

Abstract
Reactive oxygen species (ROS) comprise a range of reactive and short-lived, oxygen-containing molecules, which are dynamically interconverted or eliminated either catalytically or spontaneously. Due to the short life spans of most ROS and the diversity of their sources and subcellular localizations, a complete picture can be obtained only by careful measurements using a combination of protocols. Here, we present a set of three different protocols using OxyBurst Green (OBG)-coated beads, or dihydroethidium (DHE) and Amplex UltraRed (AUR), to monitor qualitatively and quantitatively various ROS in professional phagocytes such as Dictyostelium. We optimised the beads coating procedures and used OBG-coated beads and live microscopy to dynamically visualize intraphagosomal ROS generation at the single cell level. We identified lipopolysaccharide (LPS) from E. coli as a potent stimulator for ROS generation in Dictyostelium. In addition, we developed real time, medium-throughput assays using DHE and AUR to quantitatively measure intracellular superoxide and extracellular H2O2 production, respectively.
Keywords: Microbiology, Issue 81, Biology (general), Biochemistry, Reactive oxygen species, Superoxide, Hydrogen peroxide, OxyBurst Green, Carboxylated beads, Dihydroethidium, Amplex UltraRed, Phagocytosis, Dictyostelium discoideum
Introduction
Reactive oxygen species (ROS) are involved in a wide variety of biological processes such as host defense, signaling, tissue development and response to injury as well as hypertension and cancer. The well-studied phagosomal NADPH oxidase machinery is dedicated to rapid ROS generation, known as the oxidative burst, to kill bacteria ingested in neutrophils' phagosomes1. In addition, leakage of electrons as a byproduct of the mitochondrial respiratory chain was previously thought to be responsible only for an unregulated source of ROS. But recently, it was identified as an important mechanism to contribute to intraphagosomal killing of bacteria in mouse macrophages2. In recent years, the social amoeba, Dictyostelium, has become a powerful and popular model to study cell intrinsic mechanisms of the innate immune response. Indeed, Dictyostelium and human phagocytes share a surprisingly high level of conservation in molecular machineries responsible for bacteria sensing, engulfment and killing3,4. The homologs of proteins and enzymes related to ROS production or detoxification, such as NADPH oxidases, catalases, superoxide dismutases, can be found in both human and Dictyostelium. As the best studied amoeba, Dictyostelium presents several unique advantages over mammalian model system. They grow at room temperature without the need for CO2, with a doubling time of 8-10 hr. They can be easily kept as adherent or suspension cultures. In addition, thanks to their fully sequenced and annotated haploid genome, and to easy genetic manipulation, Dictyostelium has become a very attractive experimental model organism.
In previous studies, various chlorinated and fluorinated derivatives of fluorescein (collectively known as OxyBurst Green, OBG) that emit fluorescence after oxidation by ROS, have been used as a ROS reporter. Cell-permeant esterified derivatives are used to measure cytoplasmic ROS, whereas cell-impermeant dextran- or protein-coupled derivatives are used to measure extracellular ROS. In particular, OBG BSA-coated beads have already been used to detect phagosomal ROS production in mammalian cells5. However, the microplate reader-based approach can only give an averaged ROS generation curve from a population of cells. With the present protocol, by using Dictyostelium , a professional phagocyte, and optimized experimental conditions, we obtain stable and efficient phagocytosis without conjugating any opsonin onto the beads. DHE has been used in various model systems, such as mammalian neutrophils and macrophages, to detect ROS production6-9. Meanwhile, there are some controversies over the specificity and sensitivity of the method8,10. As an improved version of Amplex Red, the fluorescence intensity of AUR is less sensitive to pH, which makes it more suitable to measure ROS in nonneutral or weakly acidic milieus. AUR has been recently applied in several mammalian systems11,12, but its use in nonmammalian models, which quite often require weakly acidic growth media, has not been reported yet. In addition, there is no published protocol to quantitatively and dynamically measure ROS production and localization in the social amoeba Dictyostelium.
