

Abstract
Osteocytes establish an extensive intracellular and extracellular communication system via gap junction-coupled cell processes and canaliculi, through which cell processes pass throughout bone, and the communication system is extended to osteoblasts on the bone surface. To examine the osteocyte function, several mouse models were established. To ablate osteocytes, osteocytes death was induced by diphtheria toxin. However, any types of osteocyte death result in necrosis, because dying osteocytes are not phagocytosed by scavengers. After the rupture of cytoplasmic membrane, immunostimulatory molecules are released from lacunae to bone surface through canaliculi, and stimulate macrophages. The stimulated macrophages produce interleukin (IL)-1, IL-6, and tumor necrosis factor-alpha (TNF-α), which are the most important proinflammatory cytokines triggering inflammatory bone loss. Therefore, the osteocyte ablation results in necrosis-induced severe osteoporosis. In conditional knockout mice of gap junction protein alpha-1 (GJA1), which encodes connexin 43 in Gap junction, using dentin matrix protein 1 (DMP1) Cre transgenic mice, osteocyte apoptosis and enhanced bone resorption occur, because extracellular communication is intact. Overexpression of Bcl-2 in osteoblasts using 2.3 kb collagen type I alpha1 (COL1A1) promoter causes osteocyte apoptosis due to the severe reduction in the number of osteocyte processes, resulting in the disruption of both intracellular and extracellular communication systems. This mouse model unraveled osteocyte functions. Osteocytes negatively regulate bone mass by stimulating osteoclastogenesis and inhibiting osteoblast function in physiological condition. Osteocytes are responsible for bone loss in unloaded condition, and osteocytes augment their functions by further stimulating osteoclastogenesis and further inhibiting osteoblast function, at least partly, through the upregulation of receptor activator of nuclear factor-kappa B ligand (RANKL) in osteoblasts and Sost in osteocytes in unloaded condition.
Keywords: Bcl-2, Osteocyte, RANK ligand, Sost protein, Stress mechanical
INTRODUCTION
Bone mass is determined by the balance between the activities of osteoblasts, which form bone, and those of osteoclasts, which resorb bone. Osteoporosis, which is one of the major age-related diseases in our modern world, is caused by the unbalance of these two activities, which are influenced by diet, physical activities, hormonal status, cytokines, and clinical status, such as diabetes mellitus and glucocorticoid treatment.[1] Bone mass is increased by exercise but reduced by long-term bed rest, immobilization by nerve injury, and low gravity in space, causing disuse osteoporosis.[2]
Osteocytes, which are embedded in the bone matrix, establish an extensive intracellular and extracellular communication system via gap junction-coupled cell processes and canaliculi, through which cell processes pass throughout bone, and the communication system is extended to osteoblasts on the bone surface.[3] Osteocytes acquire oxygen, nutrition, and survival signals, and release various signals and soluble factors through the intracellular and extracellular communication system. The lacunocanalicular network formed by osteocytes is thought to be an ideal mechanosensory system and suitable for mechanotransduction, by which mechanical energy is converted into electrical and/or biochemical signals.[4,5,6,7,8,9]
1. Can we reveal the functions of osteocytes by inducing osteocyte death?
It has been very difficult to prove that osteocytes are responsible for sensing and transducing mechanical stress, because we need an animal model, in which osteocytes are deleted. By inducing osteocyte death, osteocyte functions could be examined. A typical example is osteocyte ablation by diphtheria toxin.[10] Transgenic mice expressing diphtheria toxin receptor under the control of dentin matrix protein 1 (DMP1) promoter, which directs the transgene expression to osteoblasts that are going to be embedded into bone matrix and osteocytes, showed enhanced bone resorption resulting in severe osteoporosis after injection of diphtheria toxin. However, we have to think about the special circumstance of osteocytes, which are embedded in bone.
