

Abstract
Cancer was seen for a long time as a strictly cell-autonomous process in which oncogenes and tumor-suppressor mutations drive clonal cell expansions. Research in the past decade, however, paints a more integrative picture of communication and interplay between neighboring cells in tissues. It is increasingly clear as well that tumors, far from being homogenous lumps of cells, consist of different cell types that function together as complex tissue-level communities. The repertoire of interactive cell behaviors and the quantity of cellular players involved call for a social cell biology that investigates these interactions. Research into this social cell biology is critical for understanding development of normal and tumoral tissues. Such complex social cell biology interactions can be parsed in Drosophila. Techniques in Drosophila for analysis of gene function and clonal behavior allow us to generate tumors and dissect their complex interactive biology with cellular resolution. Here, we review recent Drosophila research aimed at understanding tissue-level biology and social cell interactions in tumors, highlighting the principles these studies reveal.
Keywords: cell competition, compensatory proliferation, interclonal cooperation, basement membrane, tumor immunity, JNK
INTRODUCTION
Cancer development has been regarded for a long time as a cell-autonomous process in which mutations in oncogenes and tumor suppressors result in fast-dividing immortal cells. This is now clearly seen as an oversimplification. It is now widely recognized that, similar to normal development, cell interactions and tissue context hold the key to understanding most aspects of tumorigenesis, tumor development, and metastasis (11). In addition, it is increasingly clear that tumors are not homogenous lumps of cells. On the contrary, tumors behave as complex tissues consisting of many different types of cells (30). These tumor tissues contain not only highly heterogeneous populations of mutant cells but also surrounding wild-type cells and immune cells. Thus, substantial attention is shifting from the cell-autonomous properties of oncogenes and tumor-suppressor mutations to the non-cell-autonomous aspects of tumoral biology.
Social Cell Biology of Cancer
According to a popular image, cells in a tissue “talk to each other.” The connotations of harmony in these cellular conversations are perhaps appropriate to describe normal development. However, they do not capture the variety and tone of the exchanges occurring in tumors, frequently involving conflicting signals that compel cells to die or proliferate aggressively. The repertoire of possible cell behaviors and the quantity of distinct cellular players involved call for a social cell biology that examines the multiple interactions taking place inside and around tumors (Figure 1). Research into tissue-level biology and social cell interactions is critical for understanding development of normal and tumoral tissues.
Figure 1.

Social cell biology and tumors. Schematic representation of different types of social interactions that have been studied in Drosophila tumors. Abbreviation: WT, wild-type.
Drosophila has emerged as a system in which tumors can be easily induced and a range of questions can be addressed using an array of precise and powerful genetic tools. These tools can be combined to design experiments that are very difficult to perform in any other model organism. Importantly, techniques to create genetic mosaics allow production of clones of labeled mutant cells that arise from single cells in a wild-type tissue, such as an imaginal disc. This situation conveniently recapitulates the emergence and progression of solid tumors in epithelial tissues, which is the case for most human cancers. As a result, studies using the fruit fly Drosophila melanogaster are leading the way in investigating these social interactions by revealing and dissecting phenomena, including cell competition, compensatory proliferation, and interclonal cooperation. Drawing from a tradition of developmental thinking in terms of clonal behavior and cell-autonomous and nonautonomous effects, the fly is thus very well positioned to serve as a useful model to dissect cancer complexity and the social cell biology of tumors.
Cancer Genetics in Drosophila
Cancer studies in Drosophila have a long and distinguished history. The first tumor-causing mutation was identified in Drosophila. Mary Stark, a student of Thomas H. Morgan, reported in 1918 a blood neoplasm in larvae of the mutant l(1)7, which was isolated by Calvin Bridges (136). This evidence of an inherited tumor had enormous influence at a time when the idea of cancer as an ultimately genetic disease was starting to crystalize. The somatic mutation theory of cancer is often credited to Theodor Boveri, who attributed tumors to wrongly combined chromosomes resulting from abnormal mitotic segregation (15); however, it was Morgan who reinterpreted Boveri's ideas in light of Stark's work to suggest that “mammalian cancer may be due to recurrent somatic mutation of some gene” (91, p. 109). Decades later, the then very active field of comparative cancer biology took particular interest in the study of tumors in lower organisms. These tumors included many examples documented by field naturalists in wild specimens of insects and other invertebrates, as well as the Drosophila genetic tumors (50, 128). Arguments as to whether tumors in invertebrates could be deemed homologous to human neoplasias continued until Elisabeth Gateff, working with another mutation isolated by Bridges, showed that lethal giant larvae (lgl) tumors display true malignant growth, as cells from these tumors can be transplanted indefinitely from one animal to another, resulting in metastasis and death of the host (40). More recently, awareness of the astonishing conservation of most cellular pathways made Drosophila a preferred model to study signaling cascades and other cellular machineries misregulated in cancer, such as those involved in apoptosis, polarity, migration, and cell cycle and epigenetic regulation. These studies have produced many mechanistic insights into the properties of oncogenes and tumor suppressors and their normal roles during development. Finally, spectacular progress has been made recently in Drosophila in addressing the non-cell-autonomous aspects of cancer, tissue-level biology, and social cell interactions.
Tumors have been shown to develop from different tissues of the fly, such as the imaginal discs of the larva, the central nervous system, the blood cells, the male and female germ lines, and the digestive tract. This review focuses mostly on findings concerning tumors arising from imaginal discs. Excellent reviews have been published that also cover other aspects of Drosophila cancer research. We encourage readers to consult them for a broader view and appreciation of the rapid development of the field (see Related Resources at the end of the review). Imaginal discs are the precursors of the wings, the legs, the eyes, and all other structures of the adult epidermis, except for the abdomen. They are epithelial monolayer invaginations that grow during larval stages as they become increasingly patterned. Underlying the epithelial cells of imaginal discs is a basement membrane consisting mostly of Collagen IV, which is the same as found in human epithelia. This basement membrane separates the imaginal disc from the hemolymph, the blood where the cells of the fly's innate-only immune system are found in circulation. Finally, the fast growth and relatively undifferentiated status of imaginal discs are important characteristics that help the growth of tumors from this tissue.
Clonal Tools to Generate and Analyze Tumors
The past few years have seen an enormous expansion in the array of tools available to Drosophila researchers (26, 150). Among these, clonal techniques to generate genetic mosaics are perhaps the most envied by scientists working in other model organisms. These techniques can create marked patches of mutant cells in proliferating tissues (Figure 2). Clonal analysis allows assessment of the effect of mutations that would be lethal if they affected the entire animal. They are, in addition, invaluable tools for understanding cell lineage, stem cells, and cell interactions during development, and for modeling cancer, the quintessential clonal disease. Mosaic analysis has been an essential part of Drosophila research for many years (13). Early techniques of mosaic analysis included transplantation, gynandromorph chimeras, and, most importantly, mitotic recombination (137). In the 1970s, mitotic recombination induced by x-rays, in combination with newly available marker mutations, brought unprecedented cell resolution to studies on the genetic control of development (38). At present, irradiation has been replaced by the more efficient use of the tool yeast recombinase flippase (Flp) as the preferred means to induce mitotic recombination and thus produce, in its most common application, clones of homozygous mutant cells in a heterozygous animal (157, 158).
Figure 2.

Clonal tools to study tumors in Drosophila. (a) Generation of homozygous mutant clones in a heterozygous animal through mitotic recombination using the Flp/FRT technique. (b) Homozygous mutant clone (−/−) and wild-type twin (+/+) generated in the eye-antennal imaginal disc of a heterozygous animal (+/−) through the flippase/flippase recognition target (Flp/FRT) technique. The mutant clone is negatively labeled by the absence of a nuclear marker. This marker, expressed from a transgene inserted in the + chromosome arm, allows identification of the twin clone by the higher amount of the label. (c) Schematic representation of different types of mitotic recombination clones in the wing imaginal disc. In the Flp/FRT system, homozygous mutant clones are negatively labeled and the twin wild-type clone is distinguishable (see b). +/+ clones in a Minute (M)/+ animal occupy large territories through cell competition at the expense of M/+ cells (note that M/M cells do not survive). The MARCM (mosaic analysis with a repressible cell marker) technique allows expression of transgenes specifically in clones, including positive labels such as GFP (green fluorescent protein). However, +/+ twins are indistinguishable from +/− cells. Twin-spot MARCM and other systems allow positive labeling of both clone and twin with different markers. (d) Invasive tumor in a live Drosophila larva caused by interclonal cooperation between RasV12 [Ras valin 12 (glycine 12 to valine mutation)] and scrib cells (left). Tumor cells expressing RasV12 in the eye-antennal imaginal discs are marked with GFP. The eye-antennal imaginal discs, from which these tumors grow, are outlined in a wild-type larva (right).
Clone generation through mitotic recombination requires the exchange of chromosome segments between the paternal and maternal homologous chromosomes during the G2 phase of the cell cycle, when each chromosome consists of two identical sister chromatids. These exchanges are possible because mitotic chromosomes can pair in Drosophila, unlike in mammals, where pairing is restricted to meiosis. Expression of Flp in transgenic lines can mediate somatic crossing-over between FRTs (flippase recognition targets) inserted into fixed chromosomal locations (42). After the exchange, there are two alternative ways the recombinant chromosomes can segregate, known as z-segregation and x-segregation. In z-segregation, the two daughter cells remain heterozygous for the mutation of interest. In x-segregation, one daughter cell is a homozygous mutant and thus becomes the founder of a mutant clone, whereas the other daughter cell is homozygous wild type, giving rise to a twin clone. Segregation after recombination is nonrandom, and, for reasons unknown, z-segregation is rare (9, 114), thus adding to the method's effectiveness.