The goal of the presented set of protocols is to provide an easy and versatile solution to monitor various ROS and their localization, and further give insights into ROS-related cellular mechanisms. For the OBG assay, we used live microscopy to monitor the whole process of phagosomal ROS generation after uptake of OBG-coated beads by Dictyostelium cells, and provided a new approach to study the mechanism of intraphagosomal killing of bacteria. We have optimized medium-throughput DHE and AUR assays in Dictyostelium, to measure intracellular superoxide and extracellular H2O2 production, respectively, by using LPS as a potent ROS stimulator. Note that LPS was recently shown to increase the bactericidal activity of Dictyostelium towards phagocytosed bacteria13. In addition, treatment with DEDTC and catalase explicitly confirmed that these two methods specifically measure different types and subcellular localizations of ROS in Dictyostelium. Finally, the OBG assay can be adapted to any adherent phagocytic cell, by further opsonizing the beads with ligands for phagocytic receptors of animal cells. In principle, the DHE and AUR assays can also be fine-tuned for measurements in any adherent or nonadherent cell under appropriate experimental conditions.
Protocol
1. Visualization and Qualitative Measurement of ROS Production in Phagosomes
The OBG-coated beads should be prepared in advance. The beads coating procedures and agar overlay technique are adapted from published references5,14.
Add 1 ml suspension of 3.0 μm carboxylated silica beads (about 1.8 x 109 beads) into an 1.5 ml tube, wash the beads 3x with 1 ml of PBS by quick spin and vortexing.
Resuspend the beads in 1 ml of PBS containing 25 mg/ml cyanamide (to activate the carboxylated silica beads to covalently bind prelabeled BSA), incubate on a wheel for 15 min.
Wash 3x with 1 ml of coupling buffer (0.1 M sodium borate, pH 8.0) by quick spin (centrifuge at full speed in a table top mini centrifuge for 5 sec or alternatively centrifuge at 2,000 x g for 1 min in a table top centrifuge) and vortexing to remove excess cyanamide.
Mix the washed beads with 500 μl of coupling buffer containing 1 mg of OxyBurst Green (OBG) H2HFF (dihydro-2',4,5,6,7,7'-hexafluorofluorescein)-BSA, fill the tube with nitrogen gas from a standard gas-can before capping, and incubate on a wheel for 14 hr (overnight) at RT in the dark.
Wash the beads 2x with 1 ml of quenching buffer (250 mM glycin in PBS) to remove unreacted OBG, and twice with coupling buffer to remove the quenching buffer by quick spin and vortexing.
Add 1 ml of coupling buffer containing 50 μg of Alexa fluor 594 succinimidyl ester to conjugate to BSA. Fill the tube with nitrogen before capping, and incubate on a wheel for 1.5 hr at room temperature in the dark.
Wash the beads 3x with 1 ml of quenching buffer by quick spin and vortexing to stop the reaction, then wash the beads 3x with 1 ml of PBS by quick spin and vortexing.
Finally, resuspend the beads in 1 ml of PBS with 2 μl of 10% w/v azide for long-term storage. Measure the concentration of beads in the suspension with a hemocytometer (usually around 1-2 x 109 beads/ml), fill the tube with nitrogen before capping, and store at 4 °C in the dark.
The coating efficiency of the OBG fluorescein can be tested by checking the emission spectrum using excitation at 500 nm. When oxidized by H2O2 in the presence of horseradish peroxidase (HRP), the fluorescence intensity of oxidized beads, at emission peak (538 nm), is 11-12x higher than that of nonoxidized coated beads.
Harvest exponentially growing Dictyostelium cells from a 10 cm Petri dish, plate different densities of cells on 3 cm dishes with an optically clear plastic or glass bottom, and grow them overnight. Choose the dishes that are about 80% confluent for the experiment. A detailed protocol for cultivating Dictyostelium cells has been published15.
Replace the culture medium with LoFlo medium (LF medium), incubate for 2 hr before the experiment in order to decrease the extracellular and intraendosomal autofluorescence of the HL5C culture medium.
In parallel, melt 10 ml 1.5% bacto agar in LF medium, pour the agar onto a flat surface (i.e. a glass plate of 10 cm x 10 cm) in order to form an agar layer about 1 mm thick, wait for 10-15 min to solidify. Cut the agar layer into 2 cm x 2 cm squares and place in LF medium for later use.