Cells die mainly through one of three pathways, i.e., apoptosis, autophagic cell death, and necrosis, in physiological and pathological conditions.[11] If cells die through apoptosis or autophagy, the death is completed with the removal of the cells through engulfment by scavengers. In these cases, the cells are quietly removed without inflammation, because the integrity of cytoplasmic membranes is maintained when the phagocytosis occurs. In contrast, strong insults cause bioenergetic failure and rapid loss of cytoplasmic membrane integrity, which are core events of necrosis. Necrosis leads to rupture of the cytoplasmic membrane, and most of the intracellular content is released into the extracellular environment. As macrophages cannot pass through the canaliculi to engulf the dying osteocytes, apoptotic or autophagic osteocyte death will end in rupture of the cytoplasmic membrane, the process called secondary necrosis.[12] After cell rupture, immunostimulatory molecules, including the so-called damage-associated molecular pattern (DAMP) molecules, such as S100 family molecules, high-mobility group box 1 (HMGB1) protein, purine metabolites, heat-shock proteins, and uric acid, are released from lacunae through canaliculi to the bone surface and vascular channels, and facilitate the recruitment and activation of macrophages, thereby promoting the production of proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1, which are the most important proinflammatory cytokines triggering inflammatory bone loss.[13,14] Therefore, induction of osteocyte death causes drastic inflammatory reaction in whole body, which leads to strong enhancement of osteoclastogenesis and bone resorption and inhibition of bone formation (Fig. 1).[12] The effects of massive necrosis of osteocytes will not be restricted to the osteoclasts and osteoblasts, and any organs and tissues will be affected. Thus, we cannot evaluate the functions of osteocytes by inducing osteocyte death.
Fig. 1.
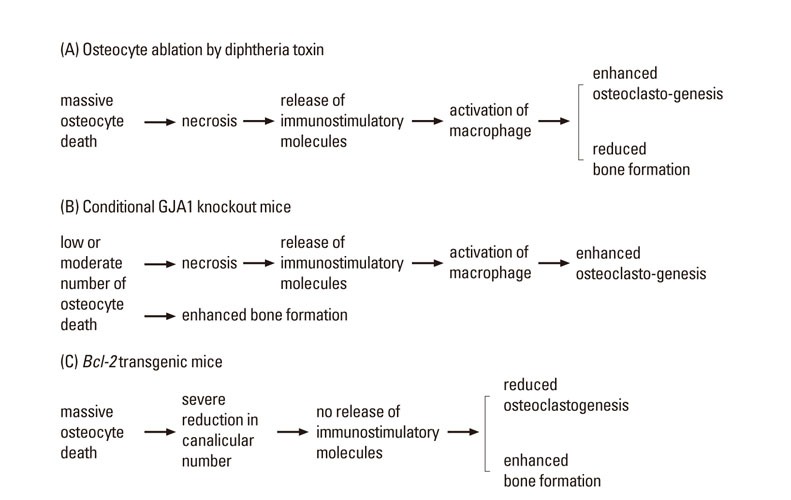
Mouse models for the evaluation of osteocyte functions. (A) Massive osteocyte necrosis enhances osteoclastogenesis and inhibits bone formation by inhibiting osteoblast maturation, resulting in severe osteoporosis. The mouse model of osteocyte ablation shows the effects of massive necrosis of osteocytes. (B) In conditional gap junction protein alpha-1 (GJA1) knockout mice, low or moderate number of osteocytes die by apoptosis. After the rupture of the cytoplasmic membrane, immunostimulatory molecules are released and osteoclastogenesis is enhanced. The increased apoptosis is associated with the enhanced bone formation. (C) In Bcl-2 transgenic mice, massive osteocyte death occurs but immunostimulatory molecules are not released due to the severe reduction in the number of canaliculi. In Bcl-2 transgenic mice, osteoclastogenesis is inhibited and bone formation is enhanced.
2. Can gap junction protein alpha-1 (GJA1) conditional knockout mice be mouse models for the evaluation of osteocyte functions?
