

Significance
The high photostability of single nucleobases is related to the rapid disposal of the UV excitation energy from high-lying electronic states into heat, preventing damaging reactions. However, in the biological important DNA strands, further long-living excited states are found. With femtosecond vibrational spectroscopy, these excited states in DNA are now identified as charge-separated states, which are delocalized along the strand. The charge separation is directed by the redox potential of the involved bases and is thus encoded in the DNA sequence. The presence of delocalized charged species in DNA strands for a considerably long time after UV light absorption may lead to reactions—oxidative or reductive damage—currently not considered in DNA photochemistry.
Keywords: DNA photophysics, DNA damage, DNA electron transfer, ultrafast vibrational spectroscopy
Abstract
Base stacking in DNA is related to long-living excited states whose molecular nature is still under debate. To elucidate the molecular background we study well-defined oligonucleotides with natural bases, which allow selective UV excitation of one single base in the strand. IR probing in the picosecond regime enables us to dissect the contribution of different single bases to the excited state. All investigated oligonucleotides show long-living states on the 100-ps time scale, which are not observable in a mixture of single bases. The fraction of these states is well correlated with the stacking probabilities and reaches values up to 0.4. The long-living states show characteristic absorbance bands that can be assigned to charge-transfer states by comparing them to marker bands of radical cation and anion spectra. The charge separation is directed by the redox potential of the involved bases and thus controlled by the sequence. The spatial dimension of this charge separation was investigated in longer oligonucleotides, where bridging sequences separate the excited base from a sensor base with a characteristic marker band. After excitation we observe a bleach of all involved bases. The contribution of the sensor base is observable even if the bridge is composed of several bases. This result can be explained by a charge delocalization along a well-stacked domain in the strand. The presence of charged radicals in DNA strands after light absorption may cause reactions—oxidative or reductive damage—currently not considered in DNA photochemistry.
DNA photophysics is crucial for the understanding of light-induced damage of the genetic code (1). The excited state of single DNA bases is known to decay extremely fast on the subpicosecond time scale, predominantly via internal conversion (2, 3). This ultrafast decay is assumed to suppress destructive decay channels, thereby protecting the DNA from photodamage and avoiding disintegration of the genetic information. In contrast to this ultrafast deactivation of single nucleobases, the biological relevant DNA strands show further long-living states (4, 5). Several explanations for these long-living states and the size of their spatial extent have been discussed in the literature (5–9). Delocalized excitons (9); excitons that decay to charge-separated states or neutral excimer states (10, 11); exciplexes located on two neighboring bases (5, 8, 12, 13); or even excited single bases, where steric interactions in the DNA strand impedes the ultrafast decay (14), have been proposed. Further computations suggest a decay of an initially populated delocalized exciton to localized neutral or charged excimer states (15–17). However, to our knowledge, a final understanding of the nature of these long-living states has not been reached. Related experiments were performed in the last decade to investigate charge transport processes in DNA, motivated by DNA electronics and oxidative damage (18, 19). Charge transport was initiated by photoexcitation of modified DNA bases or chromophores and followed by transient absorption (20–23). The transport mechanism was described by charge-hopping, superexchange, or transfer of charge along delocalized domains in DNA (18).
Until now, most experimental investigations of the long-living state were performed with transient absorption spectroscopy in the UV-visible (UV/Vis) regime (5, 9, 12) or with time-resolved fluorescence (10, 24, 25). Due to the broad, featureless, and overlapping absorption bands of the different DNA bases in this spectral region, it is difficult to investigate the molecular origin of the long-living states using these methods. A further drawback is the unselective and simultaneous excitation of several bases used in most experiments. To circumvent these problems, we used for the present study well-defined oligonucleotides, which enable selective excitation of one single base. Observation of the long-living excited states was performed via time-resolved IR spectroscopy, which can profit from the many “fingerprint” vibrational bands (26, 27). IR spectroscopy is able to distinguish between different DNA bases and their molecular states. It can also reveal changes in the electronic structure and identify charge-separated states.
