

Abstract
Allosteric modulation is a general feature of nicotinic acetylcholine receptors, yet the structural components and movements important for conversions among functional states are not well understood. In this study, we examine the communication between the binding sites for agonist and the modulator morantel (Mor) of neuronal α3β2 receptors, measuring evoked currents of receptors expressed in Xenopus oocytes with the two-electrode voltage-clamp method. We hypothesized that movement along an interface of β sheets connecting the agonist and modulator sites is necessary for allosteric modulation. To address this, we created pairs of substituted cysteines that span the cleft formed where the outer β sheet meets the β sheet constituting the (−)-face of the α3 subunit; the three pairs were L158C-A179C, L158C-G181C and L158C-K183C. Employing a disulfide trapping approach in which bonds are formed between neighboring cysteines under oxidation conditions, we found that oxidation treatments decreased the amplitude of currents evoked by either the agonist (ACh) or co-applied agonist and modulator (ACh + Mor), by as much as 51%, consistent with the introduced bond decreasing channel efficacy. Reduction treatment increased evoked currents up to 89%. The magnitude of the oxidation effects depended on whether agonists were present during oxidation and on the cysteine pair. Additionally, the cysteine mutations themselves decreased Mor potentiation, implicating these residues in modulation. Our findings suggest that these β sheets in the α3 subunit move with respect to each other during activation and modulation, and the residues studied highlight the contribution of this intramolecular allosteric pathway to receptor function.
Keywords: Neuronal nicotinic receptors, Allosteric modulation, Disulfide trapping, Xenopus oocyte expression
1. Introduction
Nicotinic acetylcholine receptors (nAChRs) are widely distributed throughout the central and peripheral nervous systems and are implicated in a range of normal and pathological functions (Albuquerque et al., 2009; Hurst et al., 2013). Neuronal nAChRs are implicated in memory loss and diminished cognitive ability associated with Alzheimer’s disease and other dementias (Haydar and Dunlop, 2010), and mediate nicotine addiction (Ortells and Arias, 2010). Although the molecular and physiological mechanisms of these disorders are not completely understood (Parri et al., 2011; Picciotto and Kenny, 2013), the known nicotinic ligands rivastigmine (e.g., Grossberg et al., 2010) and varenicline (e.g., Mills et al., 2012), for example, are currently in clinical use. Recently, interest has grown in allosteric modulators of nAChRs as possible therapeutic targets (e.g., Maelicke and Albuquerque, 2000; Williams et al., 2011).
nAChRs are pentameric, membrane-bound ligand-gated ion channels that are part of the Cys-loop superfamily (Hurst et al., 2013). Binding sites for agonists and competitive antagonists are located at subunit interfaces. Due to the radial asymmetry of subunits (Brejc et al., 2001), these sites have sidedness, with α subunits contributing (+)-face residues to the canonical site, and the (−)-face residues coming from a diverse set of neighboring subunits, depending on whether the receptor is homo- or heteromeric. Based on several crystal structures determined for the muscle-type nAChR (Unwin, 2005), bacterial homologs (Bocquet et al., 2009; Hilf and Dutzler, 2008) and the extracellular domain and homologs thereof (e.g., Brejc et al., 2001; Dellisanti et al., 2007; Hansen et al., 2005; Li et al., 2011), the general structure of nAChRs and related Cys-loop proteins is well-known. However, the diversity among ion channel subunit genes, receptor stoichiometries and subunit arrangements means that homology models are of limited use at the atomic scale for specific subtype residues (Hurst et al., 2013). Understanding how individual residues contribute to ligand binding and receptor movement at this scale is critical for rational drug design.
In a similar way, homology-based structural models give little information about the conformational changes that are the hallmark of receptor function. Unwin (2005) compared closed and putatively open forms of the muscle-type nAChR to reveal the major structural changes in the full receptor. These changes have been substantiated and refined by the comparison of the ELIC (Hilf and Dutzler, 2008) and GLIC structures (Bocquet et al., 2009; Hilf and Dutzler, 2009), bacterial homologs of the Cys-loop receptors, which are thought to correspond to closed and open forms, respectively. Many studies, using a wide range of techniques, have indicated that the C loop, a component of the canonical binding site, moves to “cap” the agonist (e.g., Hansen et al., 2005; Mukhtasimova et al., 2009; Wang et al., 2009). However, most of this work has employed the acetylcholine binding protein, a homolog of just the nAChR extracellular domain, meaning that the picture of movement for this region of the full receptor is incomplete. Disulfide trapping in receptors with substituted cysteines has been useful to deduce regional motions in both GABAA receptors (e.g., Horenstein et al., 2001; Venkatachalan and Czajkowski, 2008) and nAChRs (e.g., Mukhtamisova and Sine, 2007). On the whole, a major focus in this body of work has been to understand the allosteric communication between agonist binding sites and the channel pore (e.g., Lee and Sine, 2005; Purohit and Auerbach, 2007). Thus, while progress has been made elucidating the nature of movement in Cys-loop receptors and their intra-molecular pathways of communication, much remains to be discovered.
A related interest is the communication of allosteric modulator sites with agonist sites and the channel gate. We have demonstrated that the anthelmintics morantel (Mor) and oxantel potentiate α3β2 nAChRs from a site in the β2(+)/α3(−) interface, which is structurally homologous to the canonical agonist site (Cesa et al., 2012; Seo et al., 2009). Several small-molecule modulators, both positive and negative, specific for the α4β2 subtype have been discovered (reviewed in Pandya and Yakel, 2011), but for only a few of these have the binding sites been identified. For certain ligands of α7 nAChRs such as galanthamine and anthelmintics, evaluating the interactions of modulator and agonist sites is more complicated because each occurs at the same type of α7(+)/α7(−) interface (Bartos et al., 2006; Ludwig et al., 2010; Rayes et al., 2009). In addition, two groups have recently determined that an α4(+)/α4(−) site in the α4β2 receptors binds agonist, dictating the overall receptor sensitivity (Harpsøe et al., 2011; Mazzaferro et al., 2011). The emerging picture from these studies is the generality of the nAChR interfaces as potential ligand binding sites, which might have been predicted from the symmetry of the system and allostery theory (Wyman and Gill, 1990).
Given the spatial relationship of the Mor and ACh sites at the (−) and (+) interfaces, respectively, of the α3 subunit (Figure 1), we hypothesize that a subset of α3 residues mediates communication between these two sites. The β sandwich structure of Cys-loop receptor extracellular domains suggests intra-subunit movement might occur where the β sheets comprised of the β9-10-7 strands and β1-2-6-5/5′ strands meet. Using cysteines substituted into this region for a disulfide trapping approach, we investigated the role of these residues in channel activation, aiming to find evidence of movement in this region by discriminating among resting and active states of the system. We demonstrate that disulfide bonds formed here perturb both ACh activation and Mor modulation. Thus, the residues we studied appear to be involved in α3 intra-subunit movement that may functionally connect agonist and modulator sites.