Gap junctions, which are responsible for intracellular communication of osteocytes, are composed of GJA1 (connexin 43). In GJA1 conditional knockout mice using DMP1 Cre transgenic mice, the intracellular communication system is disrupted but the extracellular communication system through canaliculi is intact.[15] In this mouse model, osteocyte apoptosis is increased, osteoclast number and surface are increased at the endocortical surface, and the marrow cavity is enlarged. This indicates that the osteocyte apoptosis in conditional GJA1 knockout mice could induce bone resorption, probably because the intracellular content of dead osteocytes could be released through the intact canalicular network and trigger osteoclastogenesis and bone resorption (Fig. 1). Therefore, the effects of osteocyte death mask the functions of osteocytes. The response to mechanical stress was examined in three groups using conditional GJA1 knockout mice. However, the responses to mechanical stress in the GJA1 conditional knockout mice were variable. In the unloaded condition by hind limb muscle paralysis, bone resorption was enhanced in the endocortical surface of tibiae in wild-type mice but not in the conditional GJA1 knockout mice using 2.3 kb collagen type I alpha1 (COL1A1) promoter Cre transgenic mice, in which GJA1 is deleted in osteoblasts and osteocytes.[16] However, the fact that bone resorption in the endocortical surface of the conditional GJA1 knockout mice was enhanced in the physiological condition makes the evaluation difficult. Two groups reported the response to mechanical stress in the GJA1 conditional knockout mice using human osteocalcin promoter Cre transgenic mice, in which GJA1 is deleted in mature osteoblasts and osteocytes. Zhang et al.[17] showed that periosteal bone formation is enhanced by mechanical loading in the conditional GJA1 knockout mice but not in wild-type mice, while Lloyd et al.[18] showed that bone formation in both endocortical and periosteal surfaces is decreased in wild-type mice but not in the GJA1 conditional knockout mice at unloading. Therefore, further evaluation of GJA1 conditional knockout mice, including GJA1 conditional knockout mice using DMP1 Cre transgenic mice, is required to reveal the involvement of intracellular communication system in the regulation of bone mass by mechanical stress.
3. Can oeteoblast-specific Bcl-2 transgenic mice be a model mouse for the evaluation of osteocyte functions?
Unexpectedly, we found that overexpression of Bcl-2 in osteoblasts using 2.3 kb COL1A1 promoter eventually caused osteocyte apoptosis due to a reduction in the number of osteocyte processes.[19] Bcl-2 is able to form a complex with actin and gelsolin, which functions to decrease gelsolin-severing activity to increase actin polymerization, and to suppress cell adhesion, spreading, and motility.[20] Therefore, Bcl-2 seemed to alter cytoskeletal organization and reduced the number of osteoblast processes. When the osteoblasts with reduced number of processes are embedded into bone matrix and become osteocytes, the osteocytes also have a reduced number of processes, and the number of canaliculi, which the processes pass through, is also reduced. The osteocytes cannot get enough oxygen, nutrient, and survival factors through Gap junction and canaliculi and die by apoptosis.[19] Indeed, secondary necrosis occurs in these osteocytes but inflammatory reaction does not occur, because the number of canaliculi is severely reduced and immunostimulatory molecules cannot be released from lacunae to the bone surface and vascular channels (Fig. 1). Therefore, both intracellular and extracellular communication systems are disrupted in Bcl-2 transgenic mice.[12,21]
In Bcl-2 transgenic mice, osteoclastogenesis and bone resorption are reduced, indicating that osteocytes stimulate osteoclastogenesis and bone resorption in physiological condition.[21] Osteocyte death occurs during aging, after menopause, at unloading, and at pathological conditions such as microcracks, and the death of osteocytes is closely coupled with bone resorption.[22,23,24] As the death of osteocytes induces bone resorption, it has been generally considered that the physiological function of osteocytes is to inhibit bone resorption.[10,25] Therefore, it is critical to discriminate the effects of osteocyte death and the functions of osteocytes (Fig. 1).