In this study we used single-stranded DNA, in which π stacking between neighboring bases leads to structured domains, similar to the structure in a double helix (28). This interaction is known to be crucial for the long-living states (5). The investigation of single-stranded DNA enables us to construct special sequences, where only one base can be selectively excited. We used the natural bases 2′-deoxyuridine (U), 2′-deoxyadenosine (A), 5-methyl-2′-deoxycytidine (mC), and 2′-deoxyguanosine (G). The nucleobase U occurs naturally in RNA and is similar to the DNA base thymine but shows a blue-shifted absorbance spectrum. mC occurs with a frequency of 4–5% in mammalian DNA (29) and plays an important role as an epigenetic marker (30). The UV/Vis absorbance of mC and G are red-shifted in comparison with A and U, which allows selective excitation at 295 nm in oligonucleotides consisting of mC, A, and U (Fig. 1 A and B) or G and A. This selectivity can only be obtained in single-stranded DNA because G and its complementary base mC have overlapping absorbance bands in the UV range (Fig. S1). Selectivity in probing is based on the significant differences in the IR-absorption spectra of these bases, which display distinct marker bands for each base (Fig. 1 A and E).
Fig. 1.
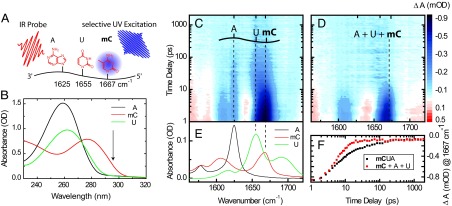
Selective excitation of mC in mCUA and probing of characteristic A, U, and mC marker bands in the IR. (A, B, and E) Picosecond UV light pulses at 295 nm allow selective excitation of mC (shown in bold) in mixed DNA sequences consisting of mC, U, and A. (B) Absorbance spectra of 2′-deoxyadenosine monophosphate (A), 2′-deoxy-5-methylcytidine (mC), and uridine monophosphate (U). (C) Time-resolved absorption difference (color-coded) plotted vs. wavenumber and delay time for mCUA and (D) for a mixture of the corresponding monomers. (E) Probing the individual contribution of each base is possible in the IR at 1,625 cm−1 (A), 1,655 cm−1 (U), and 1,667 cm−1 (mC) (marked by dashed lines). (F) Transients at 1,667 cm−1 for mCUA and the mixture of monomers.
With the combination of selective excitation and selective probing we are able to elucidate the nature of the long-living states in DNA strands. Investigation of dinucleotides clearly shows that light absorption in DNA leads to charge separation between stacked neighboring bases, which recombine on the 100-ps time scale. In longer oligonucleotides we observe simultaneous bleach of several bases, which points to a delocalization of the charges along the strand. Our results show that charge transfer in DNA is a natural process, induced by UV-light absorption of DNA.
Results and Discussion
In a first example, the trimer mCUA is used to demonstrate the feasibility of the approach. In Fig. 1C, the evolution of the absorption transients in the IR is plotted vs. wavenumber and delay time between UV excitation and IR probing pulses. After selective excitation of mC with light at 295 nm, two processes are evident: an ultrafast decay within 10 ps and a much slower process on the 100-ps time scale. The fast process can be assigned to the decay of the excited electronic state (S1) of mC (31) with concomitant vibrational cooling of the molecule. These fast dynamics resemble those found in a reference measurement of a solution containing the monomers mC, A, and U (Fig. 1 D and F). In this solution, slower transients are not observed. The slower process is only found in the trimeric sample; its absorption change clearly shows contributions of all three bases, although only the mC has initially been excited. Apparently, the slow component is the consequence of the connection of the bases in the trinucleotide. The time constant in the 100-ps range agrees well with the dynamics found in previous investigations with UV probing (5).
A global fit (Materials and Methods) confirms the qualitative view given above. The ultrafast decay of the excited electronic state and the vibrational cooling are represented by the two fast components with time constants in the one picosecond (τ0) and 5- to 10-ps range (τ1). Only in the oligonucleotide an additional kinetic process in the 100-ps range (τ2 = 90 ps) is evident. The decay-associated difference spectrum D2(ν) related to τ2 contains the spectral information on the long-living species; it gives an insight into the molecular nature of the state (see below) and allows us to estimate the fraction F2 of molecules involved.
Amplitude of the Long-Living State.