Figure 1.

Residues Lining the Edges of the β-Sandwich. A. A ribbon diagram of the α3 subunit extracellular domain is shown, with the four residues examined in this study in space-filling format. Mor and ACh indicate the relative locations of the binding sites of modulator and agonist at the β(+)/α(−) and α(+)/β(−) interfaces, respectively. Residue numbering is for the mature α3 protein; this rendering is derived from the homology model a3b2rr.pdb (Sallette et al., 2004). B. The sequences of several rat nAChR α subunits in the region of interest, and the rat GABA receptor γ2 subunit, are aligned. The gap in the alignment excludes the F loop, which varies in length across nAChR subunits and Cys-loop receptors. The solid arrows indicate the extent of the β8 and β9 strands as originally identified for the acetylcholine binding protein (Brejc et al., 2001), while the dashed arrows indicate these structures in the Torpedo model (Unwin, 2005).
2. Materials and Methods
2.1 Reagents
All chemicals used, unless otherwise noted, were reagent grade and obtained from Sigma (St. Louis, MO). Morantel (Mor) is 1,4,5,6-tetrahydro-1-methyl-2-(2-[3-methyl-2-thienyl]ethenyl)pyrimidine, tartrate salt. MTS-dansyl (dansylamidoethyl methanesthiosulfonate) was obtained from Toronto Research Chemicals (Toronto, ON, Canada); some experiments also employed MTSET (2-(trimethylammonium)ethyl methanethiosulfonate) and MTS-biotin, also from TRC.
2.2 Nicotinic Receptor Clones and Mutagenesis
Wild type rat α3 and β2 subunits in pGEMHE-based vectors were a gift from Dr. Charles Luetje (University of Miami); clones were originally isolated in the lab of Dr. Jim Patrick (Baylor University; Boulter et al., 1987). All mutant subunits were custom synthesized by GenScript (Piscataway, NJ), except α3G181C which was prepared in our laboratory using standard thermocycling methods (e.g., Seo et al., 2009). Mutations were verified by sequencing of the entire extracellular domain using capillary electrophoresis of dye-detected, dideoxy-generated fragments. Unless otherwise noted all α3 and β2 residue numbering corresponds to that in the structure a3b2rr.pdb (http://www.ebi.ac.uk/compneur-srv/LGICdb/HTML/a3b2rr.html) (Sallette et al., 2004); these position numbers are smaller by two compared with numbering used elsewhere in the literature, a discrepancy which arises due to homology modeling based on a crystal structure of a protein of different sequence. The cDNA plasmids were linearized with a unique restriction enzyme, and then made RNase-free by phenol-chloroform extraction. cRNAs were transcribed in vitro from these using the T7 kit from Ambion (Life Technologies; Carlsbad, CA), and were diluted to 0.5 μg/μL with RNase-free water and stored at −20°C.
2.3 Oocyte Preparation and Injection
Xenopus laevis oocytes were harvested from oocyte-positive frogs (obtained from Nasco, Ft. Atkinson, WI), using procedures approved by the Grinnell College Institutional Animal Care and Use Committee and in accord with National Institutes of Health guidelines; all efforts to minimize the number of animals used and minimize suffering were made. On some occasions oocytes were prepared from whole ovary tissue obtained directly from Nasco. In both cases stage V and VI oocytes were prepared by collagenase treatment and manual selection, following published procedures (Bertrand et al., 1991). Additionally, several experiments used pre-sorted stage VI oocytes obtained from Ecocyte Bioscience (Austin, TX). Oocytes were maintained at 16°C in Barth’s medium: 88 mM NaCl, 1.0 mM KCl, 2.5 mM NaHCO3, 0.30 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 15 mM HEPES, and 2.5 mM sodium pyruvate, pH 7.6, supplemented with 100 U/mL penicillin/streptomycin and 50 μg/mL gentamicin (Life Technologies; Carlsbad, CA). A Nanoject microinjector (Drummond; Broomall, PA) was used to inject each oocyte with 46 nL of a 1:1 (v:v) combination of the desired α and β subunits, prepared from the respective 0.5 μg/μL stock solutions. Following 2–3 days for protein expression, current responses could be recorded for up to 7 subsequent days. During this time the Barth’s solution was changed daily and any dead cells removed.
2.4 Voltage-Clamp Recordings
Macroscopic currents were recorded using a Geneclamp 500B amplifier and a Digidata 1322A data acquisition system (Molecular Devices; Sunnyvale, CA) using the two-electrode voltage-clamp method, as previously described (Cesa et al., 2012; Seo et al., 2009; Wu et al., 2008). Voltage was clamped at −60mV, and leak currents were generally 0–200 nA, although in some cases higher leak currents were tolerated if the baseline was stable. Recording electrodes were filled with 3.0 M KCl and selected for resistances between 0.5 and 4.0 MΩ. Perfusion and drug administration were controlled via VC-6 solenoid valve systems (Warner Instruments; Hamden, CT). Cells were perfused with oocyte Ringer’s medium (OR2; 115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 10 mM HEPES, pH 7.3) until the baseline stabilized prior to recording, typically 90–120 sec. Drug challenges lasted 5 sec unless otherwise noted, and the oocytes were typically washed with OR2 for 100 sec between challenges, sufficient time for the current to return to baseline.
In experiments involving oxidation or reduction (throughout referred to as treatments) of the putative disulfide bonds, oxidation was typically accomplished via perfusion for 5 min with a solution that was 4.4 mM H2O2, 100 μM ACh and 10 μM Mor. The solution was prepared fresh before every second or third cell from a refrigerated 30% (w/v) H2O2 stock. Reduction was typically accomplished via perfusion for 5 min with a 40 μM DTT solution, which was prepared fresh every second or third cell from frozen stock. Following treatment with either H2O2 or DTT, oocytes were washed for 100 sec with OR2 before any challenges (5-sec application of ACh or ACh + Mor) were made. As an emphasis of this study, we altered the nature of the treatments, the conditions of which are given in the figure legends and text. Typically two challenges were administered before and after a given treatment, with deviations from these regular conditions noted where appropriate. We refer to the responses prior to the treatment as Icontrol, and those after H2O2 (which may contain ACh and/or Mor) and DTT treatments as Ioxidation and Ireduction, respectively. For experiments in which the treatment also contained ACh, Mor or ACh + Mor, we refer to these conditions as with activators, to avoid listing these conditions separately.