Refinements on Flp/FRT-mediated mitotic recombination have significantly expanded the number of ways in which clonal populations can be labeled and genetically manipulated. A crucial improvement in this regard was the MARCM (mosaic analysis with a repressible cell marker) technique, making positive labeling of these clones possible (71). MARCM combines the Flp/FRT and GAL4-UAS (upstream activator sequence) systems to allow GAL4-mediated expression of transgenes after clonal loss of a GAL4 repressor. In addition to GAL4-UAS (16), two other binary systems for expression of transgenes have been adapted to clonal analysis in Drosophila: lexA/lexAop (69) and the Q system (117). These alternatives to the GAL4-UAS system, on which most Drosophila research relies heavily, expand notably the number of possible experiments that can be done by using them in combination. Most importantly, this includes positive marking of both clones and twins, for which two additional techniques have been implemented (43, 161). Also, combinatorial labeling of multiple clones, twin spots, and subclones within clones with different fluorescent proteins is now possible thanks to the dBrainbow (48) and Flybow (47) systems. The amount of different experimental conditions that can be created with the tools of clonal analysis is therefore enormous. In addition, the amount of tools for analyzing gene function has seen a great expansion with the creation of RNAi transgenic libraries and the availability of convenient techniques for tagging of proteins by modifying genes at their genomic loci (see sidebar, Gene Function Techniques). Furthermore, combining clonal analysis with recent improvements in culturing imaginal discs ex vivo allows direct observation and recording of cell behavior (2, 101). At present, the main limitation, other than imagination, when designing experiments is really the number of transgenes that need to be bred into the same fly through crosses; for example, ten are needed for positively labeled mutant clones and twins using the Q system.
COMPETITIVE CELL INTERACTIONS
In recent years, cell competition has become a very active research area because of the obvious implications for the initial stages of cancer, when interactions with surrounding cells determine whether mutant cells die or thrive. Cell competition occurs when cells of a given genotype are eliminated as a result of their interactions with cells of a different genotype. Following an easy nomenclature, the eliminated cells are called losers and the cells responsible for their demise are called winners (87). This phenomenon was first observed in flies mutant for Minute genes (86), most of them encoding ribosomal proteins. Flies heterozygous for Minute mutations (M/+) develop slowly but normally. When clones of wild-type cells (+/+) are generated in the wings of M/+ flies, the +/+ clones end up occupying disproportionately large territories at the expense of the M/+ cells without disrupting the development of the wing (Figure 2c). Such large +/+ clonal territories generated in M/+ flies had been used to prove the existence of compartment boundaries that clonal populations cannot cross (39). It was later shown that the striking behavior of +/+ clones in M/+ animals involved apoptosis of the loser M/+ cells rather than just differences in proliferation rates (89). Similarly, cells with reduced levels of Myc (dmycP0 mutant) were shown to suffer cell competition (64, 88). It must be stressed that M/+ and dmycP0/dmycP0 cells grow well, although slowly, by themselves, and M/+ and dmycP0 adults are phenotypically normal in most respects. Therefore, it is the presence of the +/+ cells that actively causes their elimination. It is this context-dependent behavior of cells that defines cell competition in all cases: Loser cells are viable by themselves but eliminated when confronted by others.
Cell Competition as a Tumor-Suppressor Mechanism
Cancer biology addresses why we get cancer. However, given the number of alterations capable of disturbing tissue structure and homeostasis, why we do not get cancer more often seems an equally pertinent question (10). In this light, cell competition could be an important homeostatic mechanism that prevents the development of tumors. A low level of apoptosis can be seen in wild-type imaginal discs, and it has been postulated that cell competition can work as a homeostatic mechanism eliminating suboptimal cells during normal development. Consistent with this, preventing cell death, although it does not dramatically affect wing development, increases size variability (25). In addition, cell competition is partially or completely responsible for the elimination of cells bearing deleterious or suboptimal mutations when clones of such cells are generated in imaginal discs. The enhanced growth of clones in M heterozygotes is routinely used to create larger clones for mutations that cause poor cell viability (x/x clones surrounded by M/x cells). Eliminating the surrounding tissue through expression of a proapoptotic protein or a cell-lethal mutation has the same effect (138). More direct evidence of the role of cell competition preventing tumor development comes from clonal studies with tumor suppressors that affect cell polarity. Germ line mutations in tumor-suppressor genes, such as scribbled (scrib) and discs large (dlg), produce large imaginal disc tumors that lose apicobasal cell polarity and overgrow in mutant larvae (49). In contrast, scrib and dlg homozygous mutant clones do not develop into tumors when generated in wild-type discs but instead are eliminated through cell competition (17, 60). Consistent with the involvement of cell competition in their elimination, scrib clones survive and produce large invasive tumors when surrounding cells are killed (17).
Triggering and Executing Cell Competition
Cell competition studies have mostly focused on competition triggered by Minute, myc, and the scrib and dlg mutations. The extent to which the mechanisms involved in cell competition converge or differ among these different set tings is far from clear. Furthermore, causal connections between the different steps in the process that lead to elimination of the loser cells in each case are unknown as well. Upstream events that lead to the apoptosis of loser cells in particular are currently the subject of intensive research and no-less-intense discussion. A requirement for JNK (c-Jun N-terminal kinase) activation in the apoptosis of loser cells has been reported in competition triggered by Minute (89), myc (88), scrib (17), and dlg (61). There is evidence showing that in cell competition triggered by Minute or myc, cells may literally compete for the growth factor decapentaplegic (Dpp/TGFβ), as increasing either endocytosis or Dpp signaling can rescue loser cells from being outcompeted (88, 89). Another upstream event in Minute, myc, and scrib competition is mediated by Flower, a conserved transmembrane protein for which winner and loser cells express different isoforms (124). Flower is a putative component of a Ca2+ channel (160), and mechanistic elucidation of its involvement in cell competition awaits further investigation. In Minute competition, winner cells have been shown to engulf the M/+ loser cells in a process that requires draper and other genes involved in the clearance of apoptotic corpses (75). The histological observation of cells inside cells, called entosis, is common in tumors and could be related to a similar engulfment mechanism (103). Truly surprising and counterintuitive, however, is the fact that the loser cells seem to survive when engulfment is prevented, and so cell competition does not take place. It follows from this that the engulfment of loser cells is not a late event but an integral part of cell competition. In other words, the engulfment of the loser cells by the winner cells is an active killing process rather than a cleanup of the battlefield. A recent study, however, found that engulfment did not affect myc or Minute cell competition and that it was recruited blood cells, rather than wild-type cells, that engulfed most debris from loser cells (79). Therefore, the role of engulfment remains a subject of heated debate.
Engulfment of loser cells by surrounding wild-type cells occurs in the outcompetition of scrib and dlg cells as well (101). In fact, elimination of scrib and dlg cells involves at least two JNK-dependent mechanisms (Figure 3) for which a connection is not clear at present. First, the Drosophila TNF (tumor necrosis factor) homolog Eiger (59, 90), expressed at low levels in imaginal discs, is differentially endocytosed by scrib and dlg cells, thus activating JNK signaling in them (61). Second, wild-type cells surrounding scrib and dlg cells activate JNK signaling as well and upregulate PVR (PDGF-VEGF receptor), resulting in ELMO (engulfment and cell motility)-dependent engulfment of the loser cells (101). The fact that JNK is required in both winner and loser cells is intriguing, suggesting that JNK is a permissive factor in the cell competition process, with the outcome being determined by a prior or separate mechanism. Alternatively, JNK could have a truly instructive role if the outcome of the competitive interaction is determined or biased by the level of JNK activation, which is presumably higher in losers. In addition to JNK signaling, JAK/STAT (janus kinase/signal transducer and activator of transcription) signaling is also required in surrounding wild-type cells for outcompetition of scrib cells to occur (129).
Figure 3.
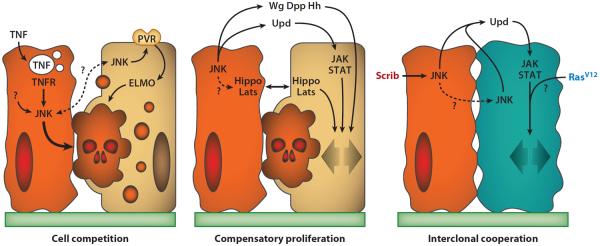
Mechanisms of social interaction in Drosophila tumors. Represented are some of the mechanisms involved in cell competition, compensatory proliferation, and interclonal cooperation. Abbreviations: ELMO, engulfment and cell motility; JAK, janus kinase; JNK, c-Jun N-terminal kinase; PVR, PDGF-VEGF receptor; STAT, signal transducer and activator of transcription; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor.
Despite the similarities in the engulfment phase and the late events of cell competition, important differences exist among the different setups in which competition has been studied, suggesting that different paths may converge onto a common cell elimination mechanism. For instance, Eiger-mediated JNK activation is not involved in the elimination of M/+ cells, as cell competition takes place normally in egr mutant flies (101). Furthermore, even when Eiger is involved, activation of JNK by increased endocytosis of Eiger may be specific to scrib and dlg cells because transcriptional upregulation of eiger has been reported in other tumors (106). Therefore, our mechanistic understanding of cell competition and JNK activation mechanisms remains fragmentary at this time, although a more general picture is likely to develop soon.
