

Significance
Growing evidence underlines the role attributed to abnormal forms of Tau in several neurodegenerative diseases known as tauopathies, including Alzheimer’s disease, which are characterized by accumulation of oligomers and filamentous Tau inclusions in the CNS. We identify a direct interaction of the immunophilin FK506-binding protein with a molecular mass of ∼52 kDa (FKBP52) and the pathological mutant of Tau containing a proline-to-leucine mutation at position 301 (Tau-P301L), inducing Tau-P301L oligomerization and assembly into filaments. This interaction has a considerable impact on the progress of the tauopathy because some early aspects of the disease are rescued, in vivo, by knocking down FKBP52. Our results open a new field for the study of FKBP52 pathophysiology in tauopathic dementias, including prediction of disease phenotype and search for new types of antipathological Tau treatments.
Keywords: FKBP, Tau assembly, Tau-P301L dementia
Abstract
The Tau protein is the major component of intracellular filaments observed in a number of neurodegenerative diseases known as tauopathies. The pathological mutant of Tau containing a proline-to-leucine mutation at position 301 (P301L) leads to severe human tauopathy. Here, we assess the impact of FK506-binding protein with a molecular mass of ∼52 kDa (FKBP52), an immunophilin protein that interacts with physiological Tau, on Tau-P301L activity. We identify a direct interaction of FKBP52 with Tau-P301L and its phosphorylated forms and demonstrate FKBP52’s ability to induce the formation of Tau-P301L oligomers. EM analysis shows that Tau-P301L oligomers, induced by FKBP52, can assemble into filaments. In the transgenic zebrafish expressing the human Tau-P301L mutant, FKBP52 knockdown is sufficient to redrive defective axonal outgrowth and branching related to Tau-P301L expression in spinal primary motoneurons. This result correlates with a significant reduction of pT181 pathological phosphorylated Tau and with recovery of the stereotypic escape response behavior. Collectively, FKBP52 appears to be an endogenous candidate that directly interacts with the pathogenic Tau-P301L and modulates its function in vitro and in vivo.
Tau is a MAP mainly found in neurons (1, 2), which becomes hyperphosphorylated in several neurodegenerative diseases known as tauopathies and forms intraneuronal aggregates called neurofibrillary tangles (NFTs) (3–5). The accumulation of Tau aggregates is a multistep process that involves transient species, and growing evidence suggests that not only the NFTs but equally small oligomeric species contribute to Tau-mediated neurotoxicity (6–8). The pathological mutant of Tau containing a proline-to-leucine mutation at position 301 (P301L) is identified in frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17) (9). Larvae of the zebrafish overexpressing the Tau-P301L mutant protein show defects in motoneuron development early on, and in this model, different Tau phosphoforms are associated with its pathological state (10).
The FK506-binding protein with a molecular mass of ∼52 kDa (FKBP52) belongs to the immunophilin family and presents rotamase activity inhibited by FK506 binding (11). This enzymatic activity catalyzes the isomerization of peptidyl-prolyl bonds between cis and trans conformations, and therefore influences target protein folding and function (12, 13). FKBP52 contains additional functional domains, such as a tetratricopeptide repeat domain that serves as a binding site for molecular chaperone heat shock protein with a molecular mass of 90 kDa (14) and for which chaperone activity was observed (15).
A novel role of FKBP52 has recently emerged because it binds directly to tubulin and induces tubulin depolymerization in vitro (16). Biochemical characterization of FKBP52 showed that it interacts with Tau, especially its hyperphosphorylated form, and demonstrated an antagonist effect of FKBP52 on Tau’s function in tubulin assembly (17). Here, we show that FKBP52 interacts with the pathological form of Tau-P301L and induces its oligomerization and assembly into filaments. Using a transgenic zebrafish model for Tau-P301L tauopathy (10), we show that FKBP52 knockdown attenuates pathological Tau activity to reestablish axonal outgrowth and branching in defective spinal primary motoneurons. This result confirms the functional implication of FKBP52 in modulating early pathological Tau activity, at least for axonal growth and motility. We propose that FKBP52 is an important regulator of Tau conformational change and assembly, and thus may be instrumental in a therapeutic approach to the disease.