Methanethiosulfonate (MTS) reagents react specifically with free sulfhydryl groups. Established procedures were followed for experiments with MTS-dansyl (Karlin and Akabas, 1998); small aliquots of the reagent were dissolved in water and kept on ice, and then diluted to the working concentration in OR2 immediately prior to use in the experiment. Reaction was accomplished by allowing 10 mL of the MTS solution to flow through the apparatus (300 μL recording chamber), then stopping flow and incubating in the reagent until a total of 2 min had elapsed from the start of MTS solution flow. Oocytes were then washed for 100 sec with OR2 before any further challenges or treatments were applied. MTS experiments typically consisted of three sets of challenges given to the cells (See Figure 5). Responses to challenges before any treatment are again referred to as Icontrol, those after an oxidation, reduction, control or other treatment as Ipost-treatment, and finally those following reaction with the MTS reagent as IMTS.
Figure 5.

MTS Probing Confirms Disulfide Bond Oxidation and Reduction. A. Sample current traces for oocytes expressing the α3L158CG181Cβ2 mutant receptor are shown for separate experiments probing oxidation (H2O2, left) and reduction (DTT, right) treatments by a sequential treatment with methanesulfonate dansyl (MTS). Responses evoked by application of 5 μM ACh (solid traces) or 5 μM ACh and 10 μM Mor (dotted traces) were measured before and after treatment, and then after a 2-min exposure to 50 μM MTS-dansyl; the treatments were 4.4 mM H2O2 + 100 μM ACh + 10 μM Mor (5 min) and 40 μM DTT (5 min). The holding potential was −60 mV. B. Collated data for a series of five experiments on the α3L158CG181Cβ2 mutant receptor similar to that described in A are shown, for responses evoked by either 5 μM ACh alone or 5 μM ACh + 10 μM Mor. The mean percentage change following MTS-dansyl treatment [(Ipost MTS/Ipre MTS −1)×100] is plotted for each experiment; error bars represent the relative SEM. Control (white bars) refers to the effects (Ipost MTS/Ipre MTS) of 5 min MTS-dansyl exposure. H2O2 w/A + M (hashed bars) is the standard treatment of 5 min 4.4 mM H2O2 + 100 μM ACh + 10 μM Mor, whereas H2O2 alone (gray bars) is simply 5 min at 4.4 mM H2O2. H2O2 w/A + M with wash (hatched bars) was an experiment in which the oxidation treatment was as described above, but oocytes were then washed for 200–300 sec with saline prior to the MTS probe (See also Figure 4). Finally, DTT (black bars) refers to treatment of a 5-min exposure at 40 μM. Replicates were n = 10 (Control); n = 20 (H2O2 w/A + M); n = 6 (H2O2 alone); n = 11 (H2O2 w/A + M with wash); n = 12 (DTT).
2.5 Data Analysis and Statistics
The concentration-response behavior for all the mutants studied was characterized across the micromolar to millimolar range for ACh alone and ACh + Mor (10 μM, unless otherwise specified). Peak currents were normalized to that evoked by the highest ACh concentration (alone) for each data set, and means and standard errors of the mean (SEM) were calculated for the collated sets of replicates. The Hill equation [fractional response = Emax/(1+(EC50/[agonist])nH)] was fit to titration data to compare the various mutants by these parameters (Table 1). EC50 is the concentration giving half-maximal response, nH is the slope of the curve at the midpoint, and Emax is the maximum response at saturating agonist concentrations. By definition, Emax = 1 for titrations of ACh alone, but Emax varied as 0.90–2.60 for titrations of ACh + 10 μM Mor. In addition, for the α3A179Cβ2 and α3L158CG181Cβ2 receptors, a secondary effect at high ACh concentrations in the presence of Mor, most likely open channel block (Wu et al., 2008), resulted in fit Emax values smaller than the absolute largest (average) response by 25% and 6%, respectively. We did not study this phenomenon further, but avoided complications from the effect by studying sub-saturating ACh concentrations.
Table 1.
Agonist and Modulator Evoked Response Characteristics
| Subtype | ACh Response a | Mor Modulation Response | |||
|---|---|---|---|---|---|
| EC50 (μM) | nH | EC50 (μM) | nH | Emax | |
| α3β2 | 66 ± 9 | 0.75 ± 0.07 | 10 ± 6 | 1.37 ± 0.80 | 2.6 ± 0.3 |
| α3L158Cβ2 | 4.7 ± 0.9 | 0.80 ± 0.11 [6] b | 0.91 ± 0.16 | 1.47 ± 0.39 | 1.6 ± 0.1 |
| α3A179Cβ2 | 37 ± 5 | 0.79 ± 0.08 [8] | 3.2 ± 1.5 | 1.79 ± 0.75 | 1.6 ± 0.1 |
| α3G181Cβ2 | 41 ± 4 | 1.10 ± 0.11 [5] | 20 ± 2 | 1.08 ± 0.12 | 2.3 ± 0.1 |
| α3K183Cβ2 | 11 ± 2 | 2.43 ± 0.78 [9] | 21 ± 2 | 1.61 ± 0.22 | 0.97 ± 0.02 |
| α3L158C·A179Cβ2 | 4.2 ± 0.6 | 1.19 ± 0.20 [7] | 1.7 ± 0.4 | 1.70 ± 0.50 | 1.1 ± 0.1 |
| α3L158C·G181Cβ2 | 2.6 ± 0.7 | 0.93 ± 0.23 [5] | 0.74 ± 0.15 | 1.18 ± 0.25 | 1.3 ± 0.1 |
| α3L158C·K183Cβ2 | 34 ± 4 | 1.11 ± 0.14 [6] | 25 ± 5 | 0.95 ± 0.16 | 0.90 ± 0.04 |
Fits to the Hill equation for experiments with ACh alone and ACh + 10 μM Mor; with fit parameters as explained in Section 2.5. Emax is the asymptote at saturating concentrations of ACh. Wild type α3β2 data are from Cesa et al., 2012. In all experiments for the three double mutants, recordings were made on naïve oocytes, having had no previous oxidation or reduction treatment.
[n] indicates the number of replicate oocytes for each measurement, and both ACh alone and ACh + 10 μM Mor experiments were determined on the same set of oocytes. Values of Emax for all mutants differ significantly from that of wild type (p < 0.05, t-test); all EC50 values differ (ACh or ACh + Mor, respectively) from wild type, except the EC50 (ACh) for α3A179Cβ2 (p = 0.072).