Escape from Cell Competition
It is a well-known fact that a single oncogenic or tumor-suppressor mutation is rarely enough to cause the development of a tumor. In Drosophila, acquisition of additional mutations that interfere with the mechanisms described above (JNK activation, engulfment, etc.) can rescue loser cells from cell competition, thus allowing tumor growth. In the case of scrib and dlg cells, increased Ras (17, 105), increased Notch (17), and decreased Hippo/Lats signaling (20, 83) have been shown to prevent cell competition. An important question so far unanswered is whether these conditions rescue scrib and dlg cells from competition by specifically interfering with the mechanisms that determine loser status in the first place or by overpowering them through parallel mechanisms. The Ras, Notch, and Hippo/Lats pathways all have potent proliferative and antiapoptotic effects, but at least in the case of Ras and Hippo/Lats it is possible that their effect on cell competition is more specifically mediated by their ability to increase myc expression (94, 118, 162). Unique among polarity tumor-suppressor genes is the case of lgl. In contrast to scrib and dlg, clones mutant for lgl are not eliminated by +/+ cells (45), and studies reporting outcompetition of lgl clones should be reconsidered (32). lgl clones have been shown to decrease Hippo/Lats signaling, which might rescue them from cell competition. In fact, Lgl has been postulated to be an integral component of the Hippo/Lats pathway (46), which inhibits proliferation by preventing activity of the transcription factor Yorkie (Yki). However, loss of scrib or dlg also decreases Hippo/Lats signaling to some extent (20, 140), and clones are still eliminated, so the mechanism by which lgl cells escape cell competition remains unclear.
Another factor that can play an important role in escaping outcompetition is the creation of a protective microenvironment, as highlighted by two recent studies. The first of these studies has demonstrated that fusion of mutant clones helps cells escape cell competition, probably through achievement of a critical mass or by minimizing the boundary where tumor cells would be most exposed to cell competition (83). The second study has shown that the secreted basement membrane component SPARC (secreted protein, acidic and rich in cysteine) is upregulated in loser cells and protects them to some extent from cell competition. When SPARC expression in loser cells is experimentally decreased or increased, loser-cell elimination is accelerated or prevented, respectively (116). SPARC is a highly conserved extracellular matrix protein that has been shown to be required for correct Collagen IV secretion in Drosophila (81). In the absence of SPARC, Collagen IV remains on the surface of secreting cells, suggesting that binding to SPARC makes Collagen IV more soluble (109). Imaginal disc cells do not secrete Collagen IV (109), so the mechanism by which SPARC could exert a protective role during cell competition remains to be elucidated.
Reversal of Cell Competition: Supercompetition
Of special interest for cancer are genetic conditions capable of not only preventing cell competition but also of reversing its usual outcome, making wild-type cells the losers in the competitive interaction. This reversal in the outcome of cell competition is termed supercompetition (88). Cells with elevated expression of the myc oncogene become supercompetitors that kill wild-type cells (25, 88). Cells mutant for negative regulators of the Hippo/Lats pathway also cause the death of surrounding wild-type cells (143). It must be noted that not all of the conditions causing overproliferation make cells supercompetitors. Ectopic PI3K activation, for instance, causes overproliferation but does not trigger competition through death of surrounding wild-type cells (25). Therefore, the trigger for cell competition cannot be just a difference in proliferation rates, supposing that a mechanism exists that allows cells to make such comparisons. Also Axin (Axn) mutant cells, displaying elevated levels of Wg signaling, become supercompetitors through a mechanism that does not involve Myc (152). Axn cells produce the secreted feedback regulator Notum, which inhibits Wg signaling in surrounding cells, essential for wing cell survival. Given that feedback mechanisms such as this are common in signaling pathways, it is likely that hyperactivation of other pathways can produce non-cell-autonomous death through a secreted inhibitor or through increased expression of receptors by tumor cells, thus depriving wild-type cells of a putative survival factor.
Cell Competition Outside Imaginal Discs and in Mammals
Although most insights into cell competition come from studies in Drosophila imaginal discs, its occurrence in other systems is well documented. Competitive interactions take place in the Drosophila ovary (63, 98, 123) and testis (132) to decide which cells occupy stem cell niches. Evidence of Myc-dependent cell competition exists in cultures of S2 (Schneider 2) cells (Drosophila blood cells) grown in suspension, suggesting the existence of secreted factors capable of mediating cell competition (130). Importantly, evidence exists that cell competition occurs in mammals. Mouse cells heterozygous for a mutation in the ribosomal protein gene RpL24 are disproportionately disadvantaged in chimeras (102), which is similar to the behavior of M/+ clones in flies. In aged rat livers, transplanted fetal hepatocytes outcompete host hepatocytes, facilitating repopulation after transplant (99). Cell competition can occur among hematopoietic stem cells upon irradiation, when lower p53 levels make cells winners (14). Finally, an important emerging system for studying cell competition in mammalian cells is MDCK (Madin-Darby canine kidney) cells, which form confluent epithelial monolayers in culture. Cells in which the Lgl partner Mahjong (141) or Scrib (96) are knocked down are eliminated from wild-type MDCK layers. Cells expressing oncogenic RasV12 [Ras valin 12 (glycine 12 to valine mutation)] (54) or Src (66) are eliminated from MDCK layers as well through extrusion from the epithelium. Recently, epithelial extrusion has been shown to allow clonal expansion of cells expressing the ERBB2 oncogene in the lumen of acini formed by cultured MCF10A mammary epithelial cells. This is another promising model to study social cell biology in human cells (73).
COMPENSATORY PROLIFERATION
Compensatory cell proliferation takes place when elimination of cells promotes proliferation of remaining cells in the tissue. For instance, irradiation of Drosophila larvae causes widespread apoptosis in imaginal wing discs, but the adult wing is normal, indicating that proliferation occurs to compensate for the enormous cell loss (51). Compensatory proliferation may operate through two types of mechanisms: local mechanisms of apoptosis-induced proliferation, in which damaged or dying cells nonautonomously stimulate proliferation of nearby cells, and more global size-control mechanisms, which probably ensure attainment of correct organ size during normal development as well (85). Comprehensive reviews on compensatory proliferation in the wider framework of regeneration have recently been published (122, 155). In a tumor context, which is most pertinent to this review, compensatory proliferation of the apoptosis-induced type can play a significant role in the interaction between wild-type and mutant cells. More specifically, if compensatory proliferation signals are triggered in or around tumor cells, those signals could promote not only proliferation of the wild-type cells but also of the tumor cells.
Compensatory Signals
Studies in imaginal discs uncovered a role for several secreted signaling proteins in compensatory proliferation triggered by apoptotic cells, irradiation, heat shock, and wounding (Figure 3). These signals include wingless (Wg/Wnt) and Dpp/TGFβ in the wing disc (126) and hedgehog (Hh/Shh) in differentiating eye tissue (34). Wg, Dpp, and Hh are known to direct the growth and patterning of these tissues during normal development as well, suggesting that their expression entails a recapitulation of development to facilitate regeneration. Other signaling proteins known to mediate compensatory proliferation are the unpaired cytokines (Upd, Upd2, and Upd3), which are the only JAK/STAT ligands in Drosophila and relate to human interleukin-6 (IL-6). Wounding of wing imaginal discs triggers expression of Upd, Upd2, and Upd3 downstream of JNK signaling (108), and JAK/STAT signaling is required for compensatory proliferation in the eye (156). In a more specifically tumoral setup, JNK signaling and JAK/STAT can promote tumor growth, as first shown in tumors caused by the loss of Csk (C-terminal src kinase) (120). Importantly, clones of cells mutant for the tumor suppressors vps25 (53, 142, 146) and ept (84) promote nonautonomous overgrowth of the surrounding wild-type tissue through elevated expression of the Upd cytokines as well. Mutant clones for some Polycomb group (PcG) genes produce similar upregulation of the Upd cytokines and nonautonomous overgrowths (21). This, together with a study reporting that JNK activity inhibits PcG function (70), suggests that expression of the Upd cytokines following JNK damage signaling could be mediated by PcG downregulation.
Another pathway that promotes nonautonomous proliferation in response to damage is the Hippo/Lats pathway, in this case through cell-cell contact rather than through secreted signals. Two recent studies have shown that downregulation of the Hippo/Lats pathway, previously known to be involved in the regeneration of cricket legs (7), also contributes to compensatory proliferation after tissue damage in the Drosophila wing disc (44, 140). In one of these studies, Hippo/Lats inhibition was shown to occur downstream of JNK signaling to promote proliferation (140). Other recent reports examined the relationship between JNK and Hippo/Lats signaling (20, 28, 33, 100, 125), but a molecular mechanism connecting both pathways is so far missing.
Apart from imaginal discs, a system in which compensatory proliferation is being intensively studied is the Drosophila adult midgut. The fly midgut, whose cells are subject to high turnover rates, is maintained by a sparse population of pluripotent stem cells. Misregulation of the stem cell niche microenvironment could be very relevant to cancer given the virtually unlimited proliferative potential of stem cells. Indeed, it is believed that tumors can arise and develop from mutations in stem cells themselves or by tumor cells acquiring stem cell properties. Many studies in the past few years have investigated the proliferation of stem cells in response to damage in the midgut. Following different insults, such as oxidative stress, tissue damage, and infection by enteric bacteria, stem cells proliferate to regenerate the midgut and restore homeostasis of the epithelium by replacing dead cells (3). Comparison of compensatory proliferation in intestinal stem cells and imaginal discs reveals that common or similar mechanisms involving the JNK, JAK/STAT, and Hippo/Lats pathways mediate the proliferative response of stem cells (12, 18, 23, 62, 67, 121, 131, 135).
