with more donut shapes (arrow).
I. Citrate synthase (CS) activity, an index of mitochondrial mass, as a function of total cellular protein in WT and Myc cells from mono-cultures.
The decrease in ATP in Myc-expressing cells suggested a defect in oxidative phosphorylation. We explored this possibility first using a mitochondrial-localized GFP (Mito-GFP) to examine mitochondria in WT and Myc-expressing cells in vivo, in wing discs and in larval fat bodies. Fat body cells are large and flat, facilitating mitochondrial visualization. Both WT wing disc and fat body cells have long, filamentous, perinuclear networks of mitochondria (Figure 1H, a; Supp. Figure 2A, a). Expression of Myc in either cell type increased mitochondrial number compared to WT cells, consistent with Myc’s regulation of mitochondrial biogenesis (Figure 1H, b; Supp. Figure 2A, b)(Li et al., 2005). This was confirmed by an increase in citrate synthase (CS) activity, a measure of mitochondrial mass, in Myc cells in culture (Figure 1I). Compared to WT cells, Myc-expressing wing disc or fat body cells had few filamentous mitochondria but many small donut shapes and globules (Figure 1H, b; Supp. Figure 2A, b). Mitochondrial morphology is controlled by metabolism (Rossignol et al., 2004; Youle and van der Bliek, 2012), thus this remodeling is likely due to altered metabolism in Myc cells.
To evaluate mitochondrial respiration in WT and Myc cells we quantified electron transport chain (ETC) function by measuring the activities of ETC complexes. Complex activities were normalized to CS activity to account for mitochondrial number. The activities of complexes I+III (NADH cytochrome c reductase) were similar, and II+III (succinate-cytochrome c reductase) slightly higher in Myc cells compared to WT (Figure 1G). The increase in complex II activity is consistent with higher sdh mRNA expression in Myc cells. However, complex IV (cytochrome c oxidase, COX) activity was significantly decreased in Myc cells (Figure 1G). Confirming these data, histochemical assays of Sdh (complex II) and COX indicated that Sdh activity was equivalent in both WT cells and Myc cells, but COX activity was reduced specifically in the Myc cells (Supp. Figure 1E and data not shown).
p53 buffers Myc-dependent glycolytic flux and promotes oxidative respiration
Scox is the sole Drosophila homolog of the mammalian synthesis of Cytochrome c Oxidase 2 (SCO2) (Porcelli et al., 2010), which is regulated by p53, a critical sensor of cellular stresses including DNA damage and oncogene deregulation (Sutcliffe and Brehm, 2004). p53 promotes mitochondrial respiration by positively regulating Sco2 and inhibiting glycolysis (Matoba et al., 2006; Vousden and Ryan, 2009). Because scox expression was increased in Myc-expressing cells we investigated the role of p53 in metabolic homeostasis. The D. melanogaster genome contains one p53 homolog (Marcel et al., 2011) that is transcriptionally up-regulated in response to tissue and DNA damage (Brodsky et al., 2004; Wells and Johnston, 2012; Wells et al., 2006). p53 expression is low in WT wing discs (Wells and Johnston, 2012) but was induced in cells expressing Myc (Figure 2A–B) (Hulf et al., 2005; Montero et al., 2008), as was a p53 activity reporter (Figure 2C) (Brodsky et al., 2000). In cell culture, p53 mRNA was induced 2.5 fold by Myc expression (Suppl. Figure 1D).
Figure 2. Homeostatic metabolic regulation by Drosophila p53.
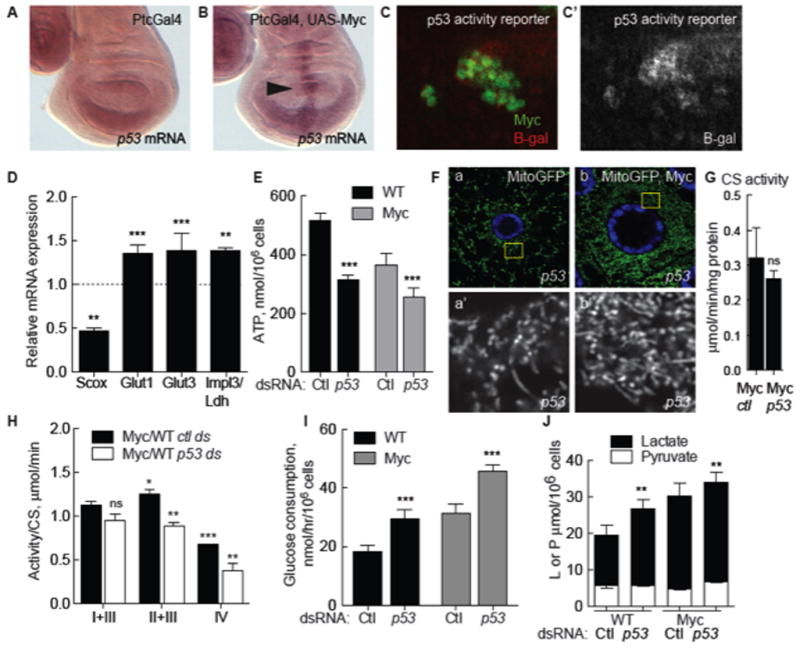
A. Expression of p53 mRNA is low in WT wing discs expressing the Ptc-Gal4 driver.
B. Myc expression under control of Ptc-Gal4 induces p53 mRNA (arrow points to the PtcGal4 domain).
C. p53 activity reported by expression from the p53 activity reporter rpr-150-lacZ (red), is induced in Myc-expressing wing disc clones (green). C′. Single channel, beta-galactosidase.
D. qRT-PCR expression of scox, glut1, glut3, and ldh/impl3 mRNA in WT S2 cells treated with treated with p53 dsRNA, expressed as a ratio to cells treated with control dsRNA. Expression of each mRNA is normalized to actin5C mRNA. All error bars in this figure are SD.
E. Steady-state ATP in WT or Myc cells from mono-cultures, treated with control or p53 dsRNA.
F. Mito-GFP in p53 mutant fat body cells. a) Mitochondria have mixed morphologies. b) p53 mutant cell expressing Myc.
G. Mitochondrial mass (CS activity) is not altered by loss of p53 in Myc cells.
H. ETC complex activity (normalized to CS activity) in Myc cells relative to WT cells, both treated with p53 dsRNA.
I. Glucose consumption in WT and Myc cell mono-cultures after p53 knock-down.
J. Intracellular lactate and pyruvate in mono-cultures of Myc cells or WT cells treated with p53 dsRNA.
To determine if Drosophila p53 functioned homeostatically to counteract the Myc-induced increase in glycolytic flux we treated WT or Myc cells with p53 dsRNA. Indeed, this significantly reduced scox expression and steady-state ATP relative to controls (Figure 2D–E and data not shown). ETC activity was uniformly reduced, affirming a role for p53 in oxidative phosphorylation (ox-phos) (Figure 2H). Using Mito-GFP in vivo, we saw disrupted mitochondrial morphology in p53 mutant wing disc cells or fat body cells with a variety of shapes including globules and donut shapes as well as the long filaments that predominated in WT cells (Figure 2F, a; Supp. Figure 2A, c). The increase in mitochondrial number induced by Myc remained in p53 mutants (Figure 2F, b; Supp. Figure 2A, d). Consistent with this, CS activity in Myc cells treated with p53 dsRNA in culture was similar to controls (Figure 2G). Thus despite morphological changes, mitochondrial mass remained the same. In contrast, p53 dsRNA stimulated expression of glut1, glut3, and ldh mRNAs and increased glucose consumption (Figure 2D, I) and intracellular lactate (Figure 2J) in WT cells and in Myc cells. Thus although virtually undetectable in steady-state conditions (Figure 2A), p53 is a physiological regulator of metabolic homeostasis in Drosophila and restrains glycolysis while promoting ox-phos. Moreover, our results suggest that the increase in sdh and scox mRNAs and CS activity in Myc cells compensates for a COX deficiency and reduced ATP.