Our primary interest was the comparison of various treatments and effects across mutant pairs. Accordingly, on indicated data sets, we performed one-way analysis of variance (ANOVA), typically on ratios of currents after and before a treatment (Ipost/Ipre), and Tukey’s post hoc test for significant differences of means; ANOVA results are given in Table 2. Due to the extensive use of a pre/post design for effects of treatments, we also used a paired comparison t-test to examine differences between the relevant set of peak amplitude currents before and after treatment; these analyses, noting the specific currents compared, are given in Table 3. We generally consider p < 0.05 to indicate statistical significance, but are cautious of over-interpreting some small differences observed in this study, given the problem of multiple comparisons (type I error).
Table 2.
Analysis of Variance (ANOVA) for Various Data Sets
| Experiment | Measurement | Challenge | Means Compared | F-statistic | p value |
|---|---|---|---|---|---|
| Figure 2 | Ioxidation/Icontrol a | 100 μM ACh | H2O2 alone, H2O2+ACh, H2O2+Mor, H2O2+ACh+Mor | 3.66 | 0.024 |
| “ | Ioxidation/Icontrol | 100 μM ACh + 10 μM Mor | H2O2 alone, H2O2+ACh, H2O2+Mor, H2O2+ACh+Mor | 0.42 | 0.737 |
| Figure 3 | Itreatment/Icontrol b | 100 μM ACh, 100 μM ACh + 10 μM Mor | DTT, H2O2+ACh+Mor | 18.2 | 9.13 × 10−5 |
| Figure 4 | Itime point/Icontrol | 100 μM ACh + 10 μM Mor | All 13 post-treatment c time points | 4.05 | 0.000102 |
| Figure 5 | IMTS/Ipost-treatment d | 5 μM ACh | Control, H2O2+ACh+Mor, H2O2 alone, H2O2+ACh+Mor*, DTT | 40.5 | 2.67 × 10−16 |
| “ | IMTS/Ipost-treatment | 5 μM ACh + 10 μM Mor | Control, H2O2+ACh+Mor, H2O2 alone, H2O2+ACh+Mor*, DTT | 2.30 | 0.069 |
| Figure 6 | Ioxidation/Icontrol | ACh | H2O2+ACh+Mor and H2O2 alone for 3 Double Cysteine Pairs e | 4.18 | 0.00593 |
| “ | Ioxidation/Icontrol | ACh + Mor | H2O2+ACh+Mor and H2O2 alone for 3 Double Cysteine Pairs | 4.25 | 0.00327 |
Four oxidation treatment conditions were as noted by Means Compared and in the Figure 2 legend.
The ratios IDTT/Icontrol and IH2O2/Icontrol for the two challenges listed were compared.
As described in the Figure 4 legend, these means were in groups from three experiments: post-oxidation, repeated challenge (100–500 sec); post-oxidation with washout (100, 400, 500 sec); 5-min ACh + Mor exposure, repeated challenge (100–500 sec).
The five treatments were as noted by Means Compared and in the Figure 5 legend.
Three double-cysteine mutants were α3L158CA179Cβ2, α3L158CG181Cβ2, α3L158CK183Cβ2.
Table 3A.
Paired t-Test for Figure 2 Data Sets
| Comparison a | Treatment | Challenge | p Value |
|---|---|---|---|
| Ioxidation vs. Icontrol | H2O2 alone | 100 μM ACh | 0.0716 |
| H2O2 + ACh | “ | 0.0021 | |
| H2O2 + Mor | “ | 0.0222 | |
| H2O2 + ACh + Mor | “ | 0.0346 | |
| Ireduction vs. Ioxidation | H2O2 alone | 100 μM ACh | 0.0137 |
| H2O2 + ACh | “ | 0.0043 | |
| H2O2 + Mor | “ | 0.1282 | |
| H2O2 + ACh + Mor | “ | 0.0276 | |
| Ioxidation vs. Icontrol | H2O2 alone | 100 μM ACh + 10 μM Mor | 0.0232 |
| H2O2 + ACh | “ | 0.0226 | |
| H2O2 + Mor | “ | 0.0105 | |
| H2O2 + ACh + Mor | “ | 0.0451 | |
| Ireduction vs. Ioxidation | H2O2 alone | 100 μM ACh + 10 μM Mor | 0.0220 |
| H2O2 + ACh | “ | 0.0151 | |
| H2O2 + Mor | “ | 0.0105 | |
| H2O2 + ACh + Mor | “ | 0.0219 |
Paired comparisons are listed as Ipost vs. Ipre; ‘oxidation’ refers to the four oxidation treatments listed.
3. Results
In this study, we sought evidence for motion in the nicotinic acetylcholine receptor α subunit underlying modulation of channel activation. We postulate that such motion is part of the allosteric communication between the β(+)/α(−) modulator and α(+)/β(−) agonist sites. Accordingly, we examined a set of double-cysteine-substituted α3 receptors (Figure 1) with a disulfide trapping approach for perturbing motion.
3.1 Oxidation and Reduction Reversibly Alter Evoked Currents
Figure 2A shows sample traces of evoked currents for the α3L158CG181Cβ2 receptor (left panel), demonstrating a key finding. Exposing the oocyte to H2O2, co-applied with activators ACh and morantel (Mor), decreased the currents evoked by both ACh alone and co-applied ACh and Mor. Following this treatment, the magnitude of the original control current was recovered by further treating with a low concentration of DTT. In contrast, oxidation did not alter currents evoked from wild type α3β2 receptors (right panel), nor did reducing treatment. Thus, we interpret the effects of oxidation and reduction as stemming from the reactivity of the cysteines introduced at the α3L158 and α3G181 positions. Importantly, the α3L158CG181Cβ2 double mutant and wild type α3β2 receptors are potentiated by 10 μM Mor at this ACh concentration (~EC95 and ~EC60, respectively; see Table 1). The degree of potentiation (IACh+Mor/IACh) of nAChRs depends on the concentrations of agonist and modulator (e.g., Wu et al., 2008), and in the present study we were interested in whether disulfide trapping might show differential effects for activation by agonist alone compared to potentiation. In most experiments on the α3L158CG181Cβ2 mutant we used 100 μM ACh + 10 μM Mor because this combination showed the maximal potentiation.
Figure 2.