Meanwhile, prepare the spinning-disk or wide field microscope, set the temperature of the environmental chamber at 22 °C and adjust the settings for this experiment. (See additional comments in the discussion).
After 2 hr of incubation, aspirate the LF medium from the 3 cm dish, but the cell monolayer should still be covered by a thin film of medium. Dilute the coated beads to 1.5 x 107 beads/ml, and add 10 μl onto the cell layer.
Take one square agar sheet, drain excess liquid but keep wet. Gently put the agar square on top of the cell layer. Do not move the agar square when it is lying on the cells. The agar overlay is used to increase contact between beads and cells, thereby improving uptake, and it also slightly compresses the cells, keeping them better in the focal plane of the objective.
Place the lid onto the dish, place it on the microscope stage, and automatically take pictures in the red, green and phase channels every 1 min for 2 hr or longer.
Select and focus on cellular events that contain the whole process of phagocytosis. Merge the optimized 3 channels and assemble the pictures into a movie using professional image processing software. Quantify fluorescence intensities of each selected beads in red and green channels, and the ratio of green/red will reflect the dynamic phagosomal ROS production of the cells.
2. Quantitative and Medium-throughput Measurement of Intracellular Superoxide Production
Collect one 80% confluent dish of Dictyostelium in 10 ml of HL5C medium. Centrifuge Dictyostelium cells at 850 x g for 5 min, carefully and completely aspirate excess medium. Resuspend cells in SS6.4 buffer [0.12 M sorbitol in Sorensen buffer (14.69 mM KH2PO4 and 6.27 mM Na2HPO4), pH 6.4], count cells and dilute to a final density of 6 x 106 cells/ml (see additional comments in the discussion).
Add 50 μl of cell suspension into each well of a white nontransparent 96 well plate (see additional comments in the discussion).
Dilute DHE stock (30 mM in DMSO) 500 fold with SS6.4, pipette 50 μl of diluted DHE into each well using a multichannel pipette if necessary. The final concentration of DHE is 30 μM. The reaction starts immediately.
Stimuli or inhibitors can be added at this point, or at other time points according to specific needs.
Use the end point "top reading" mode of a fluorescence microplate reader, with fluorescence excitation/emission at 522 nm/605 nm, read every 2 min for 1 hr, medium shake 5 sec/min, at 22 °C.
3. Quantitative and High Throughput Measurement of Extracellular H2O2 Production
Follow the same steps as described in 2.1 and 2.2.
Prepare diluted HRP by adding 5 μl of HRP stock (100 U/ml in distilled water) into 10 ml of SS6.4, vortex or invert the tube to mix well and keep on ice for later use. The diluted HRP solution is at 0.05 U/ml.
Prepare the AUR reaction mixture by mixing the AUR stock (10 mM AUR in DMSO) and the diluted HRP solution into SS6.4 buffer to the final concentrations of 6.25 μM, and 0.005 U/ml, respectively. As an example, to prepare the reaction mixture for 40 reactions, mix 1600 μl of SS6.4 with 400 μl of diluted HRP and 2.5 μl of AUR stock solution.
Add 50 μl of AUR mixture into each well using a multichannel pipette if necessary. Now the reaction starts.
Stimuli or inhibitors can be added at this point, or at other time points according to specific needs.
Use the end point "top reading" mode of a fluorescence micro plate reader, with fluorescence excitation/emission at 530 nm/590 nm, read every 2 min for 1 hr, medium shake 5 sec/min, at 22 °C.