Results
FKBP52 Binds Tau-P301L Mutant.
The interaction between FKBP52 and the Tau-P301L mutant was investigated using a dot blot assay. Purified recombinant protein (2.2 μg of Tau-P301L), phosphorylated or not phosphorylated, was spotted on nitrocellulose and incubated with purified human FKBP52 protein. A polyclonal antibody against FKBP52 was used to monitor the amount of protein retained. As shown in Fig. 1, the amount of FKBP52 retained by Tau-P301L was 18% (±4.5), whereas 55% (±10) of FKBP52 was retained by the same amount of the phosphorylated Tau-P301L. Thus, FKBP52 binds Tau-P301L, especially in its phosphorylated form.
Fig. 1.

Binding of mutant Tau-P301L to FKBP52. (A) Representative dot blot analysis: (1) 0.5 μg of FKBP52 used at 100%, (2) 2.2 μg of mutant Tau-P301L, (3) 2.2 μg of phosphorylated mutant Tau-P301L, and (4) 5 μg of GST used as negative control were dotted on nitrocellulose. The dots were developed with FKBP52 antibody. (B) Amount of FKBP52 retained by Tau proteins was quantified using GeneTools software (Syngene). These results represent the data from four separate experiments. (C) SDS/PAGE analysis of recombinant mutated Tau-P301L and P301L hyperphosphorylation (HP). IB, immunoblot.
FKBP52 Colocalizes with Tau-P301L in Human Neuronal Cells.
In light of the emerging concept of transcellular propagation of Tau (18), we seeded SH-SY5Y neuronal cells with exogenous Tau-P301L, labeled with a cyanine (Cy) 5.5 dye, and investigated the subcellular localization of Tau-P301L and endogenous FKBP52. Fluorescence microscopy and confocal microscopy analysis showed that Tau-P301L localization was concentrated in the perinuclear region, with FKBP52 showing the intracellular colocalization of both proteins (Fig. 2 and Fig. S1).
Fig. 2.

FKBP52 colocalizes with Tau-P301L in SH-SY5Y cells. Confocal microscopy analyses of the colocalization with Z-stack imaging. Tau-P301L is in the same plane as FKBP52 (arrowhead), showing the intracellular colocalization of both proteins (arrow). The fluorescence profiles confirmed that the two proteins colocalize. (Scale bars: 5 μM.)
FKBP52 Induces the Formation of Tau-P301L Oligomers in Vitro.
To investigate the ability of recombinant FKBP52 to induce structural changes to Tau-P301L, we monitored the migration profile of these proteins using native gel electrophoresis. Fixed quantities of Tau-P301L were mixed with increasing amounts of FKBP52 and incubated for 30 min at 37 °C. On Western blotting carried out with an anti-Tau antibody, titration of Tau-P301L with FKBP52 led to the formation of several bands having different electrophoretic mobility from those of isolated Tau-P301L, whereas no modification occurred upon exposure of Tau-P301L to control GST (Fig. 3A). However, the same samples analyzed with an anti-FKBP52 antibody did not reveal any modification in the migration profile, suggesting a structural modification of Tau-P301L rather than a stable complex between FKBP52 and Tau-P301L. A similar pattern of oligomer formation after incubation of FKBP52 and phosphorylated Tau-P301L was detected by anti-Tau antibodies, AT8 or AT180, that recognize specific phosphorylated forms (Fig. 3B).
Fig. 3.

FKBP52 promotes “in vitro” oligomerization of Tau-P301L. (A) Mix of Tau-P301L with increasing concentration of FKBP52 or with GST used as a control were analyzed by blue native PAGE electrophoresis and revealed by Western blotting using specific antibodies as indicated in the figure. (B) Ability of FKBP52 to induce conformational changes of Tau-P301L HP was analyzed as in A.
Tau-P301L Can Form Paired Helical-Like Filaments When Incubated with FKBP52.
