

Abstract
Aneurysms-osteoarthritis syndrome (AOS) caused by haploinsufficiency of SMAD3 is a recently described cause of syndromic familial thoracic aortic aneurysm and dissection (TAAD). We identified a novel SMAD3 mutation in a patient with hypoplastic left heart syndrome (HLHS) who developed progressive aortic aneurysm requiring surgical replacement of the neoaortic root, ascending aorta, and proximal aortic arch. Family screening for the mutation revealed that his father, who has vascular and skeletal features of AOS, and his brother, who is asymptomatic, also have the pathogenic mutation. This is the first case report of a SMAD3 mutation in a patient with hypoplastic left heart syndrome. This case highlights the importance of genetic testing for known causes of aneurysm in patients with congenital heart disease who develop aneurysmal disease as it may significantly impact the management of those patients and their family members.
1. Introduction
Familial thoracic aortic aneurysm can be divided into syndromic and nonsyndromic forms. While abdominal aortic aneurysm generally occurs sporadically, thoracic aortic aneurysm and dissection (TAAD) is inherited in an autosomal dominant manner with decreased penetrance and variable expression [1]. The genes causing syndromic and nonsyndromic forms of TAAD encode proteins that compose the structural components associated with connective tissue, key members of the TGF-β signaling pathway, or components of the contractile unit of smooth muscle cells. The genetic etiology of nonsyndromic causes of familial TAAD is largely unknown; however, several genes including, MYH11, ACTA2, and MYLK have been implicated [2–4]. The genetic cause of syndromic forms of TAAD include FBN1, the cause of Marfan syndrome, SLC2A10, the cause of arterial tortuosity syndrome, and TGFβR1, TGFβR2, and the recent identification of TGFβ2, all of which cause Loeys-Dietz syndrome [5–9].
In 2011 SMAD3, was shown to cause a new syndromic form of thoracic aortic aneurysm and dissection. The features of this condition included early onset osteoarthritis in the majority of patients and the authors proposed the name aneurysms-osteoarthritis syndrome (AOS) [10]. In addition to aneurysm and dissection, early osteoarthritis, and other systemic findings, congenital heart disease including persistent ductus arteriosus, atrial septal defect, pulmonary valve stenosis, atrial fibrillation, and bicuspid aortic valve have also been observed in patients with defects in SMAD3 [11]. SMAD3 encodes an intracellular member of the TGF-β signaling pathway that activates or represses gene transcription. Heterozygous mutations in SMAD3 lead to increased expression of several components of the TGF-β pathway including phosphorylated SMAD2, total SMAD3, TGFβ1, and connective tissue growth factor [10].
We present a 14-year-old boy born with hypoplastic left heart syndrome (HLHS) who developed significant aneurysm of the neoaorta and proximal arch after completed, staged palliation and who was found to have a novel, pathogenic SMAD3 mutation. Further testing in the family revealed additional at risk family members and who were offered appropriate cardiovascular and orthopedic screening.
2. Case Presentation
The proband, a 14-year-old male, was conceived via in vitro fertilization secondary to infertility and was diagnosed prenatally with hypoplastic left heart syndrome with mitral valve hypoplasia and aortic valve stenosis. He underwent palliative staged reconstruction including modified Fontan procedure. He developed significant aneurysmal dilatation of his reconstructed ascending aorta and neoaortic root and regurgitation of his native pulmonic valve. He underwent partial replacement of his neoaortic root with a 24 mm Hemashield graft at an outside institution. Due to significant residual neoaortic regurgitation, he underwent neoaortic valve replacement with a 25 mm On-X prosthetic. Subsequently, over a few years, he exhibited progressive dilatation of the remaining neoaorta reaching a maximal dimension of 5.2 cm. He presented to the emergency room with history of severe chest pain radiating to the neck following exercise. Computed tomography scan ruled out the presence of an aneurysmal rupture or dissection but revealed a fusiform aneurysmal dilatation of the ascending aorta with greatest dimensions of the ascending aorta of 4.1 cm (AP) × 4.4 cm (RL) (Figures 1(a) and 1(b)). He underwent aortic root, ascending aorta, and partial transverse aortic arch replacement with a 26 mm woven Dacron (Hemashield) graft, reimplantation of the mechanical prosthesis into the graft as well as reimplantation of the proximal native ascending aorta on the side of the graft. Microscopic examination of the resected ascending aorta and native pulmonary artery showed elastic artery with mucopolysaccharide rich areas and fibrosis with fragmentation and disorganization of the elastic fibers. Additionally, the aorta showed dystrophic calcification (Figures 2(a) and 2(b)).