Cell competition enhances Myc-induced metabolic flux
What is the effect of Myc-induced metabolic reprogramming on cell competition? In cell-based assays, mixed populations of WT and Myc cells foster competitive interactions mediated by diffusible factors (Senoo-Matsuda and Johnston, 2007), causing death of WT loser cells but proliferation of Myc winner cells (Supp. Figure 3B–C). Conditioned medium (CM) generated from such co-cultures contains these activities and induces winner or loser behavior in naïve cells (Supp. Figure 3A–C) (Senoo-Matsuda and Johnston, 2007). We induced competition between Myc and WT cells in co-culture assays and found that it further boosted glycolytic flux in Myc cells. In competitive co-cultures Myc cells had more lactate and less pyruvate than Myc cells in mono-culture (Figure 3G versus Figure 1E) and in the Myc cells glucose consumption doubled (Figure 3F versus 1D). Competition also enhanced glycolytic gene expression in Myc cells. Naïve WT or naïve Myc cells treated with CM from competitive co-cultures (cCM) enhanced the increase in glut1 and glut3 mRNA expression in Myc cells (Figure 3A). hexo-kinase C (HK-C) and tpi mRNA were also enhanced by competition (Figure 3A), but impl3/ldh mRNA was not. In vivo, uptake of the fluorescent glucose derivative, 2-NBDG (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose), was modest in control wing disc cells (Figure 3B) but prominently elevated in competing clones of Myc-expressing cells (Figure 3C), indicating glucose metabolism was also increased during competition in wing discs.
Figure 3. Myc-induced glycolysis is enhanced in cell competition and requires p53.
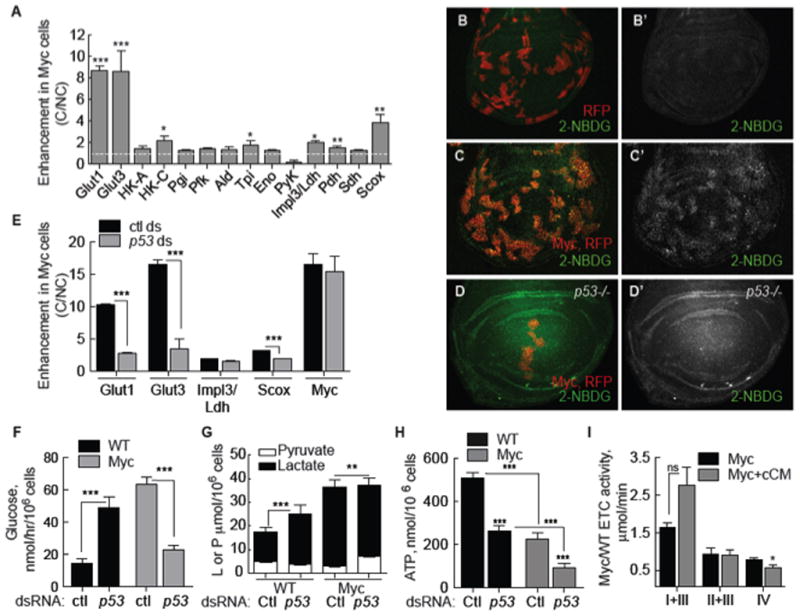
A. qRT-PCR of glycolytic gene expression (normalized to actin5C mRNA) in Myc cells grown under competitive conditions (C), expressed as fold increase over Myc cells in non-competitive conditions (NC). White dashed line indicates base level (1). Asterisks show significance relative to Myc cells in a NC context. All error bars in this figure are SD.
B–D. Glucose uptake in larval wing discs.
B. Control, RFP-expressing clones (red) with 2-NBDG uptake (green). B′. single channel of 2NBDG.
C. Uptake of 2-NBDG is increased in competing Myc, RFP-expressing cells. C′. Single channel, 2-NBDG.
D. Glucose uptake in p53 mutant wing disc containing Myc and RFP-expressing clones.
D′. Single channel of 2-NBDG.
E. qRT-PCR of Myc cells from competitive co-cultures treated with control or p53 dsRNA. Expression of each mRNA is normalized to actin5C mRNA expression and shows fold enhancement in Myc cells in C over NC conditions.
F. Glucose consumption in WT and Myc cells from competitive indirect co-cultures treated with p53 or control dsRNA.
G. Intracellular lactate and pyruvate in WT and Myc cells from competitive indirect co-cultures treated with control or p53 dsRNA.
H. Steady-state ATP in WT and Myc cells from C indirect co-cultures treated with control or p53 dsRNA.
I. ETC complex activity (normalized to CS activity) in Myc cells from mono-cultures compared to that of naïve Myc cells treated with cCM.
Expression of pdh, sdh and scox mRNA was enhanced by cCM in naïve Myc cells (Figure 3A) as well, but steady-state ATP was not improved. Rather, competition further reduced ATP to only 44% of controls (Figure 3H, Ctl). Competition-induced enhanced glycolysis and reduced ATP was specific to Myc cells, as lactate and ATP in WT loser cells remained identical to WT cell in non-competitive mono-cultures (Figure 3G–H, compare to Figure 1E). Competition mildly altered ETC activity in Myc cells and COX activity was further reduced compared to mono-culture controls (Figure 3I). Thus, confrontation between Myc and WT cells converts Myc cells into super-competitors and intensifies their already altered metabolism. The significant reduction in steady-state ATP in Myc super-competitors suggests glycolysis predominates over ox-phos as an energy source.
Enhanced metabolic flux in Myc super-competitors requires p53
To determine if p53 functions in Myc super-competition we examined cells treated with p53 dsRNA. In competitive co-cultures, p53 dsRNA reduced scox mRNA and steady-state ATP even further in Myc cells (Figure 3E, H). Unexpectedly, glycolytic flux was reduced by p53 dsRNA in Myc super-competitors, rather than stimulated (Figure 3E–G). Competition-induced enhancement of glucose consumption and of glut1 and glut3 expression was abolished by p53 dsRNA (Figure 3E, F). Enhanced expression of scox also required p53, indicating that its increase could be triggered in response to reduced ATP. Interestingly, expression of these genes remained higher than in WT cells (Figure 1B), thus glycolytic changes induced by a simple increase in Myc expression are p53-independent. p53 dsRNA did not alter impl3/ldh expression but reduced lactate in Myc cells, and did not affect Myc expression itself (Figure 3G). In contrast to Myc super-competitor cells, WT cells from the same co-cultures increased glucose consumption (Figure 3F) and lactate production (Figure 3E) in response to p53 dsRNA, as expected (Figure 2I, J). In p53 mutant wing discs, Myc-induced 2-NBDG uptake remained high in mutant discs in the absence of competition (data not shown), but was noticeably diminished in clones of Myc super-competitor cells (Figure 3D). Thus, p53 is required to enhance the Myc-stimulated increase in metabolic flux. This role is specific to Myc cells in a competitive context and distinct from p53’s normal homeostatic function in metabolic regulation.