The fraction F2 of the long-lived state is shown in Fig. 2 for different DNA oligomers. Values of F2 from 0.23 to over 0.4 have been found (Table 1). These large numbers show that the long-lived states may play a significant role in the photochemical reactions of DNA oligomers. In detail, the fraction F2 depends on the specific sequence of the compound. Longer oligonucleotides show larger values of F2 than shorter ones. Adenine in direct neighborhood of the excited mC results in a higher population of the long-living state in comparison with U (Fig. 2A). Even inverting the sequence has a major influence: adenine at the 5′ end shows a much higher fraction F2 than at the 3′ end (Fig. 2B). These observations are in line with the assumption that base stacking is the prerequisite for the appearance of the long-living component. Indeed, Kohler and coworkers (12) have shown for dinucleotides that the amplitude of the long-living state detected in UV experiments correlates well with stacking properties. In addition, CD spectroscopy (32) shows that the order of the stacking probability α of dinucleotides follows the trend αCU < αCA < αAC, which correlates exactly with the order of F2 in our study. Furthermore, the stacking probability is rising with the increasing length (8) of the DNA strand, which is also observed in our data. Apparently, the long-living state is only formed between stacked bases, whereas unstacked bases show the reported ultrafast deactivation known from single bases.
Fig. 2.

Comparison of the fraction of the long-living state in different oligonucleotides. Fraction F2 of molecules involved in the long-living state for different oligonucleotides (mC is excited, shown in bold). The values F2 are obtained from two independent experiments, which show the same trend. (A) Dependency on the type of the base neighboring to the excited mC and on the length of the oligonucleotides. (B) Dependency on the sequence of trinucleotides.
Table 1.
Fitting parameters and fraction F2 of the long-living state
| Oligonucleotide | τ0/ps | τ1/ps | τ2/ps | Fraction F2 |
| mC + A + U | 1.2 | 5 | ||
| mCA | 1.3 | 6 | 110 | 0.25 |
| mCU | 1.2 | 6 | 50 | 0.23 |
| mCG | 1.5 | 3 | 20 | |
| GA | 1.7 | 3 | 300 | |
| mCAU | 0.7 | 6 | 120 | 0.29 |
| mCUA | 1.1 | 6 | 90 | 0.24 |
| AmCU | 1.4 | 10 | 150 | 0.45 |
| UmCA | 0.7 | 8 | 110 | 0.32 |
| mCAUUUU | 1.3 | 6 | 170 | 0.34 |
| mCUAUUU | 1.4 | 8 | 120 | 0.27 |
| mCUUAUU | 1.6 | 5 | 110 | 0.32 |
| mCUUUAU | 2.0 | 5 | 90 | 0.30 |
| mCUUUUA | 1.7 | 6 | 100 | 0.28 |
| mCUUUU | 1.3 | 6 | 80 | 0.24 |
All experiments were measured in two independent experiments; time constants are an average of both experiments. Selective excited base is shown in bold. For samples with selective excitation of mC, the fraction F2 was calculated. The trend of F2 is reproduced in the two independent experiments.
Characterization of Marker Bands.
From different simple dinucleotides we obtain detailed information on the molecular properties of the long-living state via the individual decay spectra D2(ν) (Fig. 3B). Negative bands in these spectra represent the decrease of the original absorption of the bases involved in the long-living state; they compare well with the stationary absorption spectra of the corresponding dimers (dashed lines) and show that both bases of the dimer contribute to D2(ν). Positive features reflect newly formed absorption bands of the long-living state. For a possible interpretation of the nature of these states we present in Fig. 3A experimental difference spectra of G and its radical cation G∙+ (33, 34) as well as mC and its radical cation mC∙+ (35). Both spectra were obtained by two photon ionization of the bases (for details, see Materials and Methods) and show characteristic marker bands (Fig. 3A). A∙+ does not possess any characteristic absorbance band in this spectral region. The G∙+ marker bands at 1,608 cm−1 and 1,704 cm−1 are well recognized in the decay spectra of GA and mCG. The marker bands of the mC∙+ at 1,586 cm−1 and 1,692 cm−1 occur only in the long-living state of mCU and are absent in the decay spectra of mCA and mCG.
Fig. 3.

Identification of cation and anion marker bands in the decay spectra of different dinucleotides. (A) Two-photon ionization of G and mC yields the difference spectra of G/G∙+ and mC/mC∙+ with the characteristic positive marker bands at 1,608 cm−1, 1,704 cm−1 and 1,586 cm−1, 1,692 cm−1 for the radical cationic form. The corresponding inverted absorption spectra of the dinucleotides are overlaid (dashed lines). (B) Decay spectra D2(ν) of GA, mCG, mCU, and mCA [selectively excited base in bold (295 nm), mCG was unselectively excited at 266 nm]. The marker bands are highlighted in color. The position of cation marker bands in the decay spectra is marked by dashed lines. (C) mC∙− and U∙− absorption spectra calculated with density functional theory. (D) Oxidation potential of G, A, mC, and U and resulting charge-transfer states of the different dimeric samples. The species assigned in the decay spectra are highlighted by the appropriate color.