A Double-Cysteine Mutant Is Modified by Oxidation and Reduction. A. Sample current traces for an oocyte expressing the α3L158CG181Cβ2 mutant receptor (left) or the wild type α3β2 receptor (right) are shown. Each was treated with a solution of 4.4 mM H2O2 + 100 μM ACh + 10 μM Mor (5 min) followed by 40 μM DTT (5 min); before and after each treatment, responses to 100 μM ACh alone (solid traces) and 100 μM ACh and 10 μM Mor (dotted traces) were evoked. The holding potential was −60 mV. In the left group, traces are offset by 5 sec for clarity. B. Collated data for experiments like those described in A are shown for the double-cysteine mutant α3L158CG181Cβ2 in which responses were evoked by 100 μM ACh alone. The four oxidation treatments were as indicated, all with 4.4 mM H2O2; ACh was 100 μM when present and Mor was 10 μM when present. The reduction treatment was subsequent to oxidation, using 40 μM DTT in all cases. Peak currents following the two treatments were normalized to the pre-treatment control response, and error bars represent the SEM. Replicates were n = 12 (H2O2 alone); n = 5 (+ACh); n = 5 (+Mor); n = 11 (+ACh/Mor). C. Collated data for experiments like those described in A are shown for the double-cysteine mutant α3L158CG181Cβ2 in which responses were evoked by 100 μM ACh and 10 μM Mor. These were the same set of cells as the experiments in B, with ACh + Mor responses evoked after ACh-alone challenges throughout the experiment.
Reasoning that different receptor conformations might position the cysteines of α3L158CG181Cβ2 differently with respect to each other, we examined four different oxidation treatments. Results of these experiments are shown in Figure 2B and 2C. Application of 4.4 mM H2O2 alone (white bars) had no apparent effect on ACh-evoked currents, but decreased those evoked by ACh and Mor by 24%. Co-application of 100 μM ACh and 10 μM Mor with H2O2 (black bars) decreased both evoked currents by 26%. To further refine whether agonist or modulator gives rise to the differences in oxidation treatment, we also tested co-application of ACh or Mor separately with H2O2. In both cases (light gray and dark gray bars, respectively), evoked currents decreased (17–38%) following oxidation, suggesting that both ACh and Mor induce motion in the receptor that enables disulfide bond formation at these positions.
Analysis of variation (ANOVA) for these data and Tukey’s post hoc test, separated by challenge (Table 2), showed that H2O2 alone treatment differed significantly from the other three for ACh challenge, whereas the ACh + Mor challenges showed no differences. Nonetheless, all decreases in current except for H2O2 alone (ACh evoked) were significant by the paired comparison t-test (Table 3A). Because changes in IACh+Mor and IACh were generally parallel in these experiments, changes in potentiation were minimal, even for the H2O2 alone condition (p = 0.0628, paired comparison).
Following the oxidation treatment in all these experiments (Figure 2B and 2C), we tested whether the effects were reversible by treating with 40 μM DTT. In all cases, the evoked responses were at least as large as the original control responses after the DTT treatment, if not larger. All increases were significant except following H2O2 + Mor (ACh evoked; Table 3A). Together, these results indicate that if a disulfide bond is formed between cysteines at positions 158 and 181 in the α3 subunit, that bond can be reduced.
As noted above (Figure 2A), these oxidation and reduction treatments had no effects on wild type rat α3β2 evoked currents. Although these subunits contain cysteines in their extracellular domains, these are oxidized as disulfides either as the Cys-loop that defines this superfamily of receptors, or as the vicinal pair characteristic of α subunits. Replicating the experiments shown in Figure 2A as examples (right panel), we found that IH2O2/Icontrol = 1.00 ± 0.03 (n = 4) and 0.96 ± 0.02 (n = 5) for ACh- and ACh/Mor-evoked currents, respectively, followed by IDTT/IH2O2 = 1.00 ± 0.02 and 0.96 ± 0.02 for those cases. Statistical analysis (paired comparison t-test) gave p values ranging from 0.095 to 0.490, supporting the conclusion that, unlike the α3L158CG181Cβ2 mutant, wild type α3β2 receptors are not changed by the oxidation and reduction treatments used in this study.
Similarly, H2O2 and DTT treatment of the single mutants α3L158Cβ2 and α3G181Cβ2, expressed and tested separately, failed to change evoked currents: IH2O2/Icontrol = 0.78 ± 0.08 (n = 4, p = 0.133) and IDTT/IH2O2 = 0.96 ± 0.17 (n = 4, p = 0.347) for α3L158Cβ2 ACh + Mor currents; and IH2O2/Icontrol = 0.94 ± 0.03 (n = 3, p = 0.103) and IDTT/IH2O2 = 0.96 ± 0.06 (n = 3, p = 0.226) for α3G181Cβ2 ACh + Mor currents.
We were also interested in the effects of the cysteine substitutions themselves, which we characterized in a standard fashion by measuring concentration-response behavior for ACh with and without 10 μM Mor co-applied (e.g., Cesa et al., 2012). The data for these experiments are compared, along with those of wild type α3β2 receptors, in Table 1. The ACh potency is increased for the α3L158CG181Cβ2 double-cysteine mutant relative to wild type receptors (EC50 of 2.6 μM vs. 66). While there is still measurable Mor potentiation for the double mutant, with an apparent increase in potency (EC50 = 0.67 μM for ACh + 10 μM Mor) and increased efficacy (Emax = 1.5), this potentiation is much smaller than that observed for wild type. Interestingly, the double-cysteine mutant behavior appears dictated by the α3L158C, because the pharmacological profile of the double mutant is closer to this single mutant, whereas the α3G181C single mutant profile is very similar to wild type (Table 1).
We observed a similar pattern for activation by Mor alone of the two single mutants and the double mutant. These titrations were characterized by EC50 and Emax (relative to internal ACh control) values of 12 ± 7 μM and 0.021 ± 0.002 (n = 5) for α3L158Cβ2; 41 ± 26 μM and 0.32 ± 0.05 (n = 3–9) for α3G181Cβ2; and 28 ± 10 μM and 0.074 ± 0.009 (n = 3) for α3L158CG181Cβ2. Thus, Mor alone would make only a minor contribution to channel gating for these mutants, and the L158C mutant dominates the double mutant.
In some experiments, reduction following oxidation gave currents apparently larger than the original controls, suggesting the possibility that some disulfide bonding was already present in the naive (untreated) double-cysteine mutant receptors. To examine this possibility, we tested α3L158CG181Cβ2 responses first exposing oocytes to DTT, and then treating with H2O2. Results of these experiments are given in Figure 3A, and the (reverse order) oxidation-then-reduction results are plotted in the same way in Figure 3B for comparison. Even prior to H2O2 exposure, treatment with DTT increased evoked responses; ACh- (white bars) and ACh/Mor-evoked currents (gray bars) were 25% and 21% larger, respectively. Subsequently, the standard (H2O2 + ACh + Mor) oxidation decreased currents, by 52% and 46% relative to the post-reduction values, and appear smaller than the original pre-treatment controls, as expected. ANOVA and Tukey’s test revealed that the two post-reduction challenges differed significantly from the two post-oxidation challenges (Table 2). Although the current increases in these particular experiments were not significant (p > 0.05), perhaps because of a comparatively low number of replicates (Table 3B), in several others in which we first treated naive oocytes with DTT, the reduction effect was significant. In six separate experiments (data not shown), measuring both ACh and ACh + Mor responses, p values for the paired comparisons of IDTT vs. Icontrol ranged from 0.000012 to 0.0032. Thus, we are confident that reduction following control responses increases currents for the α3L158CG181Cβ2 receptor.