Enhanced metabolism supports rapid expansion of Myc super-competitor cells
Since CM from competing co-cultures contains a diffusible activity that accelerates proliferation of naïve Myc cells relative to controls (Figure 4A, Supp. Figure 3B, C) (Senoo-Matsuda and Johnston, 2007), we tested if it required p53-dependent enhanced metabolic reprogramming. We used CM generated from p53-depleted competitive co-cultures to treat naïve assay cells also pre-treated with p53 dsRNA. Indeed, cCM from p53-depleted co-cultures was unable to accelerate proliferation of naïve Myc cells, whereas ncCM from p53-depleted control co-cultures did not alter proliferation of these cells (Figure 4A).
Figure 4. Enhanced glycolysis boosts proliferation of Myc super-competitors.
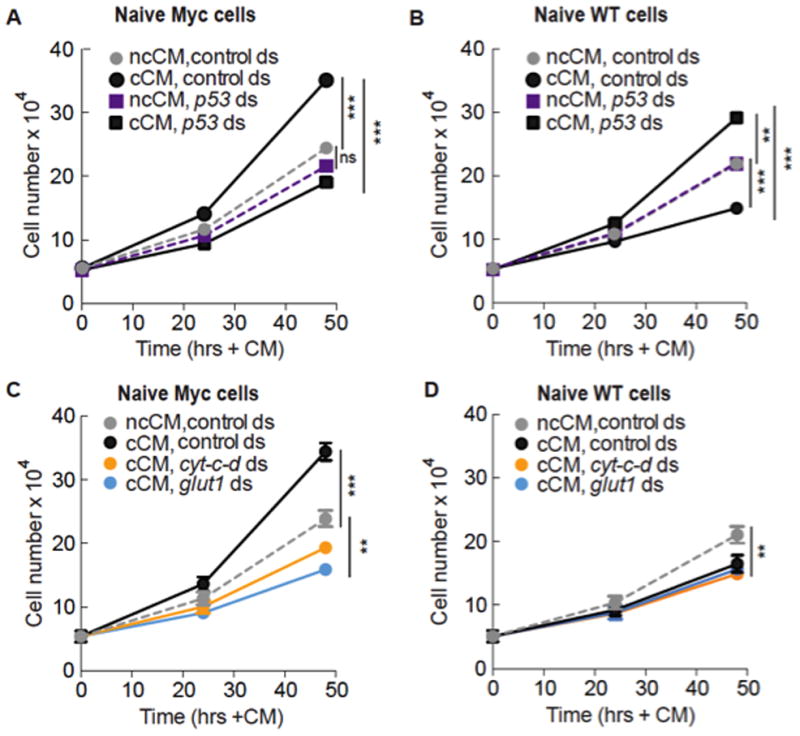
A. cCM, but not ncCM, generated from co-cultures depleted of p53 (but not control dsRNA) prevents accelerated proliferation of naïve Myc cells. Values in A and B are the mean of 5 independent experiments; cells in co-cultures and naïve cells were treated with p53 or control dsRNA in this figure. Error bars in this figure are SD.
B. Naïve WT cells treated with cCM from p53-deficient co-cultures proliferate faster than cells treated with cCM from co-cultures with control dsRNA, and faster than ncCM-treated cells.
C. Growth curve of Myc cells grown in cCM and ncCM from cyt-c-d or glut1-depleted co-cultures. Values are mean of 2 independent experiments.
D. Growth curve of WT cells grown in cCM and ncCM from cyt-c-d or glut1-depleted co-cultures. Values are mean of 2 independent experiments.
The lack of proliferation-stimulating activity in cCM from p53-depleted co-cultures could be due to loss of p53’s metabolic functions, or to another role of p53. To distinguish between these possibilities we directly disrupted metabolism by treating competitive co-cultures and naïve assay cells with dsRNA against glut1, cytochrome-c-d (cyt-c-d), or debcl/drob-1, a Bcl-2 family protein involved in mitochondrial respiration (Senoo-Matsuda et al., 2005). Like p53 dsRNA (Figures 2E, 3H), cyt-c-d or debcl dsRNA leads to reduced cellular ATP (N. S-M, unpublished data)(Senoo-Matsuda et al., 2005). cCM generated from these co-cultures did not stimulate proliferation of the naïve Myc assay cells (Figure 4C, Supp. Figure 3F–G). Knock-down of these genes does not compromise growth of Myc cells or WT cells per se, as ncCM from cyt-c-d or debcl-depleted co-cultures had little effect (Supp. Figure 3D–E). Thus, treatments that disrupt metabolism in Myc super-competitor cells suppress the proliferation-stimulating activity in cCM. As the activity is only present in cCM, and since Myc cells, but not WT cells, respond to it, these results suggest that p53-mediated enhanced metabolism promotes accelerated proliferation of Myc super-competitor cells in culture.
A second activity in cCM causes naïve WT cells to proliferate slower in response to cCM due to increased apoptosis (Figure 4B) (Senoo-Matsuda and Johnston, 2007). However, cCM from p53-depleted co-cultures stimulated naïve faster proliferation of WT cells (Figure 4B). This correlated with increased glut1, glut3 and scox expression and higher glucose consumption, as expected after p53-depletion (Figure 3G and data not shown). Thus whereas p53 loss dampens glycolysis in Myc super-competitor cells, it stimulates it in WT loser cells, indicating that p53 function in Myc cells in a competitive context is qualitatively different from its homeostatic role in regulation of metabolism. cCM from glut1, cyt-c-d, or debcl-depleted co-cultures was still as effective as control cCM at reducing the proliferation of naïve WT cells (Figure 4D and Supp. Figure 3G), due to a similar frequency of apoptosis (data not shown). The super-competitor killing activity is thus lost in p53-deficient cCM, but retained in cCM from metabolically disabled co-cultures. Thus p53-mediated enhanced metabolism in Myc cells promotes super-competitor status but cannot explain every aspect of the behavior.
During rapid growth, glutamine pools can be limiting for macromolecular biosynthesis and its uptake is increased in some cancer cells (DeBerardinis et al., 2007; Yuneva et al., 2012). We found increased expression of the glutamine synthases GS1 and GS2 and an uncharacterized gene encoding a glutaminase (CG42708) in Myc cells compared to WT cells that was enhanced in Myc super-competitors (Supp. Figure 4A). To determine if glutamine uptake was altered in Myc super-competitors we compared glutamine consumption in non-competitive versus competitive conditions in WT and Myc cells. Unexpectedly, mono-cultures of Myc cells consumed less glutamine than WT cells (Supp. Figure 4A). Glutamine consumption slightly increased in naïve Myc cells grown in cCM but remained less than naïve WT cells in cCM (Supp. Figure 4A). Thus, glutamine utilization may play a role in Myc cells, but its uptake is not rate limiting for rapid expansion of Myc super-competitor cells.
Expansion of Myc cell populations requires p53 only during competition
Our experiments indicate that without p53, WT “loser” cells became “winners” and proliferated more, seemingly reversing the outcome of competition. To determine if p53 loss in vivo had the same effect we induced clones of loser cells in p53 mutant wing discs using a competition assay in which all cells express Myc constitutively at a moderate level via a tub>myc>Gal4 gene cassette (> denotes FRT site) (de la Cova et al., 2004; Wu and Johnston, 2010). “Flp-out” of the >myc> cassette generates cell clones expressing UAS-GFP (loser GFP clones); these cells are WT with respect to Myc, but are disadvantaged because surrounded by tub>myc>Gal4 cells (de la Cova et al., 2004). WT loser GFP clones grew significantly slower than control GFP clones (Figure 5A, C). However, in either of two p53 null mutants, loser GFP clones grew to much larger sizes (Figure 5B, C and data not shown), growing as well as control clones not subject to competition (Figure 5C).