To see whether the oligomers obtained could evolve into fibrillar structures, we first used light diffraction. When measuring the light scattering at 340 nm, we indeed observed a signal when mixing FKBP52 and Tau-P301L for 30 min at 37 °C, but not with both proteins individually (Fig. 4A). We analyzed the resulting samples using transmission EM, and we did observe fibrillar structures after incubation for 1 h, whereas no such structures could be found when individual protein solutions were deposited on the grid. The resulting fibers appear to be short in length and twisted, with a width of 17 nm (Fig. 4B). As such, they resemble the filaments observed in the brains of patients with FTDP-17, most of which appear to be irregularly twisted ribbons with a width of about 15 nm (19).
Fig. 4.

Assembly of Tau-P301L into filaments. (A) Coincubation of FKBP52 and Tau-P301L, at 37 °C for 30 min, induces light scattering, whereas each protein alone gives no detectable signal. abs, absorbance. (B) EM of FKBP52/Tau-P301L after 1 h of incubation at 37 °C.
FKBP52 Knockdown Significantly Improves Axonal Growth and Branching Defects Observed in Primary Motoneurons of Tau-P301L Transgenic Fish.
To test in vivo the effect of FKBP52/Tau-P301L interaction, we used the zebrafish, cloned its full-length FKBP52 cDNA, and showed its ubiquitous expression (Fig. S2). For functional analysis, we took advantage of the transgenic Tau-P301L zebrafish model (for convenience, we use HuC-Tau in the figures), which recapitulates several neurological defects of tauopathies, including axonal growth and branching defects seen in the spinal motoneurons (10, 20) (Fig. 5 D, F, and I). We looked specifically at the primary motoneurons that undergo axonogenesis at 17 h postfertilization (hpf), using znp1 antibodies, a pan-specific marker of primary motor axons (21). We analyzed the first five outgrowing caudal primary motoneurons (CaP) anterior to the end of the yolk extension (Fig. 5).
Fig. 5.

FKBP52 modulates pathological Tau activity in spinal primary motoneurons. (A–C) Lateral views of 48-hpf control embryos and FKBP52 morphant. Lateral views of znp1 antibody staining of control (D), FKBP52 morphant (E), Huc-Tau (F), and Huc-Tau injected with either FKBP52 MO (G) or FKBP52 mRNA (H) are shown at 48 hpf. (D′–H′) Tracings of motor-axon tracts ventral to the spinal cord derived from the panels directly above. In all images, anterior is to the left and dorsal is to the top. (Scale bars: A–C, 200 μm; D′–H′, 100 μm.) (I) Bar chart depicts the difference in the percentage of axons that reach their most ventral target. No significant difference is observed between WT and Huc-Tau + FKBP52MO embryos (N.S, P > 0.05; *P < 0.05; **P < 0.01). N.S, nonsignificant.
To test the role of FKBP52 in this model, we used a knockdown approach by injecting specific antisense morpholino (MO) against FKBP52. Injection of FKBP52 MO (0.4–0.6 pmol per embryo) or FKBP52 mRNA (200 pg) into WT embryos did not lead to any defect regarding the length (ability to reach their most ventral target) or the number of branches in the CaP motoneurons (Fig. 5 E and E′). Note that FKBP52 morphants show a slightly shorter and curved body axis (Fig. 5 A–C) but have normal neurogenesis in the spinal cord (Figs. S3 and S4). CaP motor axons of control 5-mismatch morphants, FKBP52 morphants, and FKBP52 mRNA were similar to WT at 48 hpf (Fig. 5 D–E′). In the Tau-P301L transgenic model, only 57% of the CaP axons reach their most ventral target in comparison to WT (Fig. 5I; P < 0.05). They also show a randomized pattern of branching regarding the number of collateral branches or fascicles per axon (Fig. 5F). However, striking changes occurred when injecting FKBP52 MO in the Tau-P301L transgenic embryos. Here, Tau-P301L/FKBP52 morphants readjust their axonal growth, whereby 78% of the CaP axons now reach their most ventral target in comparison to HuC-Tau (Fig. 5I; P = 0.038). In this case, no significant difference is observed between WT and Tau-P301L/FKBP52 morphants (Fig. 5I; P > 0.05). They also reestablish a more close to normal distribution of collateral branching per CaP axon in comparison to Tau-P301L (t = −3.09, P = 0.01) (Fig. 5 G and G′ and Fig. S3). Injection of FKBP52 mRNA into Tau-P301L embryos aggravated their axonal defects. Here, only 44% of CaP motoneurons reach their most ventral target in comparison to WT (Fig. 5I; P < 0.01). There was also a significant difference in the collateral numbers of branches in the Tau-P301L/FKBP52 mRNA, whereby the number of CaP axons showing fewer collateral branches is significantly increased in comparison to Tau-P301L alone (Fig. 5 H and H′ and Fig. S3; t = −2.26, P = 0.01).