Representative Results
The generation of ROS in phagosomes can be visualized qualitatively and dynamically by microscopy (File S1). The red fluorescence emitted by Alexa fluor 594 is pH-insensitive and remains constant in the phagosomal environment, while oxidation of OBG increases its fluorescence in the green channel. The emission spectra of in vitro oxidized and nonoxidized OBG-coated beads are compared in Figure 1B, showing a significant increase in intensity after oxidation. To facilitate visualization of the live events, the two channels are merged and the resulting color changes from red to orange during phagocytosis by wildtype AX2 cells within 1 min of uptake. The increase in green fluorescence indicates ROS generation possibly directly in the phagosome. By using a microplate reader, medium-throughput quantitative measurement of intracellular superoxide and extracellular H2O2 production are shown in Figure 2C and Figure 3B, respectively. Basal ROS production from AX2 cells, possibly from regular oxido-reductive metabolism, can be measured. When the cells are stimulated with LPS, the signals for ROS production are significantly increased in both assays. Additionally, in the DHE assay, the decrease of blue fluorescence (dihydroethidium) and the increase of red fluorescence (ethidium) signals are inversely correlated, as expected. The localization of ROS production measured by both DHE and AUR assays are confirmed in Figure 2D and Figure 3C, respectively. Addition of DEDTC (a membrane permeant superoxide dismutase (SOD) inhibitor)16, increased the DHE signal and decreased the AUR signal in a dose-dependent manner, indicating that DEDTC caused the accumulation of the SOD substrate superoxide and the depletion of the SOD product H2O2 expected from the reaction shown in Figure 2A. Alternatively, by adding catalase (a membrane impermeant H2O2 quencher), the intracellular superoxide signal from DHE assay was not affected, while the extracellular H2O2 signal from AUR assay decreased in a dose-dependent manner.
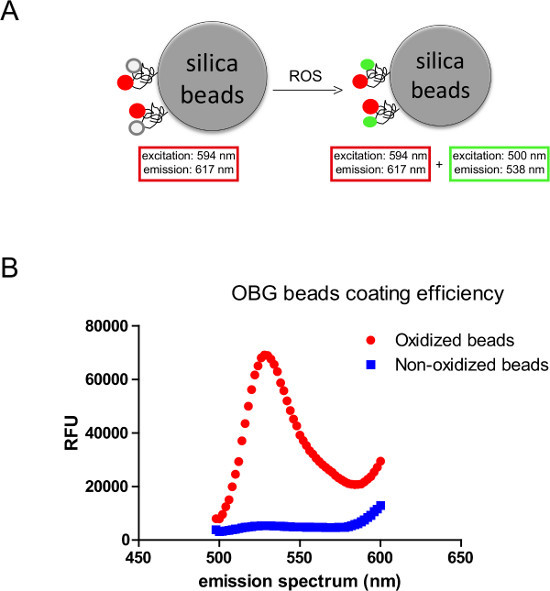 Figure 1. Principle and use of the OBG (OxyBurst Green) assay. A) OBG fluorescein and Alexa fluor 594 are covalently coupled to the surface of 3 μm silica beads via BSA. The fluorescence emission from OBG fluorescein indicates the oxidization by ROS, while the signal from Alexa fluor 594 helps localize the beads and serves as control for fluorescence quantification. B)
In vitro test for the coating efficiency. H2O2 and HRP are used to oxidize the coated beads in vitro. Under excitation at 500 nm, the oxidized beads show a significant emission peak at 538 nm compared to that of nonoxidized beads, with an intensity ratio of at least 11-12 fold.
Figure 1. Principle and use of the OBG (OxyBurst Green) assay. A) OBG fluorescein and Alexa fluor 594 are covalently coupled to the surface of 3 μm silica beads via BSA. The fluorescence emission from OBG fluorescein indicates the oxidization by ROS, while the signal from Alexa fluor 594 helps localize the beads and serves as control for fluorescence quantification. B)
In vitro test for the coating efficiency. H2O2 and HRP are used to oxidize the coated beads in vitro. Under excitation at 500 nm, the oxidized beads show a significant emission peak at 538 nm compared to that of nonoxidized beads, with an intensity ratio of at least 11-12 fold.