Figure 5. Myc super-competitor cells require p53 for genome stability and viability.
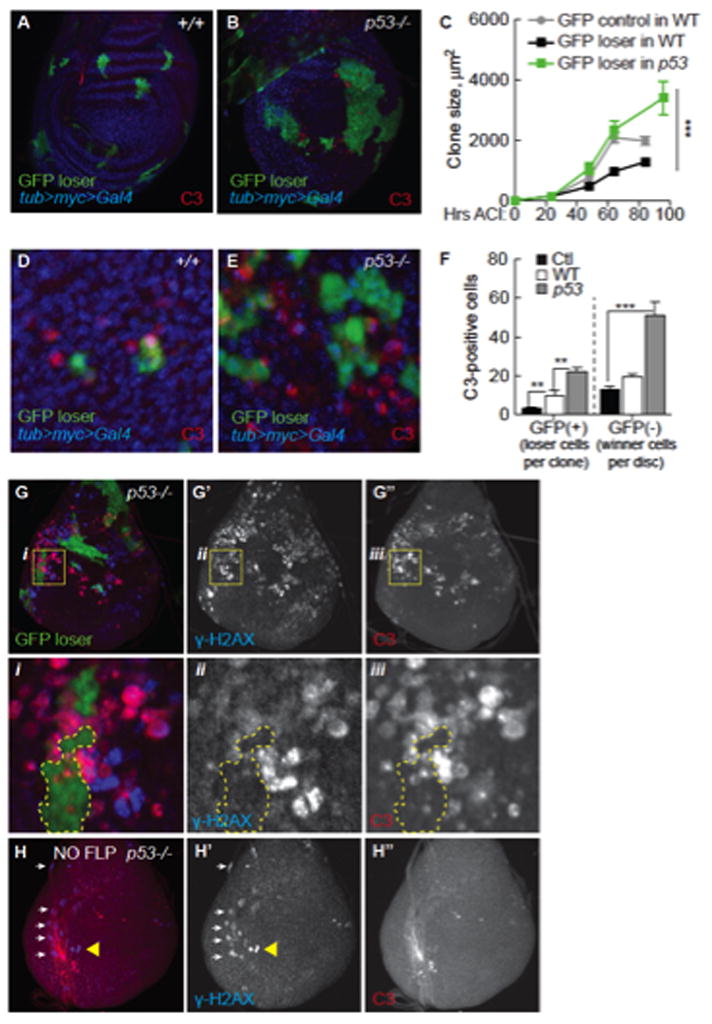
A. GFP+ loser clones (green) in a WT tub>myc>Gal4 wing disc. All GFP (−) cells express the tub>myc>Gal4 transgene. DNA is stained with Hoechst (blue). Active caspase 3 (C3, red) marks dying cells.
B. GFP+ loser clones (green) in a p53 mutant tub>myc>Gal4 wing disc. Dying cells (marked with C3) are mostly GFP(−), tub>myc-expressing cells.
C. In WT discs, loser GFP+ clones grow poorly (black line) compared to control GFP clones in a NC environment (grey line). In p53 mutant discs, loser GFP+ clones (green line) grow as well as control clones generated in parallel. Error bars in C–F are SEM.
D. In WT discs, most C3 staining is in loser GFP+ cells. Clones in D and E were allowed to grow for 24 hours.
E. C3 staining in p53 mutant, tub>myc>Gal4 winner cells competing with GFP loser clones (GFP+). Most C3-postive cells are GFP-negative (quantified in F).
F. Quantification of data from D and E. Mean number C3+ cells per GFP+ clone in NC (Ctl) or per GFP+ clone in competitive tub>dmyc>Gal4 discs in WT or p53 mutant backgrounds; or C3+ cells in GFP- tissue/wing disc.
G. GFP+ loser clones (green) in a p53 mutant wing disc. γ-H2AX (blue in G, separate channel in G′) marks cells with DNA damage. C3+ cells (red in G, single channel in G″). Yellow boxes in each panel are magnified below (i, ii, iii); H2AX is most often in Myc super-competitor cells (GFP-cells).
H. p53 mutant tub>myc>Gal4 cells do not die in the absence of cell competition. Wing discs were heat-shocked but lacked Flp, thus no loser GFP+ clones were generated. Separate channels are shown in H′, H″. Yellow triangle points to γ-H2AX-positive cells in the disc epithelium. White arrows point to overlapping trachea.
Consistent with our cell culture experiments, Myc-expressing cell clones generated in p53 mutant wing discs grew slower than controls (Supp. Figure 6C). Control GFP clones generated in either WT or p53 mutant backgrounds grew at similar rates (Supp. Figure 6C), thus loss of p53 selectively prevents expansion of competing Myc clones. Thus in the absence of p53 Myc super-competitor cells are unable to expand, while loser cells resist competition and survive.
Expansion of the Myc population requires increased ribosome biogenesis and activity, a signature of Myc activity manifested in wing discs by increased cell and nucleolar size and correlates with increased expression of the nucleolar protein, Fibrillarin (Fib) (Grewal et al., 2005; Johnston et al., 1999). We used these criteria to test if p53 was required for Myc to regulate ribosomal activity. Fib expression, nucleolar size, and cell size in Myc-expressing clones were unaffected in p53 null mutants (Supp. Figure 4B–D). We conclude that although required for Myc’s super-competitor metabolism and for expansion of the Myc super-competitor population, p53 is not required for Myc to stimulate cellular growth.
p53 promotes winner status in multiple paradigms of competition
Since confrontation between WT and Myc cells endows Myc cells with p53-dependent competitive behavior, we asked if winner cells in other competitive contexts required p53. In the classical paradigm of cell competition, clones of WT cells generated in a Minute (M) heterozygous background compete against the weaker M/+ cells and proliferate to occupy large portions of the wing disc (Morata and Ripoll, 1975; Simpson, 1979). In M(3)RpS3/+ mosaics, clones of WT cells compete to occupy the wing disc and when examined after short defined time-periods, grew significantly faster than WT clones in WT discs (Supp. Figure 5A), differing from a previous report (Martin et al., 2009). To test whether p53 mutant cells also “win” in competition with M(3)RpS3/+ cells, we generated p53 mutant clones in M(3)RpS3/+ wing discs. The clones grew faster than control p53 clones in p53 mutant wing discs and still populated a large fraction of the mature wing disc. However, the rate of p53 mutant “winner” cell proliferation was 25% slower than WT counterparts over the same time-period (Supp. Figure 5A). We conclude that although p53 loss does not abolish competition with M(3)RpS3/+ cells, it compromises the accelerated proliferation of winner cells.