Undead Cells
Through the above-mentioned proliferative signals, tissue damage and apoptosis can trigger compensatory growth that would normally aid tissue homeostasis and regeneration. However, these same homeostatic mechanisms can result in aberrant growth. A spectacular example of this is the large overgrowths that take place in imaginal discs when death of apoptotically stimulated cells is prevented. This is called the undead cell phenomenon (126). Undead cells, unable to die, seem to be locked in a situation in which they chronically induce compensatory proliferation. This is a scenario that could apply to tumor cells in many types of cancers given that apoptosis resistance is widely regarded as one of the hallmarks of cancer. Undead cells in imaginal discs potently stimulate over-growth of surrounding tissue when apoptosis is prevented by expression of the baculovirus protein p35 (57, 110, 126). Induction of Dpp and Wg by JNK signaling contributes to the nonautonomous overgrowths triggered by undead cells (126). The contribution of Dpp and Wg to compensatory proliferation that is not caused by undead cells, however, has been called into question (111, 112). Given the involvement of JNK activation in the undead cell phenomenon, roles for the Upd cytokines and Hippo/Lats signaling are expected, although not yet tested. Another situation in which the undead cell phenomenon is observed is during loss of p53 followed by irradiation, at which time JNK-dependent induction of Dpp and Wg has been shown to occur downstream of the caspase DRONC (68, 153). A positive feedback loop, however, could exist between p53, JNK, and the caspase pathway (133), making it difficult to ascertain which of these signals is the most upstream trigger of the response. Finally, clones in the eye, where JNK and Ras signaling are simultaneously activated, cause large nonautonomous overgrowth (145). Although not investigated further, these clones could represent another example of undead cells given that JNK and Ras are well-known proapoptotic and antiapoptotic signals, respectively.
Reorientation of Cell Division
Another important implication of compensatory proliferation in tumor biology stems from the fact that cell death, in addition to increasing proliferation of surrounding cells, could have an effect on those cells by reorienting their mitotic axis. Distributions of mitotic orientations in the plane of the epithelium are characteristic in each region of the wing disc (6). That orientation, given the absence of cell migration, is responsible for the normal shape of the wing. In outcompetition of M/+ clones, it has been shown that the presence of dying cells makes proliferation of surrounding wild-type cells preferentially perpendicular to the clone boundary (76). This results in higher intermingling of M/+ and +/+ cells, which maximizes the length of the boundary and might facilitate competition (83). This is in contrast to mitosis that occurs parallel to the boundary, which would produce straight borders and compact clones, and be more protected from cell competition.
INTERCLONAL COOPERATION
Tumors develop through the acquisition of multiple cooperating lesions. The canonical view on how tumors develop is a linear model of Darwinian selection, with cooperating mutations sequentially hitting cells and promoting successive clonal expansions. However, tumors exhibit a large degree of heterogeneity at both the cellular and genetic levels, and alternatives to this linear evolution model have been explored (42, 82). Recent work in Drosophila has demonstrated that tumors can grow through interclonal cooperation, i.e., through oncogenic cooperation of different mutations in separate clones of cells.
Cooperation Between RasV12 and Scrib Clones
An oncogenic cooperation model in Drosophila has been established between the RasV12 and scrib mutations. In imaginal discs, clones of cells expressing oncogenic RasV12 from a transgene and mutant at the same time as the tumor suppressor scrib can be generated using the MARCM technique (RasV12 scrib clones). When such clones are generated in the eye-antennal discs, they develop into large tumors that lose polarity, overgrow, degrade the basement membrane, metastasize, and eventually kill the animal (17, 105). Given that neither RasV12 nor scrib clones produce these effects separately, the double mutant RasV12 scrib tumors provide an example of oncogenic cooperation between two mutations that affect the same cells. Using the same clonal techniques, it is possible to simultaneously create clones of cells that express RasV12 and twin clones that are mutant for scrib. In this way, the effects of having the two mutations in adjacent cells can be analyzed. It was found that, when the RasV12 and scrib mutations affect neighboring cells, invasive tumors result (RasV12//scrib tumors), similar to when the two mutations affect the same cells (RasV12 scrib tumors) (156), providing evidence that oncogenic cooperation between RasV12 and scrib can occur not just intraclonally but also interclonally.
Interclonal cooperation between RasV12 and scrib relies on a two-step compensatory proliferation mechanism mediated by the Upd cytokines (Figure 3). First, JNK activation in the scrib clones induces production of Upd cytokines that activate JAK/STAT signaling in the RasV12 cells; this promotes the growth of RasV12 clones through a synergy between Ras and JAK/STAT signaling (52, 156). Second, JNK signaling seems to propagate from scrib clones to RasV12 clones, at which time the growth of the RasV12 cells becomes self-sustained. Neither the synergy between Ras and JAK/STAT nor the propagation of JNK is mechanistically understood so far. Another open question stems from the fact that scrib clones are eventually outcompeted by RasV12 clones, yet cell competition and apoptosis are unlikely to play a role in the development of these tumors. Proof of this is that confronting RasV12 clones with M/+ clones did not result in invasive tumors and neither did eliminating the tissue surrounding RasV12 clones through apoptosis. In contrast, scrib cells seem to behave as undead cells. Because scrib clones eventually die, this undead-like behavior must result from some property of scrib cells that plain apoptotic cells do not share, e.g., a higher level of JNK activity or comparatively long survival, which perhaps are important factors to kick-start or sustain JNK propagation.
Another example of interclonal cooperation in the eye-antennal discs involving some of the same players is the cooperation between RasV12 cells and RasV12 cells that additionally carry mutations affecting mitochondrial function and the respiratory chain (RasV12//RasV12 mito) (100). These mito mutations cause synergistic elevation in the production of reactive oxygen species in intraclonal combination with RasV12, which in turn causes JNK activation and the consequent downstream production of Upd cytokines. In a way similar to RasV12//scrib tumors, Upd cytokines from RasV12 mito cells activate JAK/STAT in neighboring RasV12 cells, which triggers uncontrolled tumor growth. This study places downregulation of Hippo/Lats signaling downstream of JNK signaling and upstream of Upd cytokine induction inside the RasV12 mito cells.
Relevance to Human Tumors
The demonstration in Drosophila that tumors can grow through interclonal cooperation has important implications for tumorigenesis and tumor progression. Sequential accumulation of independent mutations in a dominant clone is generally accepted as the driving force for cancer progression. A strictly lineal view of tumor development, however, overlooks the possible contribution of mutations in cells distinct from the dominant clone. Against this lineal view, the possibility of interclonal cooperation as a mechanism promoting development of human tumors has been postulated before (4, 80). Interclonal cooperation may be relevant to not only the initial stages of cancer but also to later stages when clonal evolution leads to the coexistence of multiple clones within the tumor mass. Indeed, evidence of persistent polyclonality in tumors has existed for a long time (97). Interclonal cooperation between mutations, therefore, may need to be included in models of tumor progression through clonal selection. Given that the probability for a tumor to acquire multiple oncogenic mutations in different cells is much higher than the probability of acquiring those same mutations in the same cell, interclonal oncogenic cooperation could play a significant role in tumor development and progression in humans. This is even more apparent in light of recent data questioning the requirement for genetic instability and increased mutation rates in cancer progression, the so-called mutator phenotype (65). To assess the importance of interclonal cooperation in human cancer, further investigation is needed. In this regard, steady progress in sequencing and other technologies might soon make it possible to reconstruct the evolutionary histories of tumors with cell resolution, for example, through single-cell sequencing (93). Recent studies are already revealing an unexpected degree of clonality and branching in the microevolution of human tumors (41, 55, 159).
INTERACTION WITH THE BASEMENT MEMBRANE
Basement membranes are polymers of extracellular matrix proteins that underlie epithelia and surround organs in all animals. The basement membrane acts as a barrier to the dissemination of tumor cells, and its breaching is a critical step in the final stages of tumor development. Collagen IV, Laminin, Nidogen, and Perlecan are the main components of basement membranes in humans, and all four of them are well conserved in flies (58). Another conserved basement membrane component is SPARC, the fly homolog of BM40/SPARC/osteonectin, whose possible involvement in tumor cell survival has been discussed above in relation to cell competition. In contrast to the conservation of basement membranes and their components, Drosophila lacks fibrillar collagens and connective tissue. Imaginal discs, for example, have no dermis. Therefore, once the basement membrane is breached, tumor cells can access the open circulatory system and potentially metastasize.
Basement Membrane Degradation
Collagen IV, the main component of basement membranes, is particularly resistant to proteolysis by most peptidases and is only efficiently degraded by matrix metalloproteases (MMPs). The family of MMPs in humans consists of 23 zinc-dependent endopeptidases that collectively can degrade all protein components of the extracellular matrix. The Drosophila genome has two genes encoding MMPs called Mmp1 and Mmp2 (77, 78). Mmp1 is a secreted metalloprotease (104), and Mmp2 is membrane bound through a GPI (glycosylphosphatidylinositol) anchor (77). This is a very simple situation compared with the 23 human MMPs. The MMP inhibitor TIMP (tissue inhibitor of metalloproteases) is conserved in flies as well (115) and is homologous to the four mammalian TIMPs.