In Bcl-2 transgenic mice, osteoblast function and bone formation are enhanced, indicating that osteocytes inhibit osteoblast function and bone formation in physiological condition.[21] The function of osteocytes in bone formation in the physiological condition has been controversial. Ablation of osteocytes by diphtheria toxin severely reduced bone formation, indicating that osteocyte death also affects bone formation (Fig. 1).[10] In contrast, osteocyte density was negatively correlated with bone formation, and both the empty lacunar density and periosteal bone apposition increase with age, suggesting a linkage between the two phenomena.[26,27,28,29,30] Mice carrying a targeted mutation of COL1A1, encoding a collagenase-resistant form of type I collagen, showed osteocyte apoptosis and increased bone formation.[31] Further, enhanced bone formation at the periosteal and endocortical surfaces is localized in the area, in which viable osteocytes are lost, in the osteocyte-specific GJA1 knockout mice (Fig. 1).[15] Therefore, many previous findings also support our conclusion that osteocytes inhibit osteoblast function and bone formation in physiological condition.
4. How do osteocytes regulate bone mass in unloaded condition?
In unloaded condition, which is obtained by tail suspension, bone mass is reduced due to the enhanced bone resorption and reduced bone formation in wild-type mice. In Bcl-2 transgenic mice, however, neither enhancement of bone resorption nor reduction of bone formation occurs and bone mass is maintained in unloaded condition, indicating that osteocyte network is responsible for bone mass regulation in unloaded condition.[21] In unloaded condition, receptor activator of nuclear factor-kappa B ligand (RANKL) expression is upregulated in osteoblasts in wild-type mice but not in Bcl-2 transgenic mice. Further, sclerostin (SOST) is locally induced at unloaded condition in osteocytes of wild-type mice but not Bcl-2 transgenic mice. Therefore, osteocyte network regulates bone mass in unloaded condition, at least in part, through the upregulation of RANKL expression in osteoblasts and upregulation of Sost expression in osteocytes.[21] RANKL and osteoprotegerin (OPG), which is a decoy receptor of RANKL, are relatively highly expressed in osteocytes.[21] RANKL is a membrane-bound protein and OPG is a secreted protein. OPG secreted by osteocytes passes through canaliculi and reaches the bone surface or vascular channels, but many OPG will be trapped by RANKL on the surface of osteocytes before reaching to the bone surface or vascular channels. As RANKL deletion using DMP1 Cre transgenic mice results in the increase of bone mass,[32,33] OPG secreted by osteocytes seems to play an important role in the regulation of bone mass in physiological condition.[12] As OPG expression in osteoblasts and osteocytes is unchanged in unloaded condition, however, OPG does not seem to be involved in the bone loss in unloaded condition.[21]
CONCLUSION
Osteocyte functions were estimated by events caused by osteocyte death, and osteocytes have been generally considered to inhibit bone resorption (Fig. 1). Using Bcl-2 transgenic mice, in which both intracellular and extracellular communication systems are disrupted, osteocyte functions have been clarified. Osteocytes stimulate osteoclastogenesis and bone resorption and inhibit osteoblast function and bone formation in physiological condition (Fig. 1). It has been also proven that osteocyte network is responsible for bone mass regulation in unloaded condition using Bcl-2 transgenic mice. Osteocyte network regulates bone mass, at least in part, through the upregulation of RANKL expression in osteoblasts and upregulation of Sost expression in osteocytes in unloaded condition. In exercise, the osteocyte network decreases the inhibitory effects on bone mass by reducing the stimulatory effect on osteoclastogenesis and the inhibitory effect on osteoblast function, leading to an increase in bone mass. Indeed, additional mechanisms will be involved in the increase of bone mass by exercise and they remained to be clarified.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bikle DD, Halloran BP. The response of bone to unloading. J Bone Miner Metab. 1999;17:233–244. doi: 10.1007/s007740050090. [DOI] [PubMed] [Google Scholar]
- 3.Marks SC, Odgren PR. Structure and development of the skeleton. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of bone biology. New York, NY: Academic Press; 2002. pp. 3–15. [Google Scholar]
- 4.Martin RB. Does osteocyte formation cause the nonlinear refilling of osteons? Bone. 2000;26:71–78. doi: 10.1016/s8756-3282(99)00242-2. [DOI] [PubMed] [Google Scholar]