In a second test, we examined competition between WT cells and dm mutant cells. As shown previously (Neto-Silva et al., 2010; Wu and Johnston, 2010), WT cells near dm mutant cells gained winner status and proliferated at a faster rate than when in a milieu of other WT cells (Supp. Figure 5B, left). In either a hypomorphic dmP0 mutant or null mutant dm4 background this was prevented by p53 loss (Supp. Figure 5B, right). Still, a p53 mutant background did not completely abolish competition, as dm mutant clones remained small. However, in three independent competitive contexts - Myc super-competitor vs WT cells, WT cells vs M/+ cells, and WT cells vs dm mutant cells – p53 is an important mediator of the rate at which the winner cell population expands, and therefore of their fitness.
p53 is selectively required in Myc super-competitors for genome stability, survival, and competitiveness
WT loser cells are eliminated from wing discs due to induction of the pro-apoptotic factor Hid (de la Cova et al., 2004). The number of loser cells expressing the active-caspase 3 (C3) apoptotic marker increases soon after induction of competition in tub>myc>Gal4 discs in either WT or p53 mutant backgrounds (Figure 5D–F, GFP(+);Supp. Figure 6A–B). However, within 24 hours after induction in p53 mutant wing discs, we observed three times more C3-positive Myc super-competitor cells than in a WT background (Figure 5D–F, GFP(−)). Many C3 (+) p53 mutant, Myc super-competitor cells were close to GFP loser clones (Figure 5E, G). In addition, many p53 mutant Myc super-competitor cells expressed the DNA damage marker, γ-H2AX, whereas in GFP loser cells γ-H2AX was rare (Figure 5G and i–iii). These findings and the fact that few competing Myc cells die in WT wing discs prompted a series of additional controls to rule out trivial causes of cell death. When all cells in p53 mutant wing discs expressed extra Myc via an intact tub>myc>Gal4 cassette (a non-competitive environment), cell death was comparable to that in WT controls (data not shown). The heat shock treatment used to induce clones also did not increase C3- or γ-H2AX-positive tub>myc>Gal4 cells (Figure 5H). Notably, C3 and γ-H2AX marks did not always correlate, suggesting γ-H2AX was not due to caspase activation (Rogakou et al., 2000). Finally, neutral GFP clones generated in p53 mutant discs did not die more frequently than in WT discs (Supp. Figure 6C, D); similar results were obtained in the S2 cell competition assay (Supp. Figure 6E). Thus, although p53 mutant, tub>myc>Gal4 cells survive and proliferate normally in wing discs for several days prior to stimulation of competition, within 24 hours of induction of GFP loser clones, genome de-stabilization in Myc cells leads to the appearance of γ-H2AX and to increased apoptosis. Accordingly, p53 selectively promotes the viability and genomic integrity of Myc super-competitors.
Without p53, the death of Myc winner cells prevents their expansion as a population and the WT losers gain the advantage. This altered outcome could be due to a requirement for p53 in either population. To determine which cells autonomously require p53 function we created p53 null FRT chromosomes wherein the mutation was either linked or unlinked to the Gal4 inhibitor, Gal80. This allowed us to separately assess the contribution of p53 to Gal4-regulated Myc cell clones and to their WT sibling cell clones, both produced by one mitotic recombination event (Figure 6) (de la Cova et al., 2004; Lee and Luo, 2001). Consistent with our previous findings, Myc clones grew faster than GFP controls at the expense of their WT sibling clones (Figure 6B, E). p53 loss specifically in loser clones (siblings of Myc super-competitor clones) did not alter their own growth or that of the Myc clones (Figure 6A–C). However, Myc clone size was reduced 40% by loss of p53 (Figure 6D), to smaller than WT control clones (Figure 6E–F). p53 mutant Myc clones were also no longer competitive, allowing WT sibling clones to grow as well as their counterparts in controls (Figure 6E–G). p53 is thus specifically and cell-autonomously required in Myc super-competitor cells.
Figure 6. Myc super-competitor status and survival require p53.
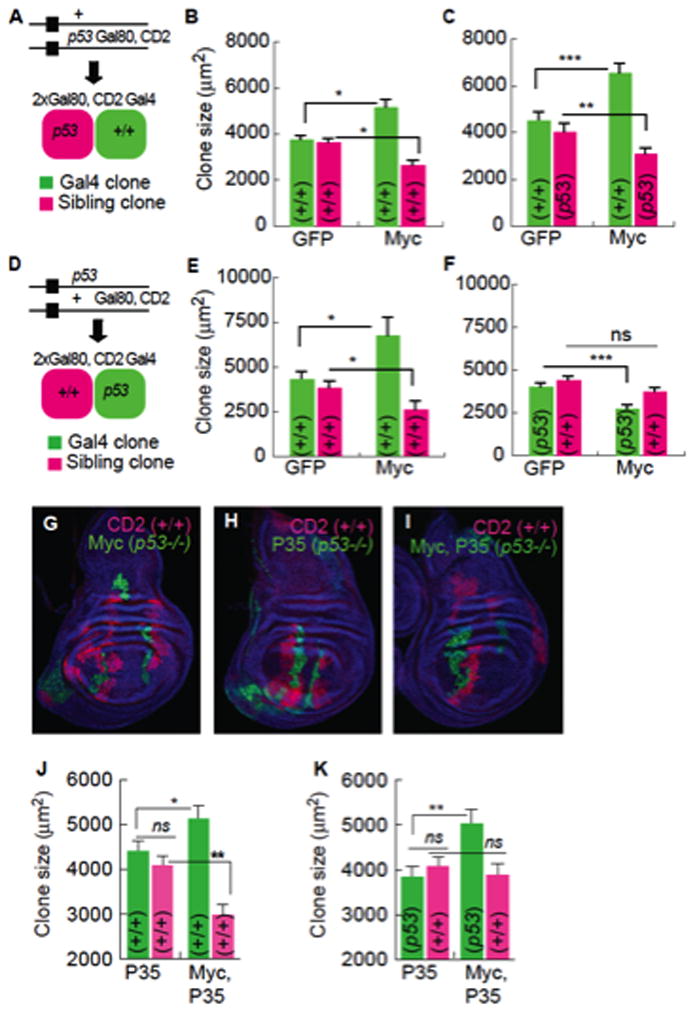
A–C. MARCM experiments in which sibling clones are p53 mutant. Error bars in this figure are SEM.
A. Scheme showing progeny generated by mitotic recombination. The p53 mutation segregates with Gal80 and CD2, yielding p53 mutant sibling clones.
B. Gal4, Myc clones compete against WT sibling clones and grow larger than Gal4 control clones.
C. WT Gal4, Myc clones compete against p53 mutant sibling clones.
D–F. MARCM experiment in which Gal4 clones are p53 mutant.
D. Scheme showing progeny generated by mitotic recombination. The p53 mutation segregates away from Gal80 and CD2 yielding p53 mutant Gal4 clones and WT sibling clones.
E. WT clones, as in B.
F. p53 mutant Myc-expressing Gal4 clones are significantly smaller than controls.
G–I Wing discs with MARCM clones of the indicated genotype. P35 is expressed in Gal4 clones (green) in I. All sibling clones (magenta) are WT, and blue cells are p53/+ (phenotypically WT).
J. P35 expression in the Gal4 clones does not affect competition between Gal4, Myc clones and WT sibling clones.
K. Inhibition of cell death of p53 mutant, Gal4, Myc clones allows them to grow equal to WT Gal4, Myc clones, but they are not competitive.
The ability of loser cells to survive when Myc super-competitors lack p53 suggested the latter had lost their killing activity. Whether this activity required cell survival, for example, for efficient signal transmission to loser cells, remained unclear. If survival and competitiveness are coupled, blocking death of p53 mutant Myc super-competitors might allow them to be competitive. We therefore protected p53 mutant Myc cells from death with the caspase inhibitor, P35 (Hay et al., 1994) and measured the size of these clones and their WT sibling clones after a defined period of growth. Expression of P35 in either WT or p53 mutant cell clones did not affect clonal growth in controls (Figure 6H, J), and co-expression of Myc and P35 did not block super-competitor status, since their sibling clones were smaller than controls (Figure 6J). Preventing apoptosis allowed p53 mutant, Myc-expressing clones to grow to the same size, and at the same rate, as WT Myc super-competitor clones (Figure 6K vs. J). Thus, the inability of the p53 mutant Myc super-competitor population to expand is due to cell death that requires the presence of nearby WT cells. Despite rescue of Myc clone size, WT sibling clones were equal in size to sibling clones in non-competitive controls (Figure 6K), thus without p53, Myc cells do not transmit a killing signal to their WT neighbors. Collectively, our results demonstrate that Myc super-competitor cells require p53 cell-autonomously for survival and super-competitor status.