FKBP52 Knockdown Does Not Modify Neuronal Cell Death in Tau-P301L Spinal Cord.
The Tau-P301L transgenic larvae show a significant increase in neuronal cell death in the spinal cord (10). To assess whether FKBP52 knockdown might modify the neuronal cell death seen in the Tau-P301L transgenic embryos, we injected FKBP52 MO and looked for cell death in the spinal cord using acridine orange (AO) labeling. No significant difference was observed in the number of AO-positive cells between Tau-P301L embryos and the Tau-P301L/FKBP52 MO siblings at 72 hpf (t = 1.15, P = 0.273; Fig. 6).
Fig. 6.

Bar chart depicts the difference in the number of AO-positive cells. No significant difference was observed between the HuC-Tau and the HuC-Tau injected with FKBP52MO. A difference is observed when comparing these two groups and the WT siblings (***P ≤ 0.0001).
FKBP52 Knockdown Differentially Regulates the Levels of Pathological Phosphorylated Forms of Tau-P301L.
To investigate a change in the phosphorylation state of pathological forms of Tau, we have analyzed, after FKBP52 knockdown, two different pathological phosphorylated forms of Tau-P301L (pT181 and pT231) by Western blotting. Although there was no significant change in the overall level of expression of Tau and the pT231 pathological form, we observed a sharp decrease in the levels of the pathological phosphorylated form of Tau recognized by AT270 (epitope pT181) (Fig. 7).
Fig. 7.
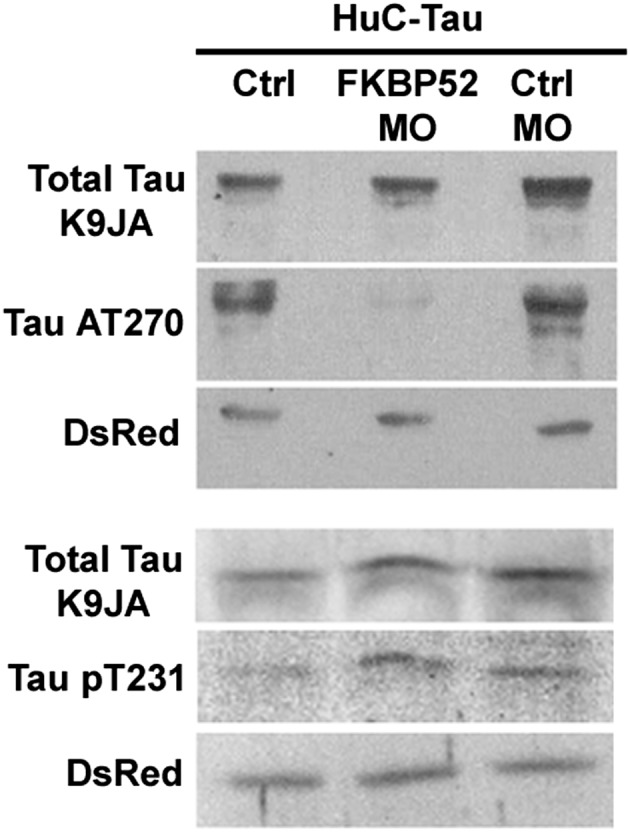
Effect of FKBP52 knockdown on total and phosphorylated Tau in 48-h-old transgenic zebrafish expressing Tau-P301L. Total protein extract (2.5–5 μL) was loaded on 10% SDS/PAGE and analyzed by Western blotting using specific antibodies as indicated. Ctrl, control.