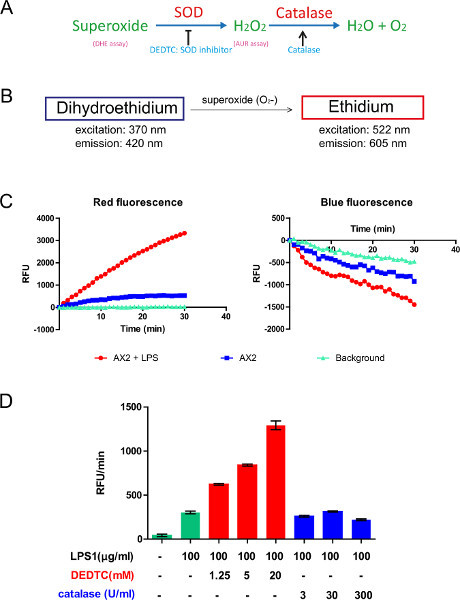 Figure 2. Principle and use of the DHE (Dihydroethedium) assay. A) Summary of the biochemical reactions related to DHE and AUR assays. B) In the cytosol, the membrane permeant DHE has excitation and emission peaks at 370/420 nm, respectively. As a result of oxidation by superoxide, ethidium is able to intercalate into nuclear and mitochondrial DNA and has shifted excitation and emission peaks at 522/605 nm, respectively. The fluorescence intensity of ethidium corresponds to the amount of superoxide production inside the cells. C) A representative experiment shows that, when stimulated with LPS, the dynamic superoxide production from AX2 cells is significantly higher than the basal level from nonstimulated AX2 cells and the background of the reaction in buffer. The decrease of blue fluorescence and the increase of red fluorescence are inversely correlated as expected. D) When treated with various concentrations of DEDTC (a membrane permeant superoxide dismutase inhibitor), the slope (RFU/min) of ethidium production increases in a dose-dependent manner, while treatment with catalase (a membrane impermeant H2O2 quencher) does not affect superoxide production. Click here to view larger figure.
Figure 2. Principle and use of the DHE (Dihydroethedium) assay. A) Summary of the biochemical reactions related to DHE and AUR assays. B) In the cytosol, the membrane permeant DHE has excitation and emission peaks at 370/420 nm, respectively. As a result of oxidation by superoxide, ethidium is able to intercalate into nuclear and mitochondrial DNA and has shifted excitation and emission peaks at 522/605 nm, respectively. The fluorescence intensity of ethidium corresponds to the amount of superoxide production inside the cells. C) A representative experiment shows that, when stimulated with LPS, the dynamic superoxide production from AX2 cells is significantly higher than the basal level from nonstimulated AX2 cells and the background of the reaction in buffer. The decrease of blue fluorescence and the increase of red fluorescence are inversely correlated as expected. D) When treated with various concentrations of DEDTC (a membrane permeant superoxide dismutase inhibitor), the slope (RFU/min) of ethidium production increases in a dose-dependent manner, while treatment with catalase (a membrane impermeant H2O2 quencher) does not affect superoxide production. Click here to view larger figure.
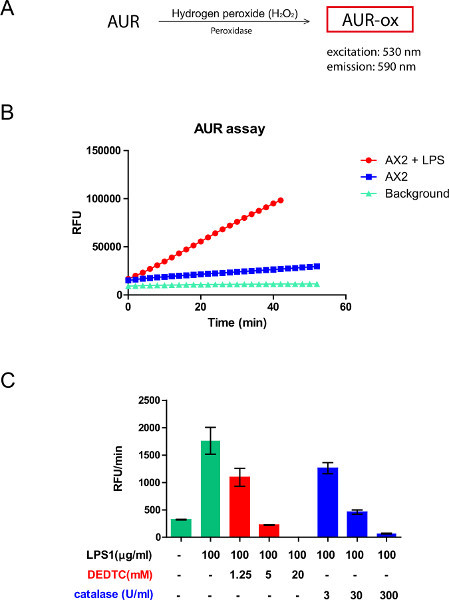 Figure 3. Principle and use of the AUR (Amplex UltraRed) assay. A) The membrane impermeant AUR can be oxidized into its fluorescent resorufin form, AUR-ox, by H2O2 in the presence of a peroxidase (excitation and emission peaks at 530 and 590 nm, respectively). The fluorescence intensity of AUR-ox corresponds to the amount of H2O2 produced outside the cells. B) A representative experiment shows that, when stimulated with LPS, the dynamic H2O2 production from AX2 cells is significantly higher than the basal level from nonstimulated AX2 cells and the background reaction. C) When treated with various concentrations of DEDTC or catalase, the slope (RFU/min) of AUR-ox production decreases in a dose-dependent manner, due to inhibition of H2O2 production from intracellular superoxide and depletion of extracellular H2O2, respectively. Click here to view larger figure.