Discussion
We have addressed the basis of cell fitness during Myc-induced super-competition, a paradigm of cell competition with parallels to mammalian tumor promotion. Here we extend these parallels with our finding that in Drosophila cells, extra Myc activity biases cells to glycolysis and lactic acid fermentation for energy production, as c-Myc does in many cancers (Dang, 1999; Shim et al., 1997). Although primarily analyzed in S2 cells, we also observed metabolic changes during competition in wing discs that suggest metabolism is similarly altered in vivo during competition. Intriguingly, our work reveals that increased Myc alters mitochondrial morphology and impairs complex IV (COX) activity, thereby weakening the ETC, which presumably causes the reduced cellular ATP in Myc cells. Together, the compromised ETC and rise in glycolysis in Myc cells echoes Warburg’s observation of flawed mitochondria and increased fermentation in cancers (Warburg, 1956). Increased glycolysis can benefit cells by freeing them from reliance on the slow but efficient energy production by ox-phos (Pfeiffer et al., 2001; Ristow and Shulz, 2009). Our finding raises the question of whether the glycolytic increase in Myc cells is independent of a faulty ETC, or is induced to compensate for defective complex IV activity and low ATP.
Our results also establish evolutionary conservation of p53’s homeostatic role in metabolic regulation in proliferating Drosophila cells (Cheung and Vousden, 2010; Matoba et al., 2006). Moreover, p53 balances internal metabolic changes in Myc cells by promoting ox-phos and curbing glycolysis. That the COX deficiency and low steady-state ATP is accompanied by a p53-dependent increase in scox mRNA suggests p53 is induced in Myc cells in a homeostatic response to protect fitness. As p53 is a sensor of numerous cellular stresses, we postulate that its activity increases in Myc cells to balance their metabolism (Figure 7A). The relationship between Myc and p53 could feasibly function as a conserved module for adaptation of cells to metabolic perturbation.
Figure 7. A model of Myc and p53-dependent reprogramming in cell competition.
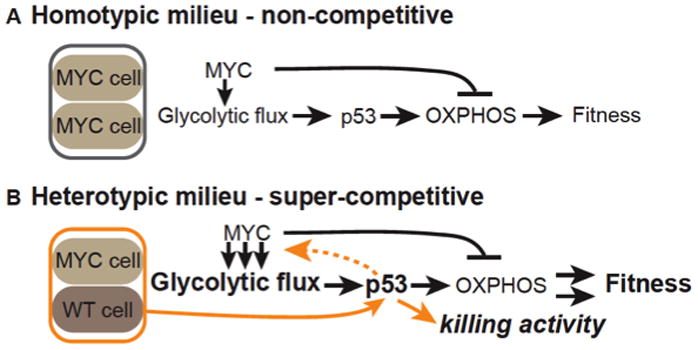
A. In a homotypic, NC milieu, high Myc activity increases glycolytic flux but inhibits oxphos, reducing ATP levels. p53 is induced and promotes oxphos, counteracting the effect of Myc and stabilizing ATP levels and thus fitness.
B. In a heterotypic, C milieu of Myc-expressing and WT cells, confrontational stress increases p53 activity, allowing Myc cells to acquire super-competitor status. As in A, p53 promotes oxphos in response to glycolytic flux induced by Myc expression. p53 also responds to confrontation with WT cells, further increasing Myc cell fitness and characterized by enhanced glycolytic flux. Super-competitor status enables them to transmit a killing signal to their WT neighbor cells, proliferate more, and expand their territory. Arrows in A and B indicate direct or indirect relationships inferred from the data.
In striking contrast to Myc cells on their own, when Myc cells co-exist with WT cells in mixed populations they undergo several additional p53-dependent changes: they produce (and also resist) a diffusible killing factor; they gain an increase in reproductive potential; and glycolytic flux is considerably enhanced. Notably, these events happen within hours (de la Cova et al., 2004)(Senoo-Matsuda and Johnston, 2007). The diverse p53-dependent changes in Myc cells imply that p53, already active to balance metabolism, detects confrontation with WT cells via a distinct role, initiating a new program that converts Myc cells into super-competitors. Enhanced glycolytic flux is clearly part of this program, but given p53 normally suppresses glycolysis we speculate that it is only indirectly regulated by p53. Even so, without p53, Myc super-competitors completely lose the traits distinguishing them from Myc cells in a homotypic environment, suggesting that p53 is a sensor of confrontation between the two populations; in its absence the cells do not “recognize” their differences.
Super-competitor status (and winner status in general) is an acquired property of cells, dictated by their local environment. Activation of Jun N-terminal kinase signaling in competing cell populations indicates that the confrontation is stressful (de la Cova et al., 2004; Moreno and Basler, 2004), which could elicit additional p53 functions. If so, the degree of p53 activation may be a proxy for the level of stress the cells receive. Thus, Myc cells lacking p53 are viable in a homotypic setting despite acute metabolic changes, but the additional stress of a heterotypic milieu leads to inviability. p53 function in Myc cells is context dependent: it is first activated to balance metabolic distress induced by Myc, and second, confrontation with WT cells activates a different function of p53 that leads to a program providing the super-competitor phenotype. Given the rapid induction of super-competitor behavior, we speculate that post-translational mechanisms are involved.
How is confrontation sensed by p53? With the exception of the killing activity, the super-competitor phenotype is an exaggeration of existing properties of Myc cells. That heterotypic confrontation causes a significant rise in metabolic flux in Myc cells raises the possibility that metabolites play a role. Like p53 depletion, direct metabolic disruption limits the fitness increase of Myc super-competitors, revealing an important metabolic underpinning to their status. However, direct metabolic disruption did not block production of the killing activity, thus metabolism cannot account for all super-competitor traits. Although metabolic functions of p53 are required for some super-competitor behaviors, our results suggest that p53’s role in promoting fitness of WT winner cells in other competitive contexts is related to the confrontational response, rather than direct metabolic regulation. Still, the evidence that p53 promotes reproductive fitness of “winner” cells in other heterotypic contexts raises the possibility that p53 generally functions as a sensor of genetic heterogeneity. Whether metabolic alterations are sensed or mediate winner behavior in these contexts remains to be seen.
Super-competitor status increases Myc cell fitness, but also comes at the cost of life-threatening vulnerability to loss of p53. Although the root cause of the vulnerability is unknown, our results suggest several possibilities. In the absence of p53, confrontation of Myc and WT cells leads to H2AX phosphorylation in Myc cells, thus DNA damage may selectively compromise survival of Myc super-competitor cells. Myc cells have increased reactive oxygen species (ROS; M. Ziosi, unpublished data), possibly arising from enhanced metabolic flux, which can cause genomic damage (Greer et al., 2013; K et al., 2006). As p53 has anti-oxidant functions (Budanov et al., 2004; Sablina et al., 2003), its loss in Myc super-competitors could intensify ROS and increase genomic instability. In addition, confrontational stress coupled with loss of p53’s ox-phos promoting capacity could lead ATP to fall below a critical threshold in the Myc cells. However, when protected from apoptosis, p53 mutant Myc super-competitors proliferate with the same kinetics and to the same extent as WT Myc super-competitors. Thus Myc remains fully functional as a growth regulator in p53 mutant cells and also still stimulates glycolytic flux.