FKBP52 Knockdown Improves the Stereotypic Escape Response Behavior in the Tau-P301L Transgenic Fish.
To test whether the improvement seen in axonal branching and outgrowth after FKBP52 knockdown translates into a functional recovery, and thus better motility, we injected FKBP52 MO into Tau-P301L transgenic embryos and analyzed the escape response behavior. Due to their altered motoneuron morphology, the Tau-P301L embryos show a slow or absent movement in most of the larvae at 48 hpf (10). Although most control fish responded to a touch stimulus with a stereotypic escape response, most mutated Tau-expressing fish showed a significantly reduced or even absent response (Fig. 8 and Movies S1 and S2). However, the Tau-P301L/FKBP52 MO embryos showed a significant improvement in their escape response (Fig. 8 and Movie S3). We saw that although Tau-P301L embryos move at an average speed of 527 units per second (n = 99), the speed of the Tau-P301L/FKBP52 MO embryos increased significantly to 1,750 units per second (n = 102) (t = 4.62, P = 0.001), whereas control embryos moved at an average speed of 4,147 units per second (n = 39).
Fig. 8.

Knockdown of FKBP52 improves larval mobility in HuC-Tau transgenics. (A–C) Tracking analyses of 48-hpf control (WT), Huc-Tau, and Huc-Tau + FKBP52MO larvae in a touch-response test. Each plot line represents the trajectory of one larva after touch stimulation. (D) Quantification of the touch-response test. Each group is divided into three categories: (a) embryos that do not respond at all, (b) embryos that move slightly upon stimulus, and (c) embryos that show a typical escape response. The locomotor defects are significantly improved in Huc-Tau larvae injected with 0.4 pmol of FKBP52 MO (Kruskal–Wallis test,***P < 0.0001).
Discussion
FKBP52/Tau-P301L Interaction Leads to Tau Oligomerization.
We show that FKBP52 is able to interact physically with the Tau-P301L mutant. In this case, FKBP52 can induce a conformational change of Tau-P301L and leads to its oligomerization and filamentous assembly. This observation is in line with previous data showing important and differential effects of FKBP52 on the conformational state of other proteins, such as citrate synthase (15) and alpha-synuclein (22). Interestingly, the Tau-P301L fibers induced by FKBP52 have an average width of 17 nm, and their ultrastructure is reminiscent of those isolated from patients with FTDP-17 (19).
However, we failed to detect a stable Tau-P301L/FKBP52 complex, suggesting a rapid and short-lived interaction between both partners that might involve the enzymatic prolyl cis/trans isomerase of FKBP52. This activity links FKBPs to the parvulin family of isomerases, such as Pin1, the importance of which has already been reported in the pathological function of Tau (23).
FKBP52/Tau-P301L Interaction Can Modulate Axonal Growth but Not Cell Death at Early Stages of Tauopathy.
We show that reducing FKBP52 levels can significantly enhance the growth of defective spinal primary motoneuron projections and branching, as well as improving the motility of zebrafish larvae. Moreover, the phosphorylation site pT181 is strongly decreased in FKBP52 morphants. This result suggests that reducing FKBP52 levels may delay the early progression of pathological axonal growth by acting on specific phosphorylation site(s) of Tau.
We propose a model whereby lowering the levels of FKBP52 in this transgenic model of FTDP-17 tauopathy prevents Tau-P301L from adopting a conformation prone to phosphorylation and aggregation, thereby reducing the neurotoxicity of the transgene. Given that the cell death phenotype is not modified in the morphants at these early stages, the impact of this interaction affects the defective microtubule dynamics and stability related to this pathological Tau model. However, further experiments should shed some light on the impact of FKBP52 knockdown on cell death at later stages.
We also cannot rule out the possibility that the absence of FKBP52 could lead to a release of endogenous physiological Tau, allowing it to execute its normal function of microtubule assembly. The latter hypothesis is in line with the one described previously in PC12 cells (17).
FKBP52/Tau Interaction: The Early vs. Late Aspects of Tauopathy.