- 5.Ehrlich PJ, Lanyon LE. Mechanical strain and bone cell function: a review. Osteoporos Int. 2002;13:688–700. doi: 10.1007/s001980200095. [DOI] [PubMed] [Google Scholar]
- 6.Knothe Tate ML. "Whither flows the fluid in bone?" An osteocyte's perspective. J Biomech. 2003;36:1409–1424. doi: 10.1016/s0021-9290(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 7.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–615. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noble BS. The osteocyte lineage. Arch Biochem Biophys. 2008;473:106–111. doi: 10.1016/j.abb.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Burger EH, Klein-Nulend J. Mechanotransduction in bone--role of the lacuno-canalicular network. FASEB J. 1999;13:S101–S112. [PubMed] [Google Scholar]
- 10.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 12.Komori T. Functions of the osteocyte network in the regulation of bone mass. Cell Tissue Res. 2013;352:191–198. doi: 10.1007/s00441-012-1546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 14.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 15.Bivi N, Condon KW, Allen MR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. 2012;27:374–389. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimston SK, Goldberg DB, Watkins M, et al. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26:2151–2160. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Paul EM, Sathyendra V, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd SA, Lewis GS, Zhang Y, et al. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res. 2012;27:2359–2372. doi: 10.1002/jbmr.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moriishi T, Maruyama Z, Fukuyama R, et al. Overexpression of Bcl2 in osteoblasts inhibits osteoblast differentiation and induces osteocyte apoptosis. PLoS One. 2011;6:e27487. doi: 10.1371/journal.pone.0027487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ke H, Parron VI, Reece J, et al. BCL2 inhibits cell adhesion, spreading, and motility by enhancing actin polymerization. Cell Res. 2010;20:458–469. doi: 10.1038/cr.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriishi T, Fukuyama R, Ito M, et al. Osteocyte network; a negative regulatory system for bone mass augmented by the induction of Rankl in osteoblasts and Sost in osteocytes at unloading. PLoS One. 2012;7:e40143. doi: 10.1371/journal.pone.0040143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble BS, Reeve J. Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol. 2000;159:7–13. doi: 10.1016/s0303-7207(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 23.Cardoso L, Herman BC, Verborgt O, et al. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emerton KB, Hu B, Woo AA, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46:577–583. doi: 10.1016/j.bone.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu G, Mulari M, Peng Z, et al. Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Biochem Biophys Res Commun. 2005;335:1095–1101. doi: 10.1016/j.bbrc.2005.06.211. [DOI] [PubMed] [Google Scholar]
- 26.Qiu S, Rao DS, Palnitkar S, et al. Relationships between osteocyte density and bone formation rate in human cancellous bone. Bone. 2002;31:709–711. doi: 10.1016/s8756-3282(02)00907-9. [DOI] [PubMed] [Google Scholar]
- 27.Metz LN, Martin RB, Turner AS. Histomorphometric analysis of the effects of osteocyte density on osteonal morphology and remodeling. Bone. 2003;33:753–759. doi: 10.1016/s8756-3282(03)00245-x. [DOI] [PubMed] [Google Scholar]
- 28.Lazenby RA. Continuing periosteal apposition. II: The significance of peak bone mass, strain equilibrium, and age-related activity differentials for mechanical compensation in human tubular bones. Am J Phys Anthropol. 1990;82:473–484. doi: 10.1002/ajpa.1330820408. [DOI] [PubMed] [Google Scholar]
- 29.Russo CR, Lauretani F, Seeman E, et al. Structural adaptations to bone loss in aging men and women. Bone. 2006;38:112–118. doi: 10.1016/j.bone.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 30.Hedgecock NL, Hadi T, Chen AA, et al. Quantitative regional associations between remodeling, modeling, and osteocyte apoptosis and density in rabbit tibial midshafts. Bone. 2007;40:627–637. doi: 10.1016/j.bone.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Zhao W, Byrne MH, Wang Y, et al. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest. 2000;106:941–949. doi: 10.1172/JCI10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 33.Xiong J, Onal M, Jilka RL, et al. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]