Figure 3.

Oxidation and Reduction of the Double-Cysteine Mutant Are Mutually Reversible. The effects of the order of sequential oxidation/reduction treatments are shown. Responses of the α3L158CG181Cβ2 mutant evoked by 100 μM ACh (white bars) and by 100 μM ACh + 10 μM Mor (gray bars) were normalized in all cases to the control response evoked by ACh alone (pre-treatment). A. Oocytes were exposed to 40 μM DTT (5 min) followed by 4.4 mM H2O2 with 100 μM ACh and 10 μM Mor (5 min). Replicates were n = 4, and error bars represent the SEM. B. Oocytes were exposed to 4.4 mM H2O2 with 100 μM ACh and 10 μM Mor (5 min) followed by 40 μM DTT (5 min). Replicates were n = 11, and error bars represent the SEM; data were also shown in Figure 2B and C.
Table 3B.
Paired t-Test for Figure 3 Data Sets
| Comparison a | Treatment | Challenge | p Value |
|---|---|---|---|
| Ireduction vs. Icontrol | H2O2 + ACh + Mor | 100 μM ACh | 0.1107 |
| H2O2 + ACh + Mor | 100 μM ACh + 10 μM Mor | 0.0805 | |
| Ioxidation vs. Ireduction | H2O2 + ACh + Mor | 100 μM ACh | 0.0982 |
| H2O2 + ACh + Mor | 100 μM ACh + 10 μM Mor | 0.0280 |
In sum, oxidation and reduction treatments alter α3L158CG181Cβ2 currents, and these treatments are mutually reversible. That is, they do not depend on the order applied. This suggests that the putative disulfide bonds mediating these effects exist in naive, untreated receptors in some proportion with free sulfhydryl groups at positions 158 and 181.
3.2 Putative Disulfide Bonds Are Labile and Resistant to Further Modification
In certain experiments, it appeared that the extent of the oxidation effect on the α3L158CG181Cβ2 mutant might depend on the time elapsed after the treatment or on the number of subsequent challenges given. We therefore designed a systematic test of these possibilities, the results of which are shown in Figure 4. Following control responses, we treated receptors with the standard oxidation condition of H2O2 + ACh + Mor. In one series of measurements, we then challenged every 100 sec following treatment (white bars). The ACh/Mor-evoked currents in this case decreased to about 60% of the control, and with five successive challenges in 500 sec these did not exceed 80% of the control. In contrast, in a separate experiment, washing for 300 sec after the first post-oxidation challenge (gray bars), returned currents to control levels (Itime point/Icontrol ≈ 1 at 400 and 500 sec). As a control for the prolonged exposure to activators ACh and Mor, we also tested a mock treatment of ACh + Mor without H2O2 (black bars). While the first post-treatment response was smaller than control by about 15%, reflecting minor desensitization, repeated challenges returned to control levels. Statistical analysis of these data (Tables 2 and 3C) indicate that the currents decreased by oxidation remain the same by repeated challenge, and differ from those allowed to recover by wash and those of the ACh + Mor control.
Figure 4.
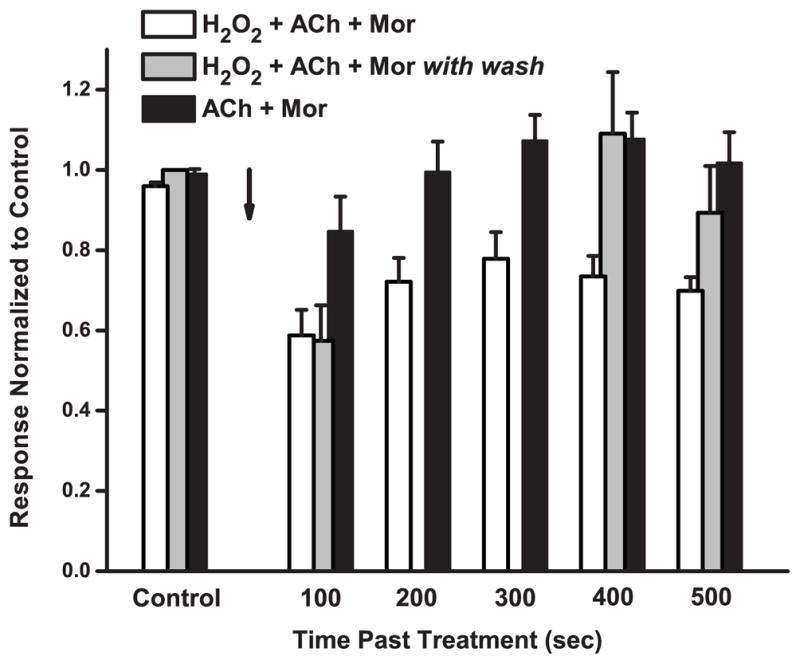
Repeated Challenges Maintain the Oxidation Effect. The effects of repeated challenges and time course on the response of α3L158CG181Cβ2 mutant receptors are shown. For three independent sets of oocytes, the responses evoked by 100 μM ACh and 10 μM Mor challenges, normalized to pre-treatment control, are plotted as a function of time following treatment; error bars represent the SEM. The arrow represents the end of the 5-min treatment period. Both the white and gray bars used 5-min treatments of 4.4 mM H2O2 with 100 μM ACh and 10 μM Mor; the white bars represent responses evoked every 100 sec past treatment, whereas the gray bars are responses with a 300-sec wash following the first post-treatment response. The black bars represent a control experiment in which the treatment was 5 min of 100 μM ACh and 10 μM Mor without H2O2 and challenges every 100 sec. Control responses are means of 4–5 trials prior to treatment, all normalized to the very first response evoked; error bars (SEM) reflect the variability of a stable response. Replicates were n = 5 (white bars), n = 8 (gray bars), n = 5 (black bars).
Table 3C.