Figure 1.
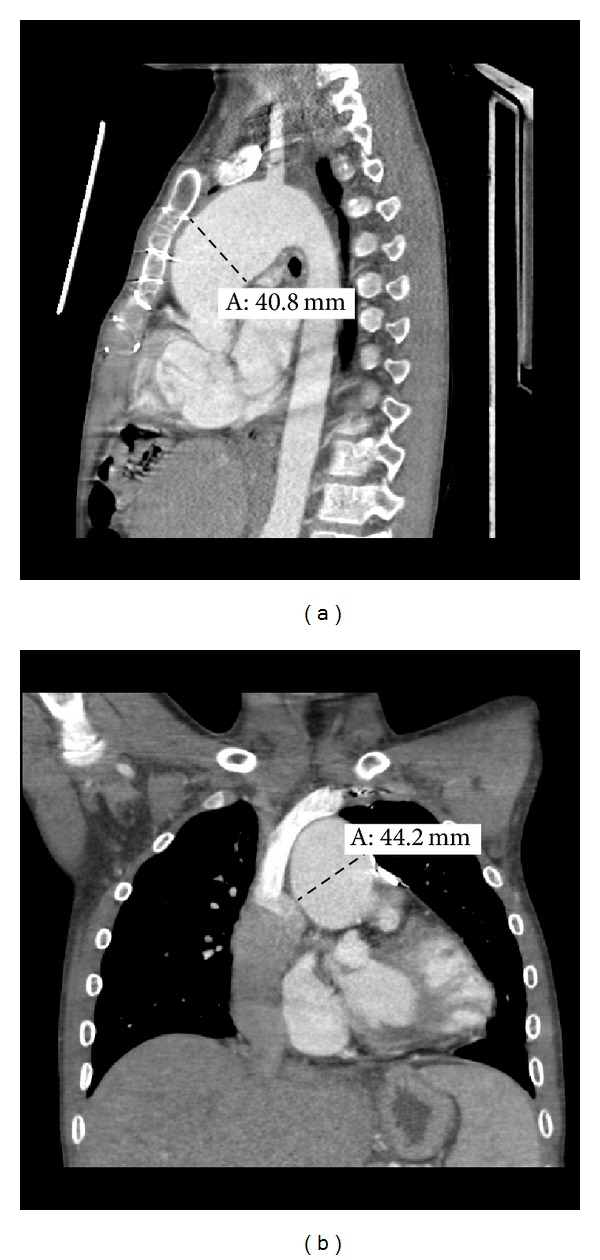
Optiray contrast-enhanced computed tomography images of the chest with sagittal (a) and coronal (b) reconstructions in the 14-year-old proband revealing aneurysmal dilation of the ascending aorta.
Figure 2.