Early evidence of the ability of fly tumors to degrade the basement membrane was found in lgl tumors when a type IV collagenase activity was found in the tumors (154). Another indication of the involvement of MMPs in Drosophila tumors was the fact that the short-range migratory ability of Csk cells in the wing disc is decreased by the loss of one copy of Mmp2 (151). A more complete understanding of this role came from three different studies showing that expression of MMPs is highly upregulated in different Drosophila models of clonally induced (134, 144) or transplanted (8) metastatic tumors. Similar to their role in human tumors, these studies showed that MMPs degrade the basement membrane in fly tumors, which is essential for the invasive and metastatic behavior of tumor cells. Two of these studies found that RasV12 scrib tumors express Mmp1 (134, 144) and Mmp2 (134) as a consequence of activation of JNK signaling (Figure 4). Indeed, expression of Mmp1, which can be efficiently detected through antibody staining, has become a widely used reporter of JNK activity in imaginal disc cells. Loss of MMP function or expression of the protease inhibitors TIMP and RECK suppress invasion by these tumors. JNK-activated expression of MMPs is also required for the eversion of imaginal discs (134), a morphogenetic process involving basement membrane degradation and cell invasion (107), which suggests that this normal developmental process is co-opted by tumors. Notably, the transplantation study found that brat tumors, although not invasive per se, are capable of metastasizing specifically to the ovary (8). In contrast to lgl tumors, brat tumor cells do not express MMPs but are capable of inducing Mmp1 expression in the host ovary, which highlights the importance of tumor-host interactions for the seeding of tumor cells and efficient metastasis. Similar specific interactions between tumor cells and target tissues may need to be invoked in the case of eyeful (36) and frazzled (147) tumors, where metastasis is observed despite the fact that tumor cells appear well differentiated.
Figure 4.

Basement membrane degradation and immune response. Expression of secreted Mmp1 and membrane-bound Mmp2 degrades the basement membrane in tumors. JAK/STAT (janus kinase/signal transducer and activator of transcription)-activating Upd cytokines promote proliferation of circulating blood cells. Expression of Upd cytokines induces more Upd expression in blood cells and the fat body, a positive feedback loop that turns a local response into a systemic one. Blood cells are recruited to tumors in regions that lack an intact basement membrane. Tumor necrosis factor (TNF) expression in tumor-associated blood cells contributes to JNK activation in tumors.
Basement Membrane Tension and Mechanical Sensing
One aspect of the tumor context that researchers have not extensively explored is physical tension, a potentially important regulator of cell signaling, growth, and motility. Matrix stiffness, enhanced in breast tumors through increased collagen cross-linking, is an important environmental cue that leads to enhanced integrin signaling and invasive behavior in tumors (74). In Drosophila, it has been shown that Collagen IV and the basement membrane exert a constricting force that shapes imaginal discs (109). The presence of Perlecan in basement membranes counters this constricting force (109). The distribution of physical tension across the tissue has been proposed to control cell proliferation during the development of the wing disc (1, 56). Intriguingly, the ability of actin and cytoskeletal components to modulate Hippo/Lats pathway activity in imaginal discs hints to a role of this growth control pathway in mechanical sensing (24, 35, 119, 127). YAP/TAZ, the mammalian homolog of Yki, has been shown to be regulated by mechanical cues from the extracellular matrix and thus plays a role in mechanotransduction (29). It will be interesting to study whether tension exerted by the basement membrane regulates normal and tumoral development, and whether the Hippo/Lats pathway plays a role.
TUMOR/IMMUNE INTERACTIONS
In mice and humans, the immune system is known to exert antitumoral and protumoral effects (27). On the one hand, there are immunosurveillance mechanisms by which the immune cells detect and fight tumors. On the other hand, the immune reaction provides an inflammatory environment that often leads to enhanced tumor growth. Interactions between tumors and immune cells are very complex, with many different types of immune cells involved in different phases of the response.
Innate Immune Response to Tumors
Insects do not have adaptive immunity or antibodies, which are evolutionary inventions of vertebrates. However, they possess a potent innate immune system that effectively defends the animal against several threats through a number of humoral and cellular mechanisms (72). The cellular branch of the immune system in Drosophila consists of three different blood cell (hemocyte) types. One of these cell types, the plasmatocytes, constitutes approximately 98% of the blood cells in a healthy animal. Recent studies have characterized an innate immune reaction to tumors in Drosophila (Figure 4) (108). In response to scrib tumors or clonal RasV12 scrib tumors, plasmatocytes increase their number circulating in the hemolymph, the blood-like liquid that fills the body cavity. This increase in circulating blood cell numbers does not involve release of new cells from the lymph glands but rather proliferation of already-circulating cells in response to JAK/STAT signaling. JAK/STAT-activating Upd cytokines are expressed first by the tumor downstream of JNK signaling and later in the fat body through a positive feedback loop that turns the local response into a systemic one. The expression of Upd cytokines, therefore, has not only autocrine effects in promoting compensatory proliferation (156), as discussed before, but also paracrine effects in blood cell proliferation. Furthermore, this response is similar to a response to tissue damage caused by direct wounding of imaginal discs in which local activation of Upd cytokines expression downstream of JNK activity is seen as well.
Tumor-Associated Blood Cells
In parallel to the increase in the number of circulating blood cells, some of these cells particularly adhere to tumors in places where the basement membrane has been degraded (108). Hemocytes have been shown to adhere to wounds in larvae (37) and embryos (139). Sessile blood cell populations exist in the larva, notably in the larval epidermis, the eye region of the eye-antennal disc, and specific regions of other late larval discs for reasons unknown. However, basement membrane degradation is sufficient to recruit blood cells to tissues, as evidenced by the fact that ectopic expression of Mmp2 causes hemocyte recruitment to regions of the imaginal discs and other organs, such as the salivary glands, where hemocytes normally are never found (108). This recruitment is likely due to the capture of blood cells by the damaged tissue, rather than directed migration, because hemocytes are in constant motion with the flow of the hemolymph in the open circulatory system. Consistent with this, in vivo imaging has shown no active migration of hemocytes to larval wounds (5), in contrast with the embryo.
In a clear parallel with the situation in mice and humans, protumoral and antitumoral roles for plasmatocytes were found in this response (22, 108). Reduction in the size of nonclonal scrib tumors is observed as a consequence of the response (108). The mechanisms for these antitumoral effects are not known, but they may involve phagocytosis of tumor cells or removal of apoptotic corpses, which can be observed in these tumors (J.C. Pastor-Pareja & T. Xu, unpublished results) and in imaginal discs following cell elimination through competition (79). Also, independent of the presence of tumors, blood cells constitutively express high levels of the TNF homolog Eiger (J.C. Pastor-Pareja & T. Igaki, unpublished results), which may affect tumor growth when blood cells become tumor-associated. Importantly, another study found that the response of blood cells to tumors has a protumoral effect in the case of RasV12 scrib tumors (22). With a blood cell transplantation assay, this study determined that expression of Eiger by tumor-associated blood cells contributes to JNK activation in the tumors (Figure 4) and thus results in enhanced tumor growth.
CONCLUDING REMARKS
The topics covered in this review represent fronts in which research done on Drosophila has produced significant insights into the non-cell-autonomous properties of tumor cells. In many cases, the conservation of key elements makes those insights not just conceptual but relevant in terms of molecular mechanisms. For example, JNK and JAK/STAT damage signaling, the role of polarity and Hippo/Lats genes as tumor suppressors, the machineries for apoptosis and cell engulfment, and basement membrane components and MMPs are all aspects of these mechanisms conserved in humans. Hopefully, findings made with the help of the tools of Drosophila genetics will continue to expand our basic knowledge of cancer and to guide research in other model organisms and humans.
GENE FUNCTION TECHNIQUES
In addition to the tools of clonal analysis, new techniques for the study of gene function are anticipated to impact progress in Drosophila cancer studies. Several large scale efforts have made resources available to facilitate the generation of loss-of-function phenotypes and characterization of gene function. Three collections of transgenic flies exist for induction of RNAi against virtually all genes in the fly genome under control of the GAL4-UAS system (113). Also, protein trapping screenings using transposons carrying artificial exons have yielded a number of transgenic fly lines in which endogenous proteins are fused to GFP and other tags (92). It is possible to use protein traps to generate loss-of-function phenotypes specific to the trapped protein by targeting the GFP sequence through RNAi [iGFPi (in vivo GFP interference)] (95, 109). This approach combines powerful imaging with functional genetic data. A similar strategy targets the GFP tag through ubiquitin-dependent degradation (19). Another technique that will likely revolutionize gene function studies in Drosophila is recombineering (149). Through recombineering, large fragments of Drosophila genomic DNA can be manipulated in bacteria by homologous recombination to, for example, delete a gene segment or introduce a tag. These modified DNA fragments can be used to create transgenic lines. Two collections of large genomic fragments allow these manipulations for most genes (31, 148).
SUMMARY POINTS
Genetic tools for the study of clonal behavior and gene function in Drosophila allow generation of tumors and analysis of social cell interactions.
Cell competition is a homeostatic mechanism capable of suppressing development of tumors.
Escape or reversal of cell competition leads to tumor formation.
Apoptosis or damage triggers production of signals that stimulate compensatory proliferation in surrounding cells.
Compensatory proliferation can lead to aberrant growth in a tumor context.
Mutations in different clones of cells can cooperate interclonally to produce tumors.
Degradation of the basement membrane through MMP upregulation allows metastasis and invasion in Drosophila.
Tumors in Drosophila cause an immune reaction that exerts both protumoral and antitu-moral effects.
FUTURE ISSUES
Which molecular mechanisms lead to JNK activation in different contexts and how do they connect with the Hippo/Lats growth control pathway?
How do compensatory proliferation signals and the undead cell phenomenon relate to developmental and regenerative processes?
What is the role of mechanical tension in normal and tumoral development?
What impact do systemic influences, including immune signals, hormones, and metabolic status, have in tumor development?