The mechanism(s) linking FKBP52 and the late stage of the tauopathy process remains to be elucidated. Previously, we have shown that FKBP52 via Tau can modulate microtubule stability in vitro (16) and that levels of FKBP52 protein, at the late stages of diseases, are dramatically decreased in the brains of patients with Alzheimer’s disease and FTDP-17 (24). Here, we show, in a preliminary stage of tauopathy, that attenuating FKBP52 expression reduces the toxicity of the transgene. Based on these findings, different roles for FKBP52 can be considered. In the early development of this tauopathy, FKBP52 may induce Tau oligomer formation that can have a significant impact on axonal outgrowth and branching. However, at much later stages of the disease, given the important accumulation of hyperphosphorylated Tau and its direct oligomerization by FKBP52, the latter is more likely to be cleared, thus reducing its overall levels. Interestingly, FKBP52/Tau-P301L colabeling could be observed particularly in a perinuclear region, where a deformation of nuclear envelope was detected (Fig. S1), suggesting that FKBP52 could be involved in spatial sequestration of Tau-P301L. This perinuclear localization of Tau has already been observed in a similar structure called an aggresome (25, 26). Whether FKBP52 is involved in the clearance of Tau at later stages is still to be investigated.
Further analyses of the FKBP52/Tau interaction may provide a basis for pursuing and evaluating potential therapies for Alzheimer’s disease, FTDP-17, and other tauopathies by fine-tuning the balance of FKBP52.
Methods
Cloning of Human and Zebrafish FKBP52 cDNA.
To clone human and zebrafish FKBP52 cDNA, RNAs were prepared from SH-SY5Y cells (human neuroblastoma cells) and from 30-hpf embryos, respectively. Additional details are provided in SI Methods.
Protein Purification.
Tau with the P301L mutation was expressed in Escherichia coli and purified as described (27). Hyperphosphorylation of Tau-P301L was performed using cytosol from the brain of a 2-mo-old rat as described previously (28). Additional details are provided in SI Methods.
Labeling of Tau-P301L with cyanine, dot blot assays, light scattering, EM, and blue native PAGE are reported in SI Methods.
Immunocytofluorescence.
Cells were grown on poly-d-lysine–coated glass coverslips for microscopy and were plated at 3.104 cells per well in a 12-well tissue culture plate. Subsequently, 20 μg of purified Cy5.5-labeled Tau-P301L was added to SH-SY5Y cell medium for 24 h of incubation. Information about antibodies used is provided in SI Methods.
Zebrafish Analysis.
Zebrafish experiments were performed as described by Tawk et al. (29, 30) and as described in SI Methods.
Supplementary Material
Acknowledgments
We thank C. Yanicostas and N. Soussi-Yanicostas for sharing the Tau-P301L fish for pilot experiments, O. Trassard and P. Leclerc for technical help with confocal imaging and analyses, N. Barois for EM, and C. Jarvis for helping with statistical analyses. We are grateful to B. Schmidt and C. Haass for providing the human Tau-P301L zebrafish line. E.S., O.D., and K.G. were funded by “Fondation Vivre Longtemps,” “Institut Mérieux,” and “Institut Baulieu.” J.G. and A.K. are supported by the Agence Nationale de la Recherche (ANR), and D.P. is supported by the Deutsche Forschungsgemeinschaft (Grant SFB 596) and European Union Seventh Framework Programme (Grant 200611, Memory Loss in Alzheimer's Disease). This work has been supported jointly by grants from ANR and Laboratory of Excellence Program Investment for the Future Development of Innovative Strategies for a Transdisciplinary Approach to Alzheimer's Disease.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402645111/-/DCSupplemental.