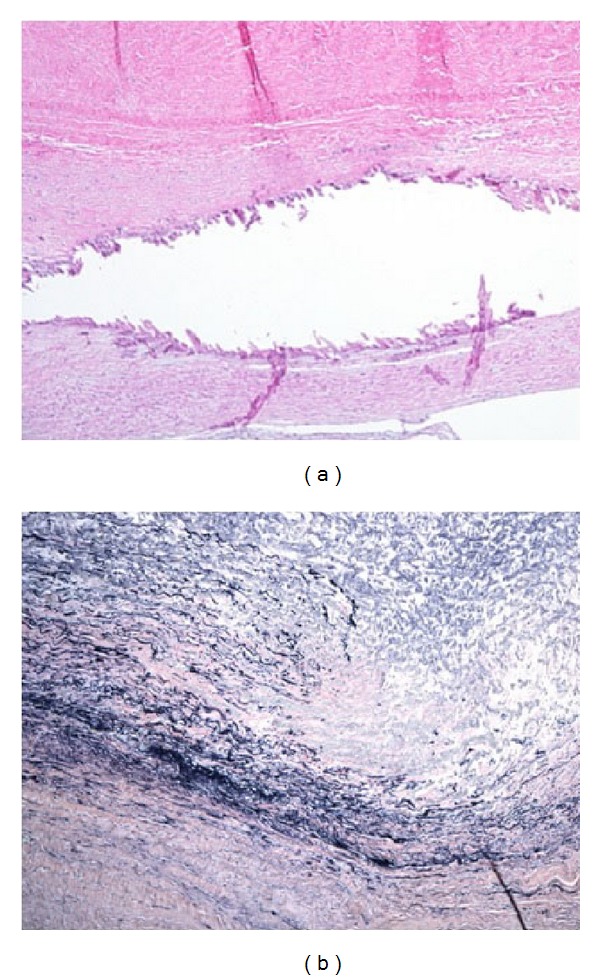
(a) Ascending aorta: medial fibrosis and dystrophic calcification (Hematoxylin and Eosin) and (b) fragmentation and disorganization of the elastic fibers (Verhoeff Van Gieson's elastic stain).
The proband underwent sequencing of genes known to cause thoracic aortic aneurysm and dissection. Next generation sequencing followed by Sanger sequencing revealed that the proband has a novel, pathogenic SMAD3 mutation, c.3G>A (p. Met1Ile) (Figure 3). First degree relatives were screened for the SMAD3 mutation. The proband's 46-year-old father and 9-year-old brother were also found to have the SMAD3 c.3G>A mutation. The affected family members underwent transthoracic echocardiography and magnetic resonance angiography (MRA) of the brain, neck, chest, abdomen, and pelvis; the proband's MRA was normal. The father, now 46 years, has intervertebral disc degeneration of his cervical spine and osteoarthritis of his knee. Echocardiogram revealed aortic root dilation with a diameter of 4.0 cm and mitral valve prolapse with mild to moderate mitral regurgitation. MRAs of the neck, chest, abdomen, and pelvis were normal. The proband's brother had a normal echocardiogram and total body MRA. His past medical history is significant for left sided inguinal hernia requiring surgical repair. Of note, the proband has no evidence of osteoarthritis. Additional screening for the SMAD3 mutation in extended relatives was recommended.
Figure 3.
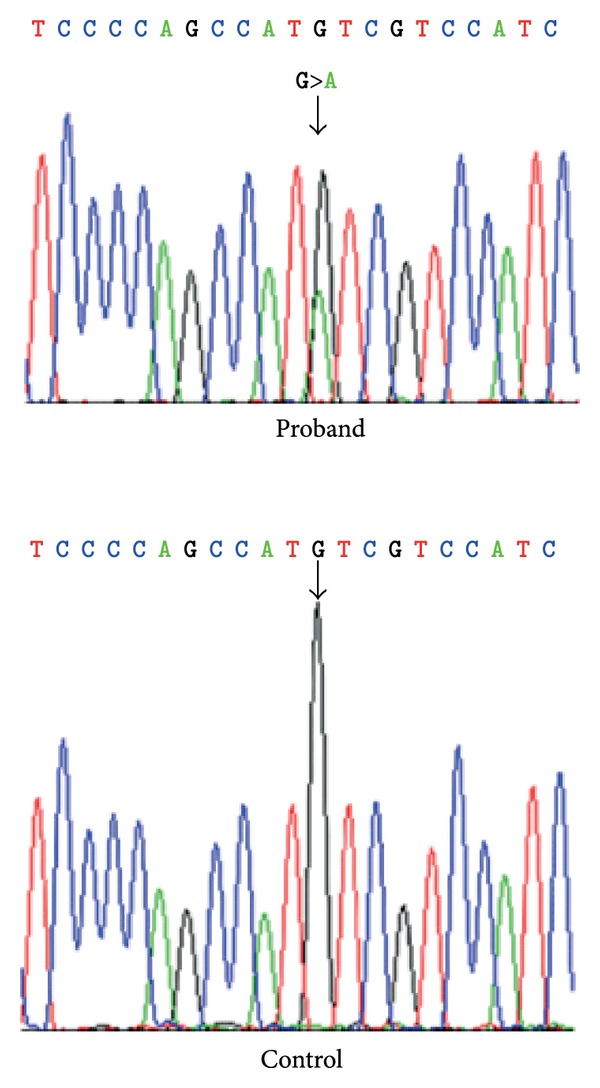
Sanger sequencing chromatogram revealing the pathogenic SMAD3 mutation, c.3G>A (p. Met1Ile) in the proband versus the normal sequence in a control.