What is the actual contribution that phenomena discovered in Drosophila make to the development of human tumors?
ACKNOWLEDGMENTS
We apologize to those whose work we could not cite because of space constraints. Thanks are owed to Tatsushi Igaki, Jinyu Lu, and Helen Rankin for comments on the manuscript. Work in our laboratories is funded by the Howard Hughes Medical Institute, NIH grants (T.X.), and the 1000 Talents program (J.C.P-P).
Glossary
- Cell competition
context-dependent elimination of cells that are viable by themselves but die when confronted with cells of a different genotype
- Compensatory proliferation
cell division stimulated by dying or damaged cells
- Interclonal cooperation
oncogenic cooperation between mutations in different clones of cells
- Basement membrane
polymer of extracellular matrix proteins that underlies epithelia in all animals, made mostly of Collagen IV
- Genetic mosaic
situation in which somatic cells of different genotypes coexist in an organism
- Mitotic recombination
chromosomal crossover in somatic cells. Depending on how chromosomes segregate, it may produce homozygous clones of cells in a heterozygote
- Flp
flippase
- FRT
flippase recognition target
- MARCM
mosaic analysis with a repressible cell marker
- JNK
c-Jun N-terminal kinase
- TNF
tumor necrosis factor
- ELMO
engulfment and cell motility
- JAK/STAT
janus kinase/signal transducer and activator of transcription
- SPARC
secreted protein, acidic and rich in cysteine
- Supercompetition
elimination of wild-type cells by mutant cells through cell competition
- MDCK
Madin-Darby canine kidney
- RasV12
Ras valin 12 (glycine 12 to valine mutation)
- Undead cells
cells apoptotically stimulated but unable to die and capable of causing nonautonomous overgrowth by chronically stimulating compensatory proliferation
- MMP
matrix metalloprotease
- Hemocytes
insect blood cells, with a central role in immune responses as macrophages and immune regulators
Footnotes
DISCLOSURE STATEMENT The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Aegerter-Wilmsen T, Aegerter CM, Hafen E, Basler K. Model for the regulation of size in the wing imaginal disc of Drosophila. Mech. Dev. 2007;124:318–26. doi: 10.1016/j.mod.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Aldaz S, Escudero LM, Freeman M. Live imaging of Drosophila imaginal disc development. Proc. Natl. Acad. Sci. USA. 2010;107:14217–22. doi: 10.1073/pnas.1008623107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apidianakis Y, Rahme LG. Drosophila melanogaster as a model for human intestinal infection and pathology. Dis. Models Mech. 2011;4:21–30. doi: 10.1242/dmm.003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc. Natl. Acad. Sci. USA. 2006;103:13474–79. doi: 10.1073/pnas.0606053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babcock DT, Brock AR, Fish GS, Wang Y, Perrin L, et al. Circulating blood cells function as a surveillance system for damaged tissue in Drosophila larvae. Proc. Natl. Acad. Sci. USA. 2008;105:10017–22. doi: 10.1073/pnas.0709951105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baena-Lopez LA, Baonza A, Garcia-Bellido A. The orientation of cell divisions determines the shape of Drosophila organs. Curr. Biol. 2005;15:1640–44. doi: 10.1016/j.cub.2005.07.062. [DOI] [PubMed] [Google Scholar]
- 7.Bando T, Mito T, Maeda Y, Nakamura T, Ito F, et al. Regulation of leg size and shape by the Dachsous/Fat signalling pathway during regeneration. Development. 2009;136:2235–45. doi: 10.1242/dev.035204. [DOI] [PubMed] [Google Scholar]
- 8.Beaucher M, Hersperger E, Page-McCaw A, Shearn A. Metastatic ability of Drosophila tumors depends on MMP activity. Dev. Biol. 2007;303:625–34. doi: 10.1016/j.ydbio.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Beumer KJ, Pimpinelli S, Golic KG. Induced chromosomal exchange directs the segregation of recombinant chromatids in mitosis of Drosophila. Genetics. 1998;150:173–88. doi: 10.1093/genetics/150.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011;17:320–29. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bissell MJ, Radisky D. Putting tumours in context. Nat. Rev. Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biteau B, Hochmuth CE, Jasper H. JNK activity in somatic stem cells causes loss of tissue homeostasis in the aging Drosophila gut. Cell Stem Cell. 2008;3:442–55. doi: 10.1016/j.stem.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair SS. Genetic mosaic techniques for studying Drosophila development. Development. 2003;130:5065–72. doi: 10.1242/dev.00774. [DOI] [PubMed] [Google Scholar]
- 14.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–22. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boveri T, Boveri M. The Origin of Malignant Tumors. Williams & Wilkins; Baltimore, MD: 1929. [Google Scholar]
- 16.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 17.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–79. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchon N, Broderick NA, Poidevin M, Pradervand S, Lemaitre B. Drosophila intestinal response to bacterial infection: activation of host defense and stem cell proliferation. Cell Host Microbe. 2009;5:200–11. doi: 10.1016/j.chom.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Caussinus E, Kanca O, Affolter M. Fluorescent fusion protein knockout mediated by anti-GFP nanobody. Nat. Struct. Mol. Biol. 2011;19(1):117–21. doi: 10.1038/nsmb.2180. [DOI] [PubMed] [Google Scholar]
- 20.Chen CL, Schroeder MC, Kango-Singh M, Tao C, Halder G. Tumor suppression by cell competition through regulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA. 2011;109(2):484–89. doi: 10.1073/pnas.1113882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Classen AK, Bunker BD, Harvey KF, Vaccari T, Bilder D. A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat. Genet. 2009;41:1150–55. doi: 10.1038/ng.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordero JB, Macagno JP, Stefanatos RK, Strathdee KE, Cagan RL, Vidal M. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell. 2010;18:999–1011. doi: 10.1016/j.devcel.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cronin SJ, Nehme NT, Limmer S, Liegeois S, Pospisilik JA, et al. Genome-wide RNAi screen identifies genes involved in intestinal pathogenic bacterial infection. Science. 2009;325:340–43. doi: 10.1126/science.1173164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das Thakur M, Feng Y, Jagannathan R, Seppa MJ, Skeath JB, Longmore GD. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr. Biol. 2010;20:657–62. doi: 10.1016/j.cub.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–16. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 26.del Valle Rodriguez A, Didiano D, Desplan C. Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods. 2012;9:47–55. doi: 10.1038/nmeth.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 28.Doggett K, Grusche FA, Richardson HE, Brumby AM. Loss of the Drosophila cell polarity regulator Scribbled promotes epithelial tissue overgrowth and cooperation with oncogenic Ras-Raf through impaired Hippo pathway signaling. BMC Dev. Biol. 2011;11:57. doi: 10.1186/1471-213X-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 30.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ejsmont RK, Sarov M, Winkler S, Lipinski KA, Tomancak P. A toolkit for high-throughput, cross-species gene engineering in Drosophila. Nat. Methods. 2009;6:435–37. doi: 10.1038/nmeth.1334. [DOI] [PubMed] [Google Scholar]
- 32.Enomoto M, Igaki T. Deciphering tumor-suppressor signaling in flies: genetic link between Scribble/Dlg/Lgl and the Hippo pathways. J. Genet. Genomics. 2011;38:461–70. doi: 10.1016/j.jgg.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Enomoto M, Igaki T. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep. 2013;14:65–72. doi: 10.1038/embor.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev. Cell. 2008;14:399–410. doi: 10.1016/j.devcel.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez BG, Gaspar P, Bras-Pereira C, Jezowska B, Rebelo SR, Janody F. Actin-capping protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development. 2011;138:2337–46. doi: 10.1242/dev.063545. [DOI] [PubMed] [Google Scholar]
- 36.Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, Bolivar J, Gutierrez-Avino FJ, Dominguez M. Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature. 2006;439:430–36. doi: 10.1038/nature04376. [DOI] [PubMed] [Google Scholar]
- 37.Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2:E239. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Bellido A. The engrailed story. Genetics. 1998;148:539–44. doi: 10.1093/genetics/148.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Bellido A, Ripoll P, Morata G. Developmental compartmentalisation of the wing disk of Drosophila. Nat. New. Biol. 1973;245:251–53. doi: 10.1038/newbio245251a0. [DOI] [PubMed] [Google Scholar]
- 40.Gateff E, Schneiderman HA. Neoplasms in mutant and cultured wild-type tissues of Drosophila. Natl. Cancer Inst. Monogr. 1969;31:365–97. [PubMed] [Google Scholar]
- 41.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golic KG, Lindquist S. The FLP recombinase of yeast catalyzes site-specific recombination in the Drosophila genome. Cell. 1989;59:499–509. doi: 10.1016/0092-8674(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 43.Griffin R, Sustar A, Bonvin M, Binari R, del Valle Rodriguez A, et al. The twin spot generator for differential Drosophila lineage analysis. Nat. Methods. 2009;6:600–2. doi: 10.1038/nmeth.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grusche FA, Degoutin JL, Richardson HE, Harvey KF. The Salvador/Warts/Hippo pathway controls regenerative tissue growth in Drosophila melanogaster. Dev. Biol. 2011;350:255–66. doi: 10.1016/j.ydbio.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Grzeschik NA, Amin N, Secombe J, Brumby AM, Richardson HE. Abnormalities in cell proliferation and apico-basal cell polarity are separable in Drosophila lgl mutant clones in the developing eye. Dev. Biol. 2007;311:106–23. doi: 10.1016/j.ydbio.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr. Biol. 2010;20:573–81. doi: 10.1016/j.cub.2010.01.055. [DOI] [PubMed] [Google Scholar]
- 47.Hadjieconomou D, Rotkopf S, Alexandre C, Bell DM, Dickson BJ, Salecker I. Flybow: genetic multicolor cell labeling for neural circuit analysis in Drosophila melanogaster. Nat. Meth. 2011;8:260–66. doi: 10.1038/nmeth.1567. [DOI] [PubMed] [Google Scholar]
- 48.Hampel S, Chung P, McKellar CE, Hall D, Looger LL, Simpson JH. Drosophila Brainbow: a recombinase-based fluorescence labeling technique to subdivide neural expression patterns. Nat. Methods. 2011;8:253–59. doi: 10.1038/nmeth.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hariharan IK, Bilder D. Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu. Rev. Genet. 2006;40:335–61. doi: 10.1146/annurev.genet.39.073003.100738. [DOI] [PubMed] [Google Scholar]
- 50.Harshbarger JC, Taylor RL. Neoplasms of Insects. Annu. Rev. Entomol. 1968;13:159–90. [Google Scholar]
- 51.Haynie JL, Bryant PJ. The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc of Drosophila melanogaster. Dev. Genes Evol. 1977;183:85–100. doi: 10.1007/BF00848779. [DOI] [PubMed] [Google Scholar]
- 52.Herranz H, Hong X, Hung NT, Voorhoeve PM, Cohen SM. Oncogenic cooperation between SOCS family proteins and EGFR identified using a epithelial transformation model. Genes Dev. 2012;26:1602–11. doi: 10.1101/gad.192021.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herz HM, Chen Z, Scherr H, Lackey M, Bolduc C, Bergmann A. vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development. 2006;133:1871–80. doi: 10.1242/dev.02356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hogan C, Dupre-Crochet S, Norman M, Kajita M, Zimmermann C, et al. Characterization of the interface between normal and transformed epithelial cells. Nat. Cell Biol. 2009;11:460–67. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 55.Hou Y, Song L, Zhu P, Zhang B, Tao Y, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–85. doi: 10.1016/j.cell.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 56.Hufnagel L, Teleman AA, Rouault H, Cohen SM, Shraiman BI. On the mechanism of wing size determination in fly development. Proc. Natl. Acad. Sci. USA. 2007;104:3835–40. doi: 10.1073/pnas.0607134104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr. Biol. 2004;14:1262–66. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 58.Hynes RO, Zhao Q. The evolution of cell adhesion. J. Cell Biol. 2000;150:F89–96. doi: 10.1083/jcb.150.2.f89. [DOI] [PubMed] [Google Scholar]
- 59.Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009–18. doi: 10.1093/emboj/cdf306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006;16:1139–46. doi: 10.1016/j.cub.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 61.Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell. 2009;16:458–65. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that TNF is required for outcompetition of scrib and dlg mutant cells.