Regardless of the mechanism, the phenotypic differences between Myc-expressing cells and Myc-super-competitor cells provide a clear demonstration of the determinative nature of cellular context, a hallmark of cell competition. Given the conserved metabolic functions of Myc and p53 we document here and the frequent activation of Myc family proteins in cancer, our findings predict that homeostatic p53 activity is induced to balance the glycolytic shift in incipient cancer cells. Moreover, in normal tissues, confrontation with an emerging patch of such pre-cancerous cells could encourage tumor growth and fitness by promoting the cells’ acquisition of super-competitor status. In this regard, the apparent cooperative relationship between Myc and p53 in regulating super-competition is particularly disquieting in terms of cancer. Two points are worth considering further. First, cancer cells commonly take advantage of glycolysis to fuel rapid energy production, but this is thought to evolve over time in response to constraints on growth (such as oxygen availability) as the tumor cells become invasive (Gatenby and Gillies, 2004). Myc-expressing cells acquire super-competitor status within a few hours of confrontation with WT cells (Senoo-Matsuda and Johnston, 2007), immediately giving them a competitive advantage, suggesting that selective pressure is not necessary for pre-cancerous cells to acquire a metabolic profile associated with super-competitor behavior. Second, although TP53 mutations are among the most frequent in cancer, their tumor-promoting effects are only evident over relatively long timescales and additional mutations are important for malignancy (Ziegler et al., 1994). We suggest that like Myc super-competitor cells, pre-cancerous cells arising among otherwise healthy cells may initially require p53 for their survival. Time and sensitivity to additional stresses could facilitate p53 loss, after which continued tumor growth would be predicted to occur only in cells successful at adapting to its loss, an idea supported by recent work in human cells (Maddocks et al., 2013). Thus our finding that p53 is critically important for the survival of super-competitor cells could be relevant when considering therapies in early stages of tumor promotion.
Experimental Procedures
Somatic clones
The tub>myc>Gal4 cassette was used to generate random GFP-marked tub>Gal4 clones (de la Cova et al., 2004). Tub>Gal4 clones were induced by heat shock (HS) of larvae at 37°C for 25 min at 48 hrs after egg-laying (AEL) and animals were allowed to grow until the indicated time point. The act>y, stop>Gal4 cassette was used to generate random GFP or Myc-expressing act>Gal4 clones in wild-type (WT) wing discs. Act>Gal4 clones were induced by larval HS at 37°C for 15 min at 48 hours AEL and animals treated as above. For p53 mutant clones we tested two null alleles, p53ns and p535-a-1-4, which behaved similarly.
Clonal growth measurements
Clonal growth was determined by measuring the two-dimensional area of clones (μm2). Clone size measurements were made using a Zeiss Axioplan 2 microscope with an Orca-100 CCD camera (Hammatsu). Clone area was measured at 200X magnification using Axiovision 4.6 software.
Histology
Fixation, antibodies and staining of imaginal discs, and RNA in situ hybridizations with digoxigenin labeled RNA probes were done as described (de la Cova et al., 2004). Images were acquired with Axiovision 4.6 or Leica SP5 confocal system software. See Supplementary materials for details.
Cell culture assays
WT cells and Myc cells were plated for mono-culture, direct co-culture, or indirect co-culture at equal density and incubated 16 hrs, after which medium was replaced with medium ±125 μM CuSO4 (Senoo-Matsuda and Johnston, 2007). Culture systems, C3 activity measurements and growth assays were as in (Senoo-Matsuda and Johnston, 2007).
ATP, lactate and pyruvate assays
WT cells and Myc cells were treated ced-9 (control) dsRNA or p53 dsRNA in the absence of CuSO4 for 96 hrs, and then plated for monoculture or indirect co-culture for 16 hrs (Senoo-Matsuda and Johnston, 2007). Cells were then treated ±125 μM CuSO4 for 24 hrs. For ATP analysis, the cells were lysed in Reporter Lysis Buffer (Promega) and lysate used in the ATP Determination Kit (Invitrogen). Lactate and Pyruvate Assay Kits (BioVision) were used as per manufacturers instructions.
Glucose consumption and 2-NBDG assays
CM was collected from single, indirect or direct co-cultures of WT and Myc cells and glucose assessed using the Glucose (GO) Assay Kit (Sigma). For in vivo assessment of glucose uptake, wing imaginal discs were monitored as follows: control RFP or RFP + Myc-expressing clones were induced at 48hrs AEL and larvae dissected at 96hrs AEL. Dissected larvae were incubated in PBS with 0.25mM 2-NBDG (Invitrogen) for 45min at 25°C, washed twice in PBS for 10min, fixed 20 min in PBS + 4% PFA and washed again twice for 10min in PBS. All washes and the fixation were done with pre-cooled PBS (4°C). Imaginal discs were stained with Hoechst, rapidly dissected and mounted in Vectashield and images immediately collected with a Leica SP5 confocal microscope. 2-NBDG fluorescence was excited at 488nm and detected at 500–520nm. RFP fluorescence was excited at 543nm and emission detected at 550–645nm (Zou et al., 2005).
Quantitative RT-PCR
WT or Myc cells were collected from mono-cultures or indirect co-cultures for analysis after 24 hours of the appropriate treatment. Reverse transcription (RT) to produce single-stranded cDNA was performed using 0.5–1 μg total RNA and SuperScript First-Strand Synthesis kit (Invitrogen). RT plus and minus controls were done in parallel. Q-PCR reactions were performed using LightCycler FastStart DNA MasterPlus SYBR Green I kit (Roche). Standard curves were produced using serially diluted cDNA made from WT cells for each primer set. Amplification was done using standard conditions in a Roche LightCycler. act5C mRNA was used to normalize data. Primer sequences are listed in Table S1.
dsRNA treatment
WT or Myc cells were pre-treated with dsRNA against ced-9 or gfp (used as controls), p53, glut1, cyt-c-d, or debcl for 72–96 hrs as described (Senoo-Matsuda and Johnston, 2007). See Supplementary materials for details.
Mitochondrial respiratory chain complex activities
Complex activities were measured according to (DiMauro et al., 1987). See Supplementary materials for details.
Statistics
All values are means of ≥ 3 independent experiments. Students t-test was used to determine significance (2-tailed, unequal variance). p-values are as follows: * p<0.05,** p<0.01, *** p<0.001. All error bars are standard deviation (SD) unless otherwise indicated in the Figure legend.
Supplementary Material
Research Highlights (character count).