Anion radical spectra of the involved bases were calculated with density functional theory. Clear marker bands are found for the mC∙− and U∙−; they are shown in Fig. 3C with their distinct positive marker bands in the investigated IR range (for additional information and calculated spectra of all cations and anions, see Fig. S2). A comparison of the anion radical marker bands with the positive bands of the long-lived components (Fig. 3B) shows that mC∙− is formed in the mCG and mCA dinucleotide, whereas the radical anion U∙− can be detected in the mCU dinucleotide. Combining the results of the anion and cation radical marker bands leads to the conclusion that the ion pairs G∙+A∙−, mC∙-G∙+, mC∙+U∙−, and mC∙-A∙+ are present after excitation of the respective dimer. These experimental results are exactly in accordance with the direction of charge separation imposed by the redox potential (36, 37) of the involved DNA bases (Fig. 3D). We can conclude that the charge separation is directed by the redox potential, i.e., the positive charge moves toward the molecule with the lower oxidation potential. The subsequent decay of the long-lived charge separated states occurs on the 100-ps (20–300 ps) time scale by charge recombination to the ground state.
Spatial Extension.
With longer oligomers we address the question about the spatial extension of the charge transfer. In the trinucleotide mCUA we observe a bleach of all three bases, although mC was solely excited (Fig. 1C); this can only be explained by charge migration or delocalization. To obtain further information about this process, longer oligonucleotides of the type mCUaAU(4 − a) with a = 0–4, were investigated (Fig. 4A). In this case, mC is excited exclusively and the bleach of the band at 1,625 cm−1 (ground state of A) is used to show the participation of A in the charge-separated state. This bleach decreases with the increasing number of U molecules between mC and approaches a constant value for a ≥3. This offset bleach may be assigned to a long-distance charge separation. However, we cannot exclude a small shift of the absorbance band of A upon stacking, which might also lead to the offset signal via direct excitation. In any case, the pronounced bleach after mC excitation, directly observable up to a = 2, reveals that charge separation occurs over a distance of more than 10 Å.
Fig. 4.

Distance dependency of the A bleach in mCUaAU(4 − a) oligonucleotides. (A) Normalized decay spectra D2(ν) of the oligomers mCUaAU(4-a), (a = 0–4). mC is selectively excited at 295 nm in all cases. Bleach of ground-state absorbance band of A at 1,625 cm−1 is highlighted by the colored area. A, mC, and U absorbance bands are marked with dashed lines. (B) Scaled transient absorbance at 1,625 cm−1 for the mCUaAU(4 − a) oligonucleotides. (C) Simple stacking model. The stacking-length probability can be calculated by assuming a noncooperative stacking probability α of 50% (red dots), which reproduces well the experimental bleach signal of A (integrated bleach signal, black triangles).
The time dependencies of the bleach at 1,625 cm−1, i.e., the transients at the position of the A band, are shown in Fig. 4B for the longer oligomers. The decay of the first excited electronic state (S1), accompanied by vibrational cooling of the hot ground state, dominates the signal during the first 5 ps. The decay of the charge-transfer state is observable after 5 ps. The normalized transient absorbance at 1,625 cm−1 (A) shows the same time dependence for all samples independent of the mC–A distance. In all cases, the absorption features due to the charge-separated state are formed within the first 5 ps. Thus, charge-hopping, which is known to occur on a much longer time scale of 10–100 ps (38, 39), cannot explain the results. In all mCUaAU(4 − a) oligonucleotides, we observe not only the mC and A bleach but also a very strong bleach of the bridging base U. As a consequence, the base U must be involved in the long-living state and a direct tunneling from the excited mC to the A base can be ruled out as the exclusive reaction mechanism. The bleach of all involved bases can only be explained by a charge delocalization over several bases. Such a behavior has been proposed in the literature, but a direct experimental evidence has been missing (40, 41).