- 62.Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009;137:1343–55. doi: 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Z, Kirilly D, Weng C, Kawase E, Song X, et al. Differentiation-defective stem cells outcompete normal stem cells for niche occupancy in the Drosophila ovary. Cell Stem Cell. 2008;2:39–49. doi: 10.1016/j.stem.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–90. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA. 2008;105:4283–88. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kajita M, Hogan C, Harris AR, Dupre-Crochet S, Itasaki N, et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J. Cell Sci. 2010;123:171–80. doi: 10.1242/jcs.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karpowicz P, Perez J, Perrimon N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development. 2010;137:4135–45. doi: 10.1242/dev.060483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol. Cell Biol. 2006;26:7258–68. doi: 10.1128/MCB.00183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lai SL, Lee T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat. Neurosci. 2006;9:703–9. doi: 10.1038/nn1681. [DOI] [PubMed] [Google Scholar]
- 70.Lee N, Maurange C, Ringrose L, Paro R. Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature. 2005;438:234–37. doi: 10.1038/nature04120. [DOI] [PubMed] [Google Scholar]
- 71.Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–61. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 72.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 73.Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482:410–13. doi: 10.1038/nature10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li W, Baker NE. Engulfment is required for cell competition. Cell. 2007;129:1215–25. doi: 10.1016/j.cell.2007.03.054. [DOI] [PubMed] [Google Scholar]; Demonstrated that winner cells engulf loser cells during cell competition.
- 76.Li W, Kale A, Baker NE. Oriented cell division as a response to cell death and cell competition. Curr. Biol. 2009;19:1821–26. doi: 10.1016/j.cub.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Llano E, Adam G, Pendas AM, Quesada V, Sanchez LM, et al. Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J. Biol. Chem. 2002;277:23321–29. doi: 10.1074/jbc.M200121200. [DOI] [PubMed] [Google Scholar]
- 78.Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C. Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J. Biol. Chem. 2000;275:35978–85. doi: 10.1074/jbc.M006045200. [DOI] [PubMed] [Google Scholar]
- 79.Lolo FN, Casas-Tinto S, Moreno E. Cell competition time line: winners kill losers, which are extruded and engulfed by hemocytes. Cell Rep. 2012;2:526–39. doi: 10.1016/j.celrep.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 80.Lyons JG, Lobo E, Martorana AM, Myerscough MR. Clonal diversity in carcinomas: its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin. Exp. Metastasis. 2008;25:665–77. doi: 10.1007/s10585-007-9134-2. [DOI] [PubMed] [Google Scholar]
- 81.Martinek N, Shahab J, Saathoff M, Ringuette M. Haemocyte-derived SPARC is required for collagen-IV-dependent stability of basal laminae in Drosophila embryos. J. Cell Sci. 2008;121:1671–80. doi: 10.1242/jcs.021931. [DOI] [PubMed] [Google Scholar]
- 82.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta Rev. Cancer. 2010;1805:105–17. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Menendez J, Perez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc. Natl. Acad. Sci. USA. 2010;107:14651–56. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev. Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 85.Mollereau B, Perez-Garijo A, Bergmann A, Miura M, Gerlitz O, et al. Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ. 2013;20:181. doi: 10.1038/cdd.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morata G, Ripoll P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Dev. Biol. 1975;42:211–21. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- 87.Moreno E. Is cell competition relevant to cancer? Nat. Rev. Cancer. 2008;8:141–47. doi: 10.1038/nrc2252. [DOI] [PubMed] [Google Scholar]
- 88.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–29. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]; Demonstrated that wild-type cells can be outcompeted by mutant supercompetitor cells. See also References 25 and 152.
- 89.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–59. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- 90.Moreno E, Yan M, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002;12:1263–68. doi: 10.1016/s0960-9822(02)00954-5. [DOI] [PubMed] [Google Scholar]
- 91.Morgan TH, Bridges CB. The origin of gynandromorphs. In: Morgan TH, editor. Contributions to the Genetics of Drosophila Melanogaster. Carnegie Inst. Wash; Washington, DC: 1919. pp. 1–112. [Google Scholar]
- 92.Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc. Natl. Acad. Sci. USA. 2001;98:15050–55. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Neto-Silva RM, de Beco S, Johnston LA. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell. 2010;19:507–20. doi: 10.1016/j.devcel.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neumuller RA, Wirtz-Peitz F, Lee S, Kwon Y, Buckner M, et al. Stringent analysis of gene function and protein-protein interactions using fluorescently tagged genes. Genetics. 2012;190(3):931–40. doi: 10.1534/genetics.111.136465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Norman M, Wisniewska KA, Lawrenson K, Garcia-Miranda P, Tada M, et al. Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci. 2012;125:59–66. doi: 10.1242/jcs.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Novelli MR, Williamson JA, Tomlinson IPM, Elia G, Hodgson SV, et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science. 1996;272:1187–90. doi: 10.1126/science.272.5265.1187. [DOI] [PubMed] [Google Scholar]
- 98.Nystul T, Spradling A. An epithelial niche in the Drosophila ovary undergoes long-range stem cell replacement. Cell Stem Cell. 2007;1:277–85. doi: 10.1016/j.stem.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 99.Oertel M, Menthena A, Dabeva MD, Shafritz DA. Cell competition leads to a high level of normal liver reconstitution by transplanted fetal liver stem/progenitor cells. Gastroenterology. 2006;130:507–20. doi: 10.1053/j.gastro.2005.10.049. quiz 90. [DOI] [PubMed] [Google Scholar]
- 100.Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, Igaki T. Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature. 2012;490:547–51. doi: 10.1038/nature11452. [DOI] [PubMed] [Google Scholar]
- 101.Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T. Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev. Cell. 2011;20:315–28. doi: 10.1016/j.devcel.2011.02.007. [DOI] [PubMed] [Google Scholar]; Demonstrated that JNK signaling is required in winner cells to promote engulfment of loser cells.
- 102.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–20. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–79. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 104.Page-McCaw A, Serano J, Sante JM, Rubin GM. Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev. Cell. 2003;4:95–106. doi: 10.1016/s1534-5807(02)00400-8. [DOI] [PubMed] [Google Scholar]
- 105.Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–31. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- 106.Pallavi SK, Ho DM, Hicks C, Miele L, Artavanis-Tsakonas S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012;31:2895–907. doi: 10.1038/emboj.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pastor-Pareja JC, Grawe F, Martin-Blanco E, Garcia-Bellido A. Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev. Cell. 2004;7:387–99. doi: 10.1016/j.devcel.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 108.Pastor-Pareja JC, Wu M, Xu T. An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis. Models Mech. 2008;1:144–54. doi: 10.1242/dmm.000950. discuss. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that Drosophila tumors cause an innate immune reaction from blood cells. See also Reference 22.