3. Discussion
In this report, we describe a proband with HLHS who developed significant aortic aneurysm in whom clinical sequencing for genetic causes of TAAD revealed a pathogenic mutation in SMAD3. Testing of the proband's family members lead to identification of a serious health risk for thus far two additional family members. Identification of the SMAD3 mutation in the proband's father also explains his orthopedic complications. While other congenital heart defects such as persistent ductus arteriosus, atrial septal defect, and pulmonary valve stenosis have been observed in patients with SMAD3 mutations, to our knowledge, this is the first case reported with HLHS.
The findings in this family are consistent with the phenotype previously described in patients with AOS. The majority of previously described mutations in SMAD3 were observed in families with TAAD, implying a bias towards an aortic aneurysm and dissection phenotype [10, 11]. The patients in this family were ascertained only after screening in the proband, who developed significant neoaortic aneurysm following extensive cardiac surgical history for his congenital heart defect.
The phenotype of AOS in this family is age dependent with the 46-year-old father exhibiting both vascular and skeletal features while the 9-year-old brother has neither. This is consistent with many previous reports; however, a recent study by Wischmeijer et al. reported pathologic aortic dilation in a patient as young as 12 months [12]. The report by Hilhorst-Hofstee et al. of an unanticipated copy number variant of chromosome 15 disrupting SMAD3 revealed a three generation family at risk for aortic dissection. This included the 12-year-old proband initially investigated for mild mental retardation and a 4-year-old asymptomatic cousin. The two children in this family had the abnormal feature of ascending aorta wider than the sinotubular junction; the authors queried if this may be the first sign of future dilation [13]. In our case, the proband's 9-year-old brother (whose transthoracic echocardiogram and MRA were normal) also had an ascending aorta wider than the sinotubular junction (ascending aorta = 2.4 cm, sinotubular junction = 2.2 cm).
Aortic dilations in congenital aortic diseases such as bicuspid aortic valve and coarctation of the aorta are well described, and of the cyanotic congenital heart diseases, Tetralogy of Fallot has been the most widely studied with dilation at the annulus and sinus occurring in 88% and 87% of patients respectively [14]. Neoaortic root dilation has also been observed in other CHD's including transposition of the great arteries following aortic switch operation, [15, 16] following Ross procedure for congenital aortic valve disease, [17] and truncus arteriosus [18]. Specific to our case, neoaortic root dilation and valve regurgitation were reported in a group of patients with HLHS following staged palliation by Cohen et al. [19] in their cohort of 53 patients; aortic root dilation and aortic valve regurgitation were observed in 98% and 61%, respectively. Transection of the main pulmonary artery followed by reconstruction, exposing the neoaortic root (native pulmonary root) at systemic pressure, is the common denominator for all surgical interventions where the pulmonary valve becomes the neoaortic valve. It has been suggested that under these circumstances, aortic dilatation could be a result of blood flow to the vessel wall being compromised [19].
While neoaortic root dilation and aortic valve regurgitation are commonly observed in HLHS patients following staged palliation, the degree of aneurysm observed in our patient is not commonly observed in this patient cohort. Additionally, significant aneurysm formation leading to dissection is not typical based on the literature. Aortic dissection was reported in one 26-year-old male with HLHS where the aortic root measured 7.8 cm by transthoracic echocardiogram [20]. Additional information regarding the phenotype of this patient or his family history was not reported.