Figure 3. Principle and use of the AUR (Amplex UltraRed) assay. A) The membrane impermeant AUR can be oxidized into its fluorescent resorufin form, AUR-ox, by H2O2 in the presence of a peroxidase (excitation and emission peaks at 530 and 590 nm, respectively). The fluorescence intensity of AUR-ox corresponds to the amount of H2O2 produced outside the cells. B) A representative experiment shows that, when stimulated with LPS, the dynamic H2O2 production from AX2 cells is significantly higher than the basal level from nonstimulated AX2 cells and the background reaction. C) When treated with various concentrations of DEDTC or catalase, the slope (RFU/min) of AUR-ox production decreases in a dose-dependent manner, due to inhibition of H2O2 production from intracellular superoxide and depletion of extracellular H2O2, respectively. Click here to view larger figure.
File S1. Movie of dynamic phagosomal ROS production in Dictyostelium cells. The time-lapse movie was recorded for 40 min by live microscopy, using a 60X magnification. During the first minute after uptake by phagocytosis, the fluorescence of OBG fluorescein-coated beads changed from red to bright orange, indicating that ROS were likely produced directly inside the phagosomes. Click here to view movie.
Discussion
Compared to the previously described methods, the OBG assay dynamically visualizes the process of phagosomal ROS generation at the single cell level, instead of measuring an average ROS signal from a population of cells. Such population-averaging methods tend to obscure critical information caused by nonsynchronous phagocytosis. We successfully adapted DHE and AUR assays to Dictyostelium and optimized the protocols. Most importantly, we specified the types and subcellular localizations of ROS measured by these two assays. Taken together, the whole set of ROS measurement protocols for Dictyostelium provides a useful and powerful system to study ROS-related cellular mechanisms, such as innate immunity, cell differentiation and intracellular signaling.
For the OBG assay, the preparation of coated beads is critical for the success of the experiment. Due to the complexity of receptor-mediated phagocytic uptake in mammalian cell, the particles need to be opsonized (e.g. with IgGs) to trigger ingestion. Low uptake efficiency in mammalian cells severely limits the visualization of individual events of phagosomal ROS generation. The rate of phagocytosis in Dicytostelium has been observed to be 10-20 fold higher than in a macrophage17, and with the present protocol we obtained robust and efficient phagocytosis without conjugating any opsonin onto the beads. This not only simplifies the bead-coating procedure, but reduces the risks of oxidation of the OBG fluorescein dye during the manipulation. Since OBG fluorescein can be oxidized by light and oxygen, the reagent has to be protected from exposure to ambient light and air as much as possible (e.g. use aluminum foil to cover the tube, and fill the tube with nitrogen or another inert gas), especially for long term storage.
For live imaging, cells can be plated on various dishes with optically clear bottoms. While Dictyostelium adheres slightly better to plastic, most mammalian cells (see below) adhere equally well to glass. Another important step for optimal imaging is to incubate Dictyostelium cells in LF medium (low fluorescence background) for at least 2 hr before proceeding to the imaging steps. Because the background green fluorescence signal of the culture medium (HL5C) needs to be depleted from their endosomal compartments to avoid interference with the signal generated from oxidized coated beads. We also highly recommend performing at least one pilot experiment to optimize the settings of the microscope (e.g. laser intensity, exposure time, etc.), especially for the green channel. The beads constantly emit strong red fluorescence during the whole experiment, but the green fluorescence increases upon reaction with ROS immediately after phagocytosis and plateaus within 5-10 min. Therefore it is better to adjust the settings for the green channel near the end of the experiment when the green fluorescence from most of the beads can be fully visualized. Once the settings are optimized, save it for future experiments in order to achieve reproducible results.
During the OBG assay, healthy cells should continue their random motility and not round up. Otherwise, this is indicative of cell damage, which can be caused either by the drying of the agar overlay or by excessive laser intensity. Therefore, keep enough liquid on the agar overlay for the duration of the whole experiment and also keep the laser intensity and exposure time as low as possible.