References
- 1.Avila J, Lucas JJ, Perez M, Hernandez F. Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004;84(2):361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- 2.Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. 1988;85(11):4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sergeant N, et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev Proteomics. 2008;5(2):207–224. doi: 10.1586/14789450.5.2.207. [DOI] [PubMed] [Google Scholar]
- 5.Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2(7):a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41(50):14885–14896. doi: 10.1021/bi026469j. [DOI] [PubMed] [Google Scholar]
- 7.Sahara N, Maeda S, Takashima A. Tau oligomerization: A role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res. 2008;5(6):591–598. doi: 10.2174/156720508786898442. [DOI] [PubMed] [Google Scholar]
- 8.Xu S, Brunden KR, Trojanowski JQ, Lee VM. Characterization of tau fibrillization in vitro. Alzheimers Dement. 2010;6(2):110–117. doi: 10.1016/j.jalz.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goedert M, Crowther RA, Spillantini MG. Tau mutations cause frontotemporal dementias. Neuron. 1998;21(5):955–958. doi: 10.1016/s0896-6273(00)80615-7. [DOI] [PubMed] [Google Scholar]
- 10.Paquet D, et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119(5):1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chambraud B, et al. Overexpression of p59-HBI (FKBP59), full length and domains, and characterization of PPlase activity. Biochem Biophys Res Commun. 1993;196(1):160–166. doi: 10.1006/bbrc.1993.2229. [DOI] [PubMed] [Google Scholar]
- 12.Göthel SF, Marahiel MA. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55(3):423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiene-Fischer C, Aumüller T, Fischer G. Peptide bond cis/trans isomerases: A biocatalysis perspective of conformational dynamics in proteins. Top Curr Chem. 2013;328:35–67. doi: 10.1007/128_2011_151. [DOI] [PubMed] [Google Scholar]
- 14.Radanyi C, Chambraud B, Baulieu EE. The ability of the immunophilin FKBP59-HBI to interact with the 90-kDa heat shock protein is encoded by its tetratricopeptide repeat domain. Proc Natl Acad Sci USA. 1994;91(23):11197–11201. doi: 10.1073/pnas.91.23.11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bose S, Weikl T, Bügl H, Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996;274(5293):1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- 16.Chambraud B, Belabes H, Fontaine-Lenoir V, Fellous A, Baulieu EE. The immunophilin FKBP52 specifically binds to tubulin and prevents microtubule formation. FASEB J. 2007;21(11):2787–2797. doi: 10.1096/fj.06-7667com. [DOI] [PubMed] [Google Scholar]
- 17.Chambraud B, et al. A role for FKBP52 in Tau protein function. Proc Natl Acad Sci USA. 2010;107(6):2658–2663. doi: 10.1073/pnas.0914957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol. 1998;153(5):1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plucińska G, et al. In vivo imaging of disease-related mitochondrial dynamics in a vertebrate model system. J Neurosci. 2012;32(46):16203–16212. doi: 10.1523/JNEUROSCI.1327-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melançon E, Liu DW, Westerfield M, Eisen JS. Pathfinding by identified zebrafish motoneurons in the absence of muscle pioneers. J Neurosci. 1997;17(20):7796–7804. doi: 10.1523/JNEUROSCI.17-20-07796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerard M, et al. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson’s disease-like pathology. J Neurosci. 2010;30(7):2454–2463. doi: 10.1523/JNEUROSCI.5983-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399(6738):784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 24.Giustiniani J, et al. Decrease of the immunophilin FKBP52 accumulation in human brains of Alzheimer’s disease and FTDP-17. J Alzheimers Dis. 2012;29(2):471–483. doi: 10.3233/JAD-2011-111895. [DOI] [PubMed] [Google Scholar]
- 25.Santa-Maria I, et al. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012;287(24):20522–20533. doi: 10.1074/jbc.M111.323279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnston JA, Ward CL, Kopito RR. Aggresomes: A cellular response to misfolded proteins. J Cell Biol. 1998;143(7):1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9(13):4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993;16(11):460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 29.Tawk M, et al. A mirror-symmetric cell division that orchestrates neuroepithelial morphogenesis. Nature. 2007;446(7137):797–800. doi: 10.1038/nature05722. [DOI] [PubMed] [Google Scholar]
- 30.Tawk M, et al. Wnt/beta-catenin signaling is an essential and direct driver of myelin gene expression and myelinogenesis. J Neurosci. 2011;31(10):3729–3742. doi: 10.1523/JNEUROSCI.4270-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.