Paired t-Test for Figure 4 Data Sets
| Comparison a | Treatment | Time | Challenge | p value |
|---|---|---|---|---|
| Itime vs. Icontrol | Post H2O2 + ACh + Mor b | 100 sec | 100 μM ACh + 10 μM Mor | 0.0060 |
| “ | 200 sec | “ | 0.0088 | |
| “ | 300 sec | “ | 0.0456 | |
| “ | 400 sec | “ | 0.0171 | |
| “ | 500 sec | “ | 0.0049 | |
| Itime vs. Icontrol | Post H2O2 + ACh + Mor c | 100 sec | 100 μM ACh + 10 μM Mor | 0.0004 |
| “ | 400 sec | “ | 0.4422 | |
| “ | 500 sec | “ | 0.0504 | |
| Itime vs. Icontrol | Post ACh + Mor d | 100 sec | 100 μM ACh + 10 μM Mor | 0.1263 |
| “ | 200 sec | “ | 0.3988 | |
| “ | 300 sec | “ | 0.2693 | |
| “ | 400 sec | “ | 0.3122 | |
| “ | 500 sec | “ | 0.4206 |
Paired comparisons are listed as Ipost vs. Ipre, with each time point compared to the original control.
Following treatment, challenges of ACh + Mor were administered every 100 sec.
Following treatment, an initial response to ACh + Mor was measured at 100 sec, then oocytes were washed for 300 sec before challenging twice more.
5-min exposure to ACh + Mor as a control experiment.
Together, the results of these experiments suggest that the disulfide bond formed between the two engineered cysteines by oxidation is better maintained when the receptor is in its ligand-bound conformations, but that the oxidation effect is not permanent. Combined with our finding that the effect of oxidation is greater when activators are present, this may indicate that the L158C and G181C are closer in the active form of the receptor and any disulfide bonds linking the two positions are under strain in the closed conformation (Damle and Karlin, 1980; Kuwajima et al., 1990).
To further support the conclusion that oxidation and reduction treatments change the proportion of disulfide bonds between the pair of cysteines, we probed α3L158CG181Cβ2 receptors for MTS reactivity following various treatments. MTS reagents form disulfide bonds with free, accessible sulfhydryl groups (Karlin and Akabas, 1998), and this method for detecting their presence has been applied in disulfide trapping studies (Hanson and Czajkowski, 2011; McLaughlin et al., 2006). Figure 5A shows sample traces from two of these experiments. Following the standard oxidation treatment, the ACh-evoked current decreased as expected (left panel). Then, exposure to 50 μM MTS-dansyl for 2 min did not further decrease the ACh current. In this particular example, the ACh/Mor-evoked currents were unchanged by oxidation. In contrast, DTT treatment in a separate experiment (right panel) increased currents as expected, but these then decreased following MTS-dansyl exposure. This MTS reagent, as discussed below, decreases currents for the α3L158CG181Cβ2 mutant. Thus, these results are consistent with the conclusion that disulfide bonds formed by oxidation are resistant to further modification, but that when these bonds are intentionally reduced, substantial free sulfhydryls are available for modification.
Systematic experiments along these lines, probing α3L158CG181Cβ2 for MTS reactivity under several conditions, further confirm this conclusion. These results are shown in Figure 5B as a percent change in evoked current following MTS-dansyl treatment. In the same set of oocytes, we tested these treatments using currents evoked both by 5 μM ACh (~EC65, left) and by 5 μM ACh + 10 μM Mor co-application (right). As noted above, the 2-min MTS-dansyl exposure of naive oocytes decreased ACh- and ACh/Mor-evoked currents by 33% and 14%, respectively (white bars). If some portion of L158C-G181C pairs is not bonded as a disulfide, there are of course two free sulfhydryl groups. Importantly, MTS-dansyl treatment decreased evoked currents for both α3L158Cβ2 [e.g., 5 μM ACh currents decreased 44 ± 2%, n = 8; p = 0.00245, paired t-test] and α3G181Cβ2 single-cysteine mutants [100 μM ACh currents decreased 21 ± 4%, n = 5; p = 0.046]. Thus, we interpret the decrease in current following MTS-dansyl treatment to indicate the presence of sulfhydryl groups.
We compared the standard (H2O2 + ACh + Mor) and H2O2 alone oxidation treatments with the MTS probe. H2O2 alone showed some MTS reactivity, with currents decreased by 27% and 21%, respectively, for ACh and ACh + Mor responses (gray bars), consistent with the results of Figure 2A. Where we might expect no MTS reactivity for the H2O2 + ACh + Mor treatment (hashed bars), we observed that the ACh currents increased by 13% following MTS exposure. Nonetheless, the currents were smaller following oxidation in these experiments (Ipost-oxidation/Icontrol = 0.66 ± 0.05), as expected. In fact, the post-MTS response was still smaller than the control (IMTS/Icontrol = 0.77 ± 0.05). The series of challenges and MTS exposure (12 min total) following oxidation may have allowed for some recovery from desensitization, especially because in all other cases MTS treatment decreased currents. In contrast, the ACh/Mor-evoked currents for H2O2 + ACh + Mor oxidation were decreased slightly (8%) by MTS.
We also decreased currents by the standard oxidation treatment, but then washed for 200–300 sec prior to MTS probing (hatched bars). In agreement with our other results showing that oxidation of α3L158CG181Cβ2 is not permanent (Figure 4), MTS decreased ACh- and ACh/Mor-evoked currents by 19% and 14%, respectively. Finally, MTS probing of α3L158CG181Cβ2 receptors following DTT treatment (black bars) was among the largest of the effects, at 39% and 18%, respectively, for ACh and ACh + Mor currents, consistent with reduction of some disulfide bonds formed in the receptor population as natively expressed (Figure 3).
For ACh-evoked currents, the H2O2 + ACh + Mor treatment and H2O2 + ACh + Mor followed by washout differed significantly from the others, whereas for ACh + Mor responses, the five treatments were not different (Table 2). Similar results were obtained in MTS probing experiments in which responses were elicited by 100 μM ACh with and without 10 μM Mor (data not shown). In addition, the changes in current upon MTS exposure were highly significant for all treatments and challenges shown in Figure 5 (Table 3D). Thus, treating α3L158CG181Cβ2 with MTS-dansyl as a probe of free sulfhydryl groups indicates that oxidation creates disulfide bonds between engineered cysteines.
Table 3D.
Paired t-Test for Figure 5 Data Sets
| Comparison a | Treatment | Challenge | p Value |
|---|---|---|---|
| IMTS vs. Ipost-treatmeant | Control | 5 μM ACh | 4.48 × 10−5 |
| H2O2 + ACh + Mor b | “ | 0.000375 | |
| H2O2 alone | “ | 5.33 × 10−5 | |
| H2O2 + ACh + Mor with wash c | “ | 1.29 × 10−5 | |
| DTT | “ | 9.26 × 10−5 | |
| IMTS vs. Ipost-treatmeant | Control | 5 μM ACh + 10 μM Mor | 0.000265 |
| H2O2 + ACh + Mor | “ | 0.00187 | |
| H2O2 alone | “ | 6.74 × 10−5 | |
| H2O2 + ACh + Mor with wash | “ | 1.10 × 10−5 | |
| DTT | “ | 0.00075 |
Paired comparisons are listed as Ipost vs. Ipre; ‘post-treatment’ refers to the conditions tested by the MTS probing as explained in the text.