In the AUR and DHE assays, the slope of the fluorescence reading curves shows a dose-dependent relationship to the amount of cells used in each well within a certain range. In order to achieve the most reproducible results, it is essential to use identical number of cells for each condition of an experiment. In addition, due to variations in cell size between different cell types, it is highly suggested that during the pilot experiments, different dilutions are tested to obtain a cell monolayer after attachment at the well bottom. For Dictyostelium AX2 cells, 3 x 105 cells form a monolayer and this cell density also applies to many other Dictyostelium wildtype and mutant strains. In order to obtain accurate cell counts, it is best to count after centrifugation, not before, since the centrifugation and pellet resuspension steps can cause some cell loss. We use an automated cell counter to count after resuspending the cell pellet (works best with cell densities above 6 x 106 cells/ml), then precisely dilute the cell samples to 6 x 106 cells/ml.
For the DHE and AUR assays, low signals might be due to use of nonoptimal 96-well plates, and thus, we recommend using white nontransparent 96 well plates. Compared to black or transparent plates, the captured fluorescent signal is intensified by reflection on the white surface, greatly increasing the signal-to-noise ratio. The drawback of white nontransparent plates is, sometimes, strong fluorescent signals may leak into neighboring wells. However, by using 100 μl of reaction volume and the described probe concentration, we did not detect any leaking signal in both DHE and AUR assays. In addition, we recommend performing some pilot experiments to optimize the fluorescence sensitivity of the plate reader. Taking the SynergyMX plate reader as an example, sensitivity 100 and 70 work best for the DHE and AUR assay, respectively. Moreover, it is important to monitor the specific excitation and emission spectra in the conditions of the experiment, with cells. Indeed, we have observed a shift of the emission maximum for AUR to 590 nm, compared to the 581 nm mentioned by the manufacturer. Finally, we obtained slightly better results by exciting at 530 nm instead of 568 nm suggested by the manufacturer.
Compared to an AUR stock solution, the DHE stock solution is much more prone to oxidation, even when protected from light at -20 °C. Thus, it is better to use the DHE stock within 10 days after dissolving from powder. If the color of the stock solution changes from gray to purple or pink, discard it and prepare fresh from powder18.
In the AUR assay, both AUR and HRP might be taken up by macropinocytosis. Therefore the fluorescent signal resulting from oxidation by H2O2 can also partially be generated inside the macropinosomes. However, since the AUR and HPR do not penetrate cellular membranes, the signal measured inside the macropinosomes by the AUR assay is still topologically equivalent to an extracellular signal, as opposed to a signal generated in the cytosol.
Based on a series of preliminary tests for both DHE and AUR assays, it appears important to use a simple buffer that does not contain glucose or other biological media components, because they strongly interfere with both assays and result in a high artefactual fluorescent signal. Therefore, we use Sorensen phosphate buffer complemented with 0.12 M sorbitol, a nondigestible osmolyte used to set an osmolarity similar to the Dictyostelium culture medium19. Note that the high salt content of iso-osmotic buffers used for mammalian cells, such as PBS, is detrimental to Dictyostelium.
The protocols presented here cannot measure all possible ROS, such as hypochlorite, hydroxyl radicals and singlet oxygen, and their subcellular localizations. A more comprehensive picture of ROS generation is still limited by the availability and development of specific probes.
This set of protocols can be easily adapted to other cells, including mammalian phagocytes, with the following modification. For example in the OBG assay, medium without FCS and phenol red can be used to obtain a low background fluorescence. In DHE and AUR assays, PBS buffer can be used instead of SS6.4, and the number of cells plated in each well to obtain a confluent layer can be adapted by trial and error, according to their size. The concentrations of HRP, AUR and DHE can also be optimized in pilot experiments in order to achieve the most sensitive results. However, possible adverse effects of DMSO should be tested when using increased concentrations of AUR and/or DHE reagents. We have tested DHE and AUR assays successfully with another protozoan, Acanthamoeba castellanii and with a mouse BV2 microglia cell line. In addition, the DHE and AUR assays can also be used to measure ROS generation during host-pathogen interactions, for example during infection of Dictyostelium or Acanthamoeba by Mycobacteria marinum.
Disclosures
No conflicts of interest declared.
Acknowledgments
We are grateful to Drs Karl-Heinz Krause and Vincent Jaquet for help and advice to set up these protocols, and also thank Christoph Bauer and Jérôme Bosset from the Bioimaging Platform of the NCCR and Dr. Navin Gopaldass for their technical support. The research is supported by a ProDoc grant of the Swiss National Science Foundation.