The phenotype of the proband and his father is consistent with what has been described thus far for the aneurysms-osteoarthritis syndrome, which leads us to conclude that the c.3G>A mutation in SMAD3 found in this family is pathogenic and is an explanation for their clinical findings. Identification of AOS in this family allowed for the screening of at risk family members, identification of early vascular disease, and it offers an explanation for the aneurysmal history in the proband and degenerative disk disease and arthritic history in his father. This case highlights the importance of screening patients with congenital heart defects for genetic causes of known aneurysmal disease.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Milewicz DM, Chen H, Park E-S, et al. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. American Journal of Cardiology. 1998;82(4):474–479. doi: 10.1016/s0002-9149(98)00364-6. [DOI] [PubMed] [Google Scholar]
- 2.Zhu L, Vranckx R, Van Kien PK, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nature Genetics. 2006;38(3):343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 3.Guo D-C, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genetics. 2007;39(12):1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Guo D-C, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. American Journal of Human Genetics. 2010;87(5):701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 6.Coucke PJ, Willaert A, Wessels MW, et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nature Genetics. 2006;38(4):452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 7.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 8.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nature Genetics. 2012;44(8):916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature Genetics. 2012;44(8):922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Laar IMBH, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nature Genetics. 2011;43(2):121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 11.van de Laar IMBH, van der Linde D, Oei EHG, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. Journal of Medical Genetics. 2012;49(1):47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 12.Wischmeijer A, Van Lear L, Tortora G, et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. American Journal of Medical Genetics Part A. 2013;161(5):1028–1035. doi: 10.1002/ajmg.a.35852. [DOI] [PubMed] [Google Scholar]
- 13.Hilhorst-Hofstee Y, Scholte AJ, Rijlaarsdam ME, et al. An unanticipated copy number variant of chromosome 15 disrupting SMAD3 reveals a three-generation family at serious risk for aortic dissection. Clinical Genetics. 2013;83(4):337–344. doi: 10.1111/j.1399-0004.2012.01931.x. [DOI] [PubMed] [Google Scholar]
- 14.Chong W-Y, Wong WHS, Chiu CSW, Cheung Y-F. Aortic root dilation and aortic elastic properties in children after repair of tetralogy of Fallot. American Journal of Cardiology. 2006;97(6):905–909. doi: 10.1016/j.amjcard.2005.09.141. [DOI] [PubMed] [Google Scholar]
- 15.Hourihan M, Colan SD, Wernovsky G, Maheswari U, Mayer JE, Jr., Sanders SP. Growth of the aortic anastomosis, annulus, and root after the arterial switch procedure performed in infancy. Circulation. 1993;88(2):615–620. doi: 10.1161/01.cir.88.2.615. [DOI] [PubMed] [Google Scholar]
- 16.Marino BS, Wernovsky G, McElhinney DB, et al. Neo-aortic valvar function after the arterial switch. Cardiology in the Young. 2006;16(5):481–489. doi: 10.1017/S1047951106000953. [DOI] [PubMed] [Google Scholar]
- 17.Elkins RC, Thompson DM, Lane MM, Elkins CC, Peyton MD. Ross operation: 16-year experience. Journal of Thoracic and Cardiovascular Surgery. 2008;136(3):623–630. doi: 10.1016/j.jtcvs.2008.02.080. [DOI] [PubMed] [Google Scholar]
- 18.Carlo WF, Mckenzie ED, Slesnick TC. Root dilation in patients with truncus arteriosus. Congenital Heart Disease. 2011;6(3):228–233. doi: 10.1111/j.1747-0803.2011.00520.x. [DOI] [PubMed] [Google Scholar]
- 19.Cohen MS, Marino BS, McElhinney DB, et al. Neo-aortic root dilation and valve regurgitation up to 21 years after staged reconstruction for hypoplastic left heart syndrome. Journal of the American College of Cardiology. 2003;42(3):533–540. doi: 10.1016/s0735-1097(03)00715-0. [DOI] [PubMed] [Google Scholar]
- 20.Egan M, Phillips A, Cook SC. Aortic dissection in the adult fontan with aortic root enlargement. Pediatric Cardiology. 2009;30(4):562–563. doi: 10.1007/s00246-009-9435-0. [DOI] [PubMed] [Google Scholar]