- 109.Pastor-Pareja JC, Xu T. Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and perlecan. Dev. Cell. 2011;21:245–56. doi: 10.1016/j.devcel.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–98. doi: 10.1242/dev.01432. [DOI] [PubMed] [Google Scholar]
- 111.Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–77. doi: 10.1242/dev.034017. [DOI] [PubMed] [Google Scholar]
- 112.Herrera SC, Martín R, Morata G. Tissue homeostasis in the wing disc of Drosophila melanogaster: immediate response to massive damage during development. PLoS Genet. 2013;9(4):e1003446. doi: 10.1371/journal.pgen.1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Perrimon N, Ni JQ, Perkins L. In vivo RNAi: today and tomorrow. Cold Spring Harb. Perspect. Biol. 2010;2:a003640. doi: 10.1101/cshperspect.a003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pimpinelli S, Ripoll P. Nonrandom segregation of centromeres following mitotic recombination in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 1986;83:3900–3. doi: 10.1073/pnas.83.11.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pohar N, Godenschwege TA, Buchner E. Invertebrate tissue inhibitor of metalloproteinase: structure and nested gene organization within the synapsin locus is conserved from Drosophila to human. Genomics. 1999;57:293–96. doi: 10.1006/geno.1999.5776. [DOI] [PubMed] [Google Scholar]
- 116.Portela M, Casas-Tinto S, Rhiner C, Lopez-Gay JM, Dominguez O, et al. Drosophila SPARC is a self-protective signal expressed by loser cells during cell competition. Dev. Cell. 2010;19:562–73. doi: 10.1016/j.devcel.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 117.Potter CJ, Tasic B, Russler EV, Liang L, Luo L. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 2010;141:536–48. doi: 10.1016/j.cell.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prober DA, Edgar BA. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002;16:2286–99. doi: 10.1101/gad.991102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rauskolb C, Pan G, Reddy BV, Oh H, Irvine KD. Zyxin links fat signaling to the Hippo pathway. PLoS Biol. 2011;9:e1000624. doi: 10.1371/journal.pbio.1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Read RD, Bach EA, Cagan RL. Drosophila C-terminal Src kinase negatively regulates organ growth and cell proliferation through inhibition of the Src, Jun N-terminal kinase, and STAT pathways. Mol. Cell Biol. 2004;24:6676–89. doi: 10.1128/MCB.24.15.6676-6689.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ren F, Wang B, Yue T, Yun EY, Ip YT, Jiang J. Hippo signaling regulates Drosophila intestine stem cell proliferation through multiple pathways. Proc. Natl. Acad. Sci. USA. 2010;107:21064–69. doi: 10.1073/pnas.1012759107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Repiso A, Bergantinos C, Corominas M, Serras F. Tissue repair and regeneration in Drosophila imaginal discs. Dev. Growth Differ. 2011;53:177–85. doi: 10.1111/j.1440-169X.2010.01247.x. [DOI] [PubMed] [Google Scholar]
- 123.Rhiner C, Diaz B, Portela M, Poyatos JF, Fernandez-Ruiz I, et al. Persistent competition among stem cells and their daughters in the Drosophila ovary germline niche. Development. 2009;136:995–1006. doi: 10.1242/dev.033340. [DOI] [PubMed] [Google Scholar]
- 124.Rhiner C, Lopez-Gay JM, Soldini D, Casas-Tinto S, Martin FA, et al. Flower forms an extracellular code that reveals the fitness of a cell to its neighbors in Drosophila. Dev. Cell. 2010;18:985–98. doi: 10.1016/j.devcel.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 125.Robinson BS, Moberg KH. Drosophila endocytic neoplastic tumor suppressor genes regulate Sav/Wts/Hpo signaling and the c-Jun N-terminal kinase pathway. Cell Cycle. 2011;10:4110–18. doi: 10.4161/cc.10.23.18243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]; Demonstrated that compensatory proliferation through Dpp and Wg causes the undead cells phenomenon. See also References 32 and 111.
- 127.Sansores-Garcia L, Bossuyt W, Wada K, Yonemura S, Tao C, et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011;30:2325–35. doi: 10.1038/emboj.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scharrer B, Lochhead MS. Tumors in the invertebrates: a review. Cancer Res. 1950;10:403–19. [PubMed] [Google Scholar]
- 129.Schroeder MC, Chen CL, Gajewski K, Halder G. A non-cell-autonomous tumor suppressor role for Stat in eliminating oncogenic scribble cells. Oncogene. 2012 doi: 10.1038/onc.2012.476. doi:10.1038/onc.2012.476. [DOI] [PubMed] [Google Scholar]
- 130.Senoo-Matsuda N, Johnston LA. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc. Natl. Acad. Sci. USA. 2007;104:18543–48. doi: 10.1073/pnas.0709021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shaw RL, Kohlmaier A, Polesello C, Veelken C, Edgar BA, Tapon N. The Hippo pathway regulates intestinal stem cell proliferation during Drosophila adult midgut regeneration. Development. 2010;137:4147–58. doi: 10.1242/dev.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sheng XR, Brawley CM, Matunis EL. Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell. 2009;5:191–203. doi: 10.1016/j.stem.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shlevkov E, Morata G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2011;19:451–60. doi: 10.1038/cdd.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc. Natl. Acad. Sci. USA. 2007;104:2721–26. doi: 10.1073/pnas.0611666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Staley BK, Irvine KD. Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol. 2010;20:1580–87. doi: 10.1016/j.cub.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stark MB. A hereditary tumor in the fruit fly. J. Cancer Res. 1918;3:279–301. [Google Scholar]
- 137.Stern C. Somatic crossing over and segregation in Drosophila melanogaster. Genetics. 1936;21:625–730. doi: 10.1093/genetics/21.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–39. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stramer B, Wood W, Galko MJ, Redd MJ, Jacinto A, et al. Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J. Cell Biol. 2005;168:567–73. doi: 10.1083/jcb.200405120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun G, Irvine KD. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev. Biol. 2011;350:139–51. doi: 10.1016/j.ydbio.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that Hippo/Lats signaling inhibition by JNK is involved in regeneration and tumoral growth. See also References 20, 28, and 125.
- 141.Tamori Y, Bialucha CU, Tian AG, Kajita M, Huang YC, et al. Involvement of Lgl and Mahjong/VprBP in cell competition. PLoS Biol. 2010;8:e1000422. doi: 10.1371/journal.pbio.1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Thompson BJ, Mathieu J, Sung H-H, Loeser E, Rørth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev. Cell. 2005;9:711–20. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 143.Tyler DM, Li W, Zhuo N, Pellock B, Baker NE. Genes affecting cell competition in Drosophila. Genetics. 2007;175:643–57. doi: 10.1534/genetics.106.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006;25:5294–304. doi: 10.1038/sj.emboj.7601401. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that invasive behavior of Ras scrib tumors is mediated by upregulation of MMP expression. See also References 8 and 134.
- 145.Uhlirova M, Jasper H, Bohmann D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl. Acad. Sci. USA. 2005;102:13123–28. doi: 10.1073/pnas.0504170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell. 2005;9:687–98. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]; Demonstrated that vps25 cells promote nonautonomous growth through expression of the JAK/STAT activating Upd cytokines. See also References 53, 84, and 142.
- 147.VanZomeren-Dohm A, Sarro J, Flannery E, Duman-Scheel M. The Drosophila Netrin receptor frazzled/DCC functions as an invasive tumor suppressor. BMC Dev. Biol. 2011;11:41. doi: 10.1186/1471-213X-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, et al. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat. Methods. 2009;6:431–34. doi: 10.1038/nmeth.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–51. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- 150.Venken KJT, Simpson JH, Bellen HJ. Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron. 2011;72:202–30. doi: 10.1016/j.neuron.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vidal M, Larson DE, Cagan RL. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell. 2006;10:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 152.Vincent JP, Kolahgar G, Gagliardi M, Piddini E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell. 2011;21:366–74. doi: 10.1016/j.devcel.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr. Biol. 2006;16:1606–15. doi: 10.1016/j.cub2006.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Woodhouse E, Hersperger E, Stetler-Stevenson WG, Liotta LA, Shearn A. Increased type IV collagenase in lgl-induced invasive tumors of Drosophila. Cell Growth Differ. 1994;5:151–59. [PubMed] [Google Scholar]
- 155.Worley MI, Setiawan L, Hariharan IK. Regeneration and transdetermination in Drosophila imaginal discs. Annu. Rev. Genet. 2012;46:289–310. doi: 10.1146/annurev-genet-110711-155637. [DOI] [PubMed] [Google Scholar]
- 156.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–48. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that oncogenic cooperation can occur between mutations affecting different nearby cells (interclonal cooperation). See also Reference 100.
- 157.Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–37. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 158.Xu T, Rubin GM. The effort to make mosaic analysis a household tool. Development. 2012;139:4501–3. doi: 10.1242/dev.085183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu X, Hou Y, Yin X, Bao L, Tang A, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–95. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yao CK, Lin YQ, Ly CV, Ohyama T, Haueter CM, et al. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell. 2009;138:947–60. doi: 10.1016/j.cell.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yu HH, Chen CH, Shi L, Huang Y, Lee T. Twin-spot MARCM to reveal the developmental origin and identity of neurons. Nat. Neurosci. 2009;12:947–53. doi: 10.1038/nn.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ziosi M, Baena-Lopez LA, Grifoni D, Froldi F, Pession A, et al. dMyc functions downstream of Yorkie to promote the supercompetitive behavior of Hippo pathway mutant cells. PLoS Genet. 2010;6:e1001140. doi: 10.1371/journal.pgen.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]