References
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey P, Gaudet P, Pilcher KE, Franke J, Chisholm RL. dictyBase and the Dicty Stock Center. Methods Mol. Biol. 2006;346:51–74. doi: 10.1385/1-59745-144-4:51. [DOI] [PubMed] [Google Scholar]
- Basu S, et al. dictyBase 2013: integrating multiple Dictyostelid species. Nucleic Acids Res. 2013;41:676–683. doi: 10.1093/nar/gks1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVen BC, Yates RM, Russell DG. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic. 2009;10:372–378. doi: 10.1111/j.1600-0854.2009.00877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn CA, Simon SR, Schoonen MA. Comparison of fluorescence-based techniques for the quantification of particle-induced hydroxyl radicals. Part. Fibre Toxicol. 2008;5:2. doi: 10.1186/1743-8977-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyrychova I, Ayaydin F, Hideg E. Detecting hydrogen peroxide in leaves in vivo - a comparison of methods. Physiol. Plant. 2009;135:1–18. doi: 10.1111/j.1399-3054.2008.01176.x. [DOI] [PubMed] [Google Scholar]
- Rodrigues JV, Gomes CM. Enhanced superoxide and hydrogen peroxide detection in biological assays. Free Radic. Biol. Med. 2010;49:61–66. doi: 10.1016/j.freeradbiomed.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Chen J, Rogers SC, Kavdia M. Analysis of Kinetics of Dihydroethidium Fluorescence with Superoxide Using Xanthine Oxidase and Hypoxanthine Assay. Ann. Biomed. Eng. 2012. [DOI] [PMC free article] [PubMed]
- Lee CW, Chen YC, Ostafin A. The accuracy of Amplex Red assay for hydrogen peroxide in the presence of nanoparticles. J. Biomed. Nanotechnol. 2009;5:477–485. doi: 10.1166/jbn.2009.1055. [DOI] [PubMed] [Google Scholar]
- Guimaraes-Ferreira L, et al. Short-term creatine supplementation decreases reactive oxygen species content with no changes in expression and activity of antioxidant enzymes in skeletal muscle. Eur. J. Appl. Physiol. 2012;112:3905–3911. doi: 10.1007/s00421-012-2378-9. [DOI] [PubMed] [Google Scholar]
- Peng D, et al. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut. 2012;61:1250–1260. doi: 10.1136/gutjnl-2011-301078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walk A, et al. Lipopolysaccharide enhances bactericidal activity in Dictyostelium discoideum cells. Dev. Comp. Immunol. 2011;35:850–856. doi: 10.1016/j.dci.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, Yumura S, Yumura TK. Agar-overlay immunofluorescence: high-resolution studies of cytoskeletal components and their changes during chemotaxis. Methods Cell Biol. 1987;28:347–356. doi: 10.1016/s0091-679x(08)61655-6. [DOI] [PubMed] [Google Scholar]
- Fey P, Kowal AS, Gaudet P, Pilcher KE, Chisholm RL. Protocols for growth and development of Dictyostelium discoideum. Nat. Protoc. 2007;2:1307–1316. doi: 10.1038/nprot.2007.178. [DOI] [PubMed] [Google Scholar]
- Bloomfield G, Pears C. Superoxide signalling required for multicellular development of Dictyostelium. J. Cell. Sci. 2003;116:3387–3397. doi: 10.1242/jcs.00649. [DOI] [PubMed] [Google Scholar]
- Dieckmann R, Gopaldass N, Escalera C, Soldati T. Autophagosomes and Phagosomes. In: Deretic V, editor. Methods in Molecular Biology. Vol. 445. Totowa, NJ: Humana Press; 2008. pp. 317–328. [DOI] [PubMed] [Google Scholar]
- Amir Y, Edward O-A, Utpal B. A protocol for in vivo detection of reactive oxygen species. Proto. Exch. 2008.
- Neuhaus EM, Soldati T. A myosin I is involved in membrane recycling from early endosomes. J. Cell Biol. 2000;150:1013–1026. doi: 10.1083/jcb.150.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]