Super-competitor status of Myc cells enhances metabolism and reproductive success. (84)
Confrontation between Myc-expressing cells and WT cells requires p53 as a sensor. (83)
Without p53, Myc super-competitor cells no trigger elimination of nearby WT cells. (84)
Pre-cancerous cells in healthy tissues may initially require p53 for survival. (80)
Acknowledgments
We thank our colleagues for sharing reagents, the Bloomington Drosophila Stock Center at Indiana University for fly stocks, the Developmental Studies Hybridoma Bank for antibodies, members of the Johnston lab for constructive comments on the manuscript, and V. Emmanuele for advice for SDH and COX histochemistry. Supported by grants from the NIH (to LAJ and to CQ) and the CIRC “Giorgio Prodi”, University of Bologna (MZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Assaily W, Benchimol S. Differential utilization of two ATP-generating pathways is regulated by p53. Cancer Cell. 2006;10:4–6. doi: 10.1016/j.ccr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Baker NE. Cell competition. Current biology : CB. 2011;21:R11–15. doi: 10.1016/j.cub.2010.11.030. [DOI] [PubMed] [Google Scholar]
- Baker NE, Li W. Cell competition and its possible relation to cancer. Cancer Research. 2008;68:5505–5507. doi: 10.1158/0008-5472.CAN-07-6348. [DOI] [PubMed] [Google Scholar]
- Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–113. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, Rio DC, Rubin GM. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004;24:1219–1231. doi: 10.1128/MCB.24.3.1219-1231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol. 2010;22:186–191. doi: 10.1016/j.ceb.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Claveria C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44. doi: 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and cellular biology. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Research. 2010;70:859–862. doi: 10.1158/0008-5472.CAN-09-3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejosez M, Ura H, Brandt VL, Zwaka TP. Safeguards for cell cooperation in mouse embryogenesis shown by genome-wide cheater screen. Science. 2013;341:1511–1514. doi: 10.1126/science.1241628. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Servidei S, Zeviani M, DiRocco M, DeVivo DC, DiDonato S, Uziel G, Berry K, Hoganson G, Johnsen SD, et al. Cytochrome c oxidase deficiency in Leigh syndrome. Annals of neurology. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Greer C, Lee M, Westerhof M, Milholland B, Spokony R, Vijg J, Secombe J. Myc-dependent genome instability and lifespan in Drosophila. PLoS One. 2013;8:e74641. doi: 10.1371/journal.pone.0074641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hulf T, Bellosta P, Furrer M, Steiger D, Svensson D, Barbour A, Gallant P. Whole-genome analysis reveals a strong positional bias of conserved dMyc-dependent E-boxes. Mol Cell Biol. 2005;25:3401–3410. doi: 10.1128/MCB.25.9.3401-3410.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA. Competitive interactions between cells: death, growth and geography. Science. 2009;324:1679–1682. doi: 10.1126/science.1163862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA. Socializing with Myc: Cell competition in Development and as a Model for Premalignant Cancer. In: Eisenman RN, Dang CV, editors. Myc. New York: CSHL Press; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KCS, Carcamo JM, Golde DW. Antioxidants prevent oxidative DNA damage and cellular transformation elicited by the over-expression of c-MYC. Mutation research. 2006;593:64–79. doi: 10.1016/j.mrfmmm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz-Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P, et al. Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ. 2011;18:1815–1824. doi: 10.1038/cdd.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin FA, Herrera SC, Morata G. Cell competition, growth and size control in the Drosophila wing imaginal disc. Development. 2009;136:3747–3756. doi: 10.1242/dev.038406. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8:e1000324. doi: 10.1371/journal.pbio.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Montero L, Muller N, Gallant P. Induction of apoptosis by Drosophila Myc. Genesis. 2008;46:104–111. doi: 10.1002/dvg.20373. [DOI] [PubMed] [Google Scholar]
- Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- Moreno E. Is cell competition relevant to cancer? Nat Rev Cancer. 2008;8:141–147. doi: 10.1038/nrc2252. [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- Neto-Silva RM, de Beco S, Johnston LA. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev Cell. 2010;19:507–520. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Porcelli D, Oliva M, Duchi S, Latorre D, Cavaliere V, Barsanti P, Villani G, Gargiulo G, Caggese C. Genetic, functional and evolutionary characterization of scox, the Drosophila melanogaster ortholog of the human SCO1 gene. Mitochondrion. 2010;10:433–448. doi: 10.1016/j.mito.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Portela M, Casas-Tinto S, Rhiner C, López-Gay JM, Domínguez O, Soldini D, Moreno E. Drosophila SPARC is a self-protective signal expressed by loser cells during cell competition. Dev Cell. 2010;19:562–573. doi: 10.1016/j.devcel.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Rhiner C, Lopez-Gay JM, Soldini D, Casas-Tinto S, Martin FA, Lombardia L, Moreno E. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev Cell. 2010;18:985–998. doi: 10.1016/j.devcel.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Ristow M, Shulz TJ. Warburg and his Legacy. In: Singh K, Costello L, editors. Mitochondria and Cancer. Springer Science; 2009. [Google Scholar]
- Rodrigues AB, Zoranovic T, Ayala-Camargo A, Grewal S, Reyes-Robles T, Krasny M, Wu DC, Johnston LA, Bach EA. Activated STAT regulates growth and induces competitive interactions independently of Myc, Yorkie, Wingless and ribosome biogenesis. Development. 2012;139:4051–4061. doi: 10.1242/dev.076760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Sablina AA, Chumakov PM, Kopnin BP. Tumor suppressor p53 and its homologue p73alpha affect cell migration. J Biol Chem. 2003;278:27362–27371. doi: 10.1074/jbc.M300547200. [DOI] [PubMed] [Google Scholar]
- Sancho M, Di-Gregorio A, George N, Pozzi S, Sanchez JM, Pernaute B, Rodriguez TA. Competitive interactions eliminate unfit embryonic stem cells at the onset of differentiation. Dev Cell. 2013;26:19–30. doi: 10.1016/j.devcel.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Igaki T, Miura M. Bax-like protein Drob-1 protects neurons from expanded polyglutamine-induced toxicity in Drosophila. EMBO J. 2005;24:2700–2713. doi: 10.1038/sj.emboj.7600721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Johnston LA. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc Natl Acad Sci U S A. 2007;104:18543–18548. doi: 10.1073/pnas.0709021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P. Parameters of cell competition in the compartments of the wing disc of Drosophila. Dev Biol. 1979;69:182–193. doi: 10.1016/0012-1606(79)90284-7. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JE, Brehm A. Of flies and men; p53, a tumour suppressor. FEBS Lett. 2004;567:86–91. doi: 10.1016/j.febslet.2004.03.122. [DOI] [PubMed] [Google Scholar]
- Tyler DM, Li W, Zhuo N, Pellock B, Baker NE. Genes affecting cell competition in Drosophila. Genetics. 2007;175:643–657. doi: 10.1534/genetics.106.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JP, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger myc-independent competitive cell interactions. Dev Cell. 2011;21:366–374. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Wells BS, Johnston LA. Maintenance of imaginal disc plasticity and regenerative potential in Drosophila by p53. Developmental biology. 2012;361:263–276. doi: 10.1016/j.ydbio.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16:1606–1615. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DM, Johnston LA. Control of Wing Size and Proportions by Drosophila Myc. Genetics. 2010;184:199–211. doi: 10.1534/genetics.109.110379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Brosel S, Acin-Perez R, Slavkovich V, Nishino I, Khan R, Goldberg IJ, Graziano J, Manfredi G, Schon EA. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum Mol Genet. 2010;19:170–180. doi: 10.1093/hmg/ddp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CD, Anderson SM. Sugar and fat - that’s where it’s at: metabolic changes in tumors. Breast Cancer Res. 2008;10:202. doi: 10.1186/bcr1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- Ziosi M, Baena-López LA, Grifoni D, Froldi F, Pession A, Garoia F, Trotta V, Bellosta P, Cavicchi S, Pession A. dMyc functions downstream of Yorkie to promote the supercompetitive behavior of hippo pathway mutant cells. PLoS Genet. 2010:6. doi: 10.1371/journal.pgen.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Biophys Methods. 2005;64:207–215. doi: 10.1016/j.jbbm.2005.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.