The decrease of the bleach signal of A with increasing distance can be modeled using a heterogeneous ensemble of oligomers with stacked and unstacked bases in the strands. Assuming a fixed probability α for the stacking of two neighboring bases, the probability of longer stacked parts rapidly decreases with increasing length. For α = 0.5, the occurrence of longer stacked strands is shown in Fig. 4C, red dots. In a model with delocalization of the charges over the stacked parts the bleach of A should reflect the probability of the corresponding stacked parts. Indeed, the integrated bleach signal of the A band at 1,625 cm−1 (black triangles) closely follows the behavior expected from stacking; it also shows that the observation of charge delocalization is limited due to the small occurrence of longer stacked domains.
The spectra of Fig. 4A display different marker bands of the involved anions and cations. Though it is straightforward to deduce the charge distribution for dinucleotides, the stacking heterogeneity in the longer oligonucleotides prevents a quantitative analysis. Because the experiment averages over the differently stacked subensembles, a ready disentanglement of the heterogeneity with a defined assignment of charge distributions is not possible; this is further complicated by the larger number of involved bases, which leads to an increased overlap of the bands.
Conclusion
Selective UV excitation of DNA multimers combined with femtosecond IR probing has been used to obtain interesting information on the nature of long-living electronic states in DNA strands (Fig. 5). Excitation of unstacked DNA bases by UV light is followed by ultrafast deactivation via internal conversion, which is known to be the dominating deactivation mechanism for single bases in solution. However, DNA single strands contain a considerable amount of well-stacked domains. Within a few picoseconds, the excitation of these bases leads to a long-living charge-separated state formed with high probability. This efficient charge transfer requires stacked bases, and the direction of the charge movement is governed by the oxidation potentials and thus by the base sequence. The charges are delocalized in the stacked domains. These long-living ionic states decay by charge recombination to the neutral ground-state on the 100-ps time scale. The mechanism is related to the charge transport observed in DNA double strands after excitation of modified bases (18). However, the present investigation reveals that charge transfer in DNA oligonucleotides is a natural process, occurring to a high probability after absorption of UV light.
Fig. 5.

Reaction model for light absorption in DNA. Model for photoexcitation of DNA: DNA bases are arranged in domains with well-orientated stacked bases (gray background). If a base absorbs light in this domain (in this case the first base), charge separation occurs during the first 5 ps. The direction of charge transfer depends on the redox potential of the involved bases and is delocalized in the domain. Only one possible charge distribution is shown. Charge recombination occurs within 20–300 ps, depending on the sequence.
The presence of charges along a DNA strand may have severe consequences for the integrity of a DNA strand, because charged base radicals form starting points for oxidative (42) and reductive (43) DNA damage. Thus, the presented observation of charged states with relatively long lifetime adds an important element to the discussion of DNA photolesions and mutational hot spots (44).
Materials and Methods
Oligodeoxyribonucleotides.
All oligonucleotides were purchased from Metabion AG. The lyophilized samples were dissolved in 50 mM phosphate buffer in D2O. The final concentration was ∼3–6 mM, depending on the solubility of the sample. The corresponding absorbance in the time-resolved experiments was 0.1–0.2 OD at 295 nm in a cuvette with 100-μm path length.
Femtosecond UV-Pump IR-Probe Measurements.
All time-resolved measurements are based on a Ti:Sapphire laser amplifier system (Spitfire Pro; Spectra-Physics) with 100-fs pulses at 800 nm and a repetition rate of 1 kHz. The pump pulses at 295 nm were generated with a frequency-doubled two-stage noncollinear optical parametric amplifier (45). The excitation energy was ∼800 nJ with a beam diameter at the sample position of 150 μm.
The mid-IR probe light was generated by a combination of a noncollinear and a collinear optical parametric amplifier and subsequent difference frequency mixing in a AgGaS2 crystal. The transmitted IR pulse was spectrally dispersed (Chromex 250IS; Bruker) and detected on a 64-channel MCT array (IR-0144; Infrared Systems Development). All experiments were performed at room temperature and under magic-angle conditions. The excited sample volume was exchanged between consecutive excitation pulses via a BaF2 flow cuvette.
Data Handling.
The data are collected as an array of absorption changes for different probing frequencies ν and delay times tD. The absorption changes ΔA(ν, tD) of all investigated oligonucleotides were globally fitted for delay times >1 ps with three exponentials and a constant offset representing long-lasting absorption changes from irreversible processes or triplet states.
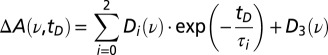 |
For each exponential component, amplitude spectra Di(ν) are determined in the fit, which represent the absorption changes related with this process [decay-associated difference spectra (DADS)]. The time constants determined by the fitting procedure are given in Table 1. For the mixture of single bases (Fig. 1), two exponentials were sufficient to model the data. For the oligonucleotides, three time constants are required. The DADS D2(ν) related to the time constant τ2 in the 20- to 300-ps range contains the information on the charge-separated long-living states. For all oligonucleotides with selective excitation via mC we estimated the fraction F2 of the molecules in the long-living state by dividing the fitting amplitude D2 by the initial bleach signal at tD = 1 ps (representing the amount of excited molecules) at the position of the mC absorption band at 1,667 cm−1.
Radical Cation Spectra.
The cation difference spectra were obtained by exciting solutions of mC and G at 266 nm with a pulse energy of 2–4 μJ (pulse length 300 fs). This excitation leads to ionization of the base, which is stable in both cases in the observed time window (1 ns). The decay spectra D∞ yields the difference spectra used in Fig. 3A. The G cation difference spectrum has been published previously (33, 34). The ionization conditions and the characterization of the mC cation are in ref. 35.
Stationary Spectroscopy.
FTIR measurements were performed with a Bruker IFS 66 FT spectrophotometer in 100-μm CaF2 cuvettes. The UV/Vis spectra were recorded with a PerkinElmer spectrophotometer (Lambda 750).
Density Functional Theory Calculations.
Becke3Lyp 6-311-G** functional with the solvent model PCM was used to calculate the harmonic vibrational frequencies with the Gaussian 03 software (46). For simplicity, all calculations were done for the 1-methyl-substituted nucleobases where all exchangeable hydrogen atoms in the structure were substituted with deuterium. Each vibrational frequency analysis was preceded by a geometry optimization. The frequencies were scaled with a factor of 0.9669 (47).
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft through the Sonderforschungsbereich Dynamics and Intermediates of Molecular Transformations SFB 749, the Clusters of Excellence Center for Integrated Protein Science Munich, and Munich-Centre for Advanced Photonics.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323700111/-/DCSupplemental.
References
- 1.Taylor JS. Unraveling the molecular pathway from sunlight to skin cancer. Acc Chem Res. 1994;27(3):76–82. [Google Scholar]
- 2.Crespo-Hernández CE, Cohen B, Hare PM, Kohler B. Ultrafast excited-state dynamics in nucleic acids. Chem Rev. 2004;104(4):1977–2019. doi: 10.1021/cr0206770. [DOI] [PubMed] [Google Scholar]
- 3.Pecourt J-ML, Peon J, Kohler B. DNA excited-state dynamics: Ultrafast internal conversion and vibrational cooling in a series of nucleosides. J Am Chem Soc. 2001;123(42):10370–10378. doi: 10.1021/ja0161453. [DOI] [PubMed] [Google Scholar]
- 4.Middleton CT, et al. DNA excited-state dynamics: From single bases to the double helix. Annu Rev Phys Chem. 2009;60(1):217–239. doi: 10.1146/annurev.physchem.59.032607.093719. [DOI] [PubMed] [Google Scholar]
- 5.Crespo-Hernández CE, Cohen B, Kohler B. Base stacking controls excited-state dynamics in A.T DNA. Nature. 2005;436(7054):1141–1144. doi: 10.1038/nature03933. [DOI] [PubMed] [Google Scholar]
- 6.Kohler B. Nonradiative decay mechanisms in DNA model systems. J Phys Chem Lett. 2010;1(13):2047–2053. [Google Scholar]
- 7.Markovitsi D, et al. Molecular spectroscopy: Complexity of excited-state dynamics in DNA. Nature. 2006;441(7094):E7–, discussion E8. doi: 10.1038/nature04903. [DOI] [PubMed] [Google Scholar]
- 8.Su C, Middleton CT, Kohler B. Base-stacking disorder and excited-state dynamics in single-stranded adenine homo-oligonucleotides. J Phys Chem B. 2012;116(34):10266–10274. doi: 10.1021/jp305350t. [DOI] [PubMed] [Google Scholar]
- 9.Buchvarov I, Wang Q, Raytchev M, Trifonov A, Fiebig T. Electronic energy delocalization and dissipation in single- and double-stranded DNA. Proc Natl Acad Sci USA. 2007;104(12):4794–4797. doi: 10.1073/pnas.0606757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vayá I, Gustavsson T, Douki T, Berlin Y, Markovitsi D. Electronic excitation energy transfer between nucleobases of natural DNA. J Am Chem Soc. 2012;134(28):11366–11368. doi: 10.1021/ja304328g. [DOI] [PubMed] [Google Scholar]
- 11.Banyasz A, et al. Multi-pathway excited state relaxation of adenine oligomers in aqueous solution: A joint theoretical and experimental study. Chemistry. 2013;19(11):3762–3774. doi: 10.1002/chem.201202741. [DOI] [PubMed] [Google Scholar]
- 12.Takaya T, Su C, de La Harpe K, Crespo-Hernández CE, Kohler B. UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases. Proc Natl Acad Sci USA. 2008;105(30):10285–10290. doi: 10.1073/pnas.0802079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doorley GW, et al. Tracking DNA excited states by picosecond-time-resolved infrared spectroscopy: Signature band for a charge-transfer excited state in stacked adenine-thymine systems. J Phys Chem Lett. 2013;4(16):2739–2744. [Google Scholar]
- 14.Conti I, Altoè P, Stenta M, Garavelli M, Orlandi G. Adenine deactivation in DNA resolved at the CASPT2//CASSCF/ AMBER level. Phys Chem Chem Phys. 2010;12(19):5016–5023. doi: 10.1039/b926608a. [DOI] [PubMed] [Google Scholar]
- 15.Improta R, Barone V. Interplay between neutral and charge-transfer- excimers rules the excited state decay in adenine-rich polynucleotides. Angew Chem Int Ed Engl. 2011;50(50):12016–12019. doi: 10.1002/anie.201104382. [DOI] [PubMed] [Google Scholar]
- 16.Santoro F, Barone V, Improta R. Influence of base stacking on excited-state behavior of polyadenine in water, based on time-dependent density functional calculations. Proc Natl Acad Sci USA. 2007;104(24):9931–9936. doi: 10.1073/pnas.0703298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olaso-González G, Merchán M, Serrano-Andrés L. The role of adenine excimers in the photophysics of oligonucleotides. J Am Chem Soc. 2009;131(12):4368–4377. doi: 10.1021/ja808280j. [DOI] [PubMed] [Google Scholar]
- 18.Genereux JC, Barton JK. Mechanisms for DNA charge transport. Chem Rev. 2010;110(3):1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382(6593):731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 20.Wan C, et al. Femtosecond dynamics of DNA-mediated electron transfer. Proc Natl Acad Sci USA. 1999;96(11):6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wan C, Fiebig T, Schiemann O, Barton JK, Zewail AH. Femtosecond direct observation of charge transfer between bases in DNA. Proc Natl Acad Sci USA. 2000;97(26):14052–14055. doi: 10.1073/pnas.250483297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis FD, et al. Direct measurement of hole transport dynamics in DNA. Nature. 2000;406(6791):51–53. doi: 10.1038/35017524. [DOI] [PubMed] [Google Scholar]
- 23.O’Neill MA, Becker H-C, Wan C, Barton JK, Zewail AH. Ultrafast dynamics in DNA-mediated electron transfer: Base gating and the role of temperature. Angew Chem Int Ed Engl. 2003;42(47):5896–5900. doi: 10.1002/anie.200352831. [DOI] [PubMed] [Google Scholar]
- 24.Markovitsi D, Gustavsson T, Vayá I. Fluorescence of DNA duplexes: From model helices to natural DNA. J Phys Chem Lett. 2010;1(22):3271–3276. [Google Scholar]
- 25.Schwalb NK, Temps F. Base sequence and higher-order structure induce the complex excited-state dynamics in DNA. Science. 2008;322(5899):243–245. doi: 10.1126/science.1161651. [DOI] [PubMed] [Google Scholar]
- 26.Schreier WJ, et al. Thymine dimerization in DNA is an ultrafast photoreaction. Science. 2007;315(5812):625–629. doi: 10.1126/science.1135428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harpe KD (2011) Femtosecond UV and infrared time-resolved spectroscopy of DNA: From well-ordered sequences to genomic DNA. PhD dissertation (The Ohio State University, Columbus, Ohio)
- 28.Saenger W. Principles of Nucleic Acid Structure. Berlin: Springer; 1984. [Google Scholar]
- 29.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith ZD, Meissner A. DNA methylation: Roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 31.Malone RJ, Miller AM, Kohler B. Singlet excited-state lifetimes of cytosine derivatives measured by femtosecond transient absorption. Photochem Photobiol. 2003;77(2):158–164. doi: 10.1562/0031-8655(2003)077<0158:sesloc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Brahms J, Maurizot JC, Michelson AM. Conformational stability of dinucleotides in solution. J Mol Biol. 1967;25(3):481–495. doi: 10.1016/0022-2836(67)90200-8. [DOI] [PubMed] [Google Scholar]
- 33.Kuimova MK, et al. Monitoring the direct and indirect damage of DNA bases and polynucleotides by using time-resolved infrared spectroscopy. Proc Natl Acad Sci USA. 2006;103(7):2150–2153. doi: 10.1073/pnas.0506860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker AW, Lin CY, George MW, Towrie M, Kuimova MK. Infrared characterization of the guanine radical cation: Finger printing DNA damage. J Phys Chem B. 2010;114(10):3660–3667. doi: 10.1021/jp9106958. [DOI] [PubMed] [Google Scholar]
- 35.Bucher DB, Pilles BM, Pfaffeneder T, Carell T, Zinth W. Fingerprinting DNA oxidation process: IR characterization of the 5-methyl-2′-deoxycytidine radical cation. ChemPhysChem. 2014;15(3):420–423. doi: 10.1002/cphc.201300954. [DOI] [PubMed] [Google Scholar]
- 36.Seidel CAM, Schulz A, Sauer MHM. Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J Phys Chem. 1996;100(13):5541–5553. [Google Scholar]
- 37.Paukku Y, Hill G. Theoretical determination of one-electron redox potentials for DNA bases, base pairs, and stacks. J Phys Chem A. 2011;115(18):4804–4810. doi: 10.1021/jp201281t. [DOI] [PubMed] [Google Scholar]
- 38.Park MJ, Fujitsuka M, Kawai K, Majima T. Direct measurement of the dynamics of excess electron transfer through consecutive thymine sequence in DNA. J Am Chem Soc. 2011;133(39):15320–15323. doi: 10.1021/ja2068017. [DOI] [PubMed] [Google Scholar]
- 39.Takada T, et al. Charge separation in DNA via consecutive adenine hopping. J Am Chem Soc. 2004;126(4):1125–1129. doi: 10.1021/ja035730w. [DOI] [PubMed] [Google Scholar]
- 40.Shao F, Augustyn K, Barton JK. Sequence dependence of charge transport through DNA domains. J Am Chem Soc. 2005;127(49):17445–17452. doi: 10.1021/ja0563399. [DOI] [PubMed] [Google Scholar]
- 41.Renaud N, Berlin YA, Ratner MA. Impact of a single base pair substitution on the charge transfer rate along short DNA hairpins. Proc Natl Acad Sci USA. 2013;110(37):14867–14871. doi: 10.1073/pnas.1309139110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanvah S, et al. Oxidation of DNA: Damage to nucleobases. Acc Chem Res. 2010;43(2):280–287. doi: 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- 43.Wang C-R, Nguyen J, Lu Q-B. Bond breaks of nucleotides by dissociative electron transfer of nonequilibrium prehydrated electrons: A new molecular mechanism for reductive DNA damage. J Am Chem Soc. 2009;131(32):11320–11322. doi: 10.1021/ja902675g. [DOI] [PubMed] [Google Scholar]
- 44.You Y-H, Szabó PE, Pfeifer GP. Cyclobutane pyrimidine dimers form preferentially at the major p53 mutational hotspot in UVB-induced mouse skin tumors. Carcinogenesis. 2000;21(11):2113–2117. doi: 10.1093/carcin/21.11.2113. [DOI] [PubMed] [Google Scholar]
- 45.Riedle E, et al. Generation of 10 to 50 fs pulses tunable through all of the visible and the NIR. Appl Phys B. 2000;71(3):457–465. [Google Scholar]
- 46. Frisch MJ, et al. (2004) Gaussian 03. Revision D.01 (Gaussian, Inc., Wallingford CT)
- 47.Irikura KK, Johnson RD, 3rd, Kacker RN. Uncertainties in scaling factors for ab initio vibrational frequencies. J Phys Chem A. 2005;109(37):8430–8437. doi: 10.1021/jp052793n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.