The treatment was the standard oxidation of H2O2 + ACh + Mor, followed immediately by MTS probing.
The treatment was the standard oxidation of H2O2 + ACh + Mor, followed by 200–300 sec washout prior to MTS probing.
3.3 Positions Along the β9 Strand Are Differentially Reactive
Given our results suggesting that positions 158 and 181 in the α3 subunit move with respect to each other as the receptor changes conformations, we sought further evidence of such movement by studying other double-cysteine mutant pairs. As depicted in Figure 1, we chose A179 and K183 in the β9 strand because these are predicted in the homology model to point toward L158 (as G181 would do if it had an L side chain). The results of experiments with all three double mutants treated with H2O2, in both the presence and absence of ACh and Mor, are shown in Figure 6. As with the α3L158CG181Cβ2 receptor, the main effect of oxidation for α3L158CA179Cβ2 and α3L158CK183Cβ2 was decreased current amplitudes, whether evoked by ACh or by ACh and Mor (Figures 6A and 6B, respectively). For the α3L158CA179Cβ2 mutant, the two oxidation conditions as tested by the two challenges decreased currents by 25–28%. For the α3L158CK183Cβ2 mutant, the H2O2 + ACh + Mor treatment trended toward a larger effect than for H2O2 alone, especially for ACh-evoked currents (28% and 13% decreases, respectively). By comparison, oxidation of the α3L158CG181Cβ2 receptor was more distinctive in that the differences between the two conditions were larger than the other two pairs.
Figure 6.

Oxidation Modifies Double-Cysteine Mutants Differentially. A. The effect of two oxidation treatments on the responses evoked by ACh alone for three double-cysteine mutant pairs is shown. Currents were evoked with ACh concentrations of 24 μM (~EC80) for α3L158CA179Cβ2, 5 μM (~EC50) for α3L158CG181Cβ2 and 125 μM (~EC80) for α3L158CK183Cβ2, respectively. In one set of experiments, treatment with H2O2 (4.4 mM for 5 min) alone was tested (black bars). In a separate experiment, the treatment was 4.4 mM H2O2 + 100 μM ACh + 10 μM Mor (white bars); 125 μM ACh was used for the α3L158CK183Cβ2 experiment. The ratio of peak evoked currents (Ipost/Ipre) is plotted, and error bars represent the SEM. Replicates were as follows (H2O2/ACh/Mor and H2O2 alone): n = 6 and 8 for α3L158CA179Cβ2; n = 5 and 4 for α3L158CG181Cβ2 and n = 9 and 8 for α3L158CK183Cβ2. B. Experiments were the same as described in A. above, with data collected in the same experiment, except that responses were evoked with the ACh concentration indicated and 10 μM Mor.
All but three of the decreases in currents (for H2O2 only treatments) shown in Figure 6 were significant (Table 3E). While ANOVA revealed some differences in these effects across the three mutants (Table 2), such differences do not support a particular pattern of reactivity. Together, these results suggest that activation-induced movement is a property of this region of α3, and not specific to the L158-G181 pair.
Table 3E.
Paired t-Test for Figure 6 Data Sets
| Mutant Pair | Comparison a | Treatment | Challenge | p Value |
|---|---|---|---|---|
| α3L158CA179Cβ2 | Ioxidation vs. Icontrol | H2O2 + ACh + Mor | 24 μM ACh | 0.0108 |
| H2O2 alone | 24 μM ACh | 0.0254 | ||
| H2O2 + ACh + Mor | 24 μM ACh + 10 μM Mor | 0.0024 | ||
| H2O2 alone | 24 μM ACh + 10 μM Mor | 0.0179 | ||
| α3L158CG181Cβ2 | Ioxidation vs. Icontrol | H2O2 + ACh + Mor | 5 μM ACh | 0.000745 |
| H2O2 alone | 5 μM ACh | 0.336 | ||
| H2O2 + ACh + Mor | 5 μM ACh + 10 μM Mor | 0.0238 | ||
| H2O2 alone | 5 μM ACh + 10 μM Mor | 0.0367 | ||
| α3L158CK183Cβ2 | Ioxidation vs. Icontrol | H2O2 + ACh + Mor | 125 μM ACh | 0.00108 |
| H2O2 alone | 125 μM ACh | 0.0386 | ||
| H2O2 + ACh + Mor | 125 μM ACh + 10 μM Mor | 0.00209 | ||
| H2O2 alone | 125 μM ACh + 10 μM Mor | 0.0699 |
Paired comparisons are listed as Ipost vs. Ipre; ‘oxidation’ refers to the two oxidation treatments listed.
We also characterized the ACh and ACh + Mor concentration-response behaviors for these double mutants, the parameters of which are shown in Table 1. The α3L158CA179Cβ2 and α3L158CG181Cβ2 mutants are similar in their increased ACh potency relative to wild type α3β2 (greater than 10-fold), while the α3L158CK183Cβ2 mutant has just a two-fold increase. However, while these first two are potentiated by Mor, albeit not to the extent of wild type, the α3L158CK183Cβ2 mutant shows effectively no potentiation, including an Emax of 0.90. In this regard, the α3L158CK183Cβ2 mutant seems dominated by the α3K183C substitution, because that single mutant also showed negligible Mor potentiation. The changes in ACh activation and Mor potentiation for these mutants (Table 1) together suggest a role for connecting the distant Mor and ACh sites.
Although we did not study them as exhaustively as we did the α3L158CG181Cβ2 receptor, we also measured a variety of other attributes of the α3L158CA179Cβ2 and α3L158CK183Cβ2 receptors. For example, for the α3L158CA179Cβ2 receptor, 40 μM DTT increased currents evoked by both ACh alone and by ACh + Mor: IDTT/Icontrol = 1.30 ± 0.08 (24 μM ACh) and IDTT/Icontrol = 1.26 ± 0.07 (24 μM ACh + 10 μM Mor) (n = 10; p = 0.00057 and 0.00076 for respective paired comparisons). We tested the reducing agent tris(2-carboxyethyl)phosphine (TCEP, 40 μM), which also increased currents [ITCEP/Icontrol = 1.21 ± 0.04 (ACh) and ITCEP/Icontrol = 1.24 ± 0.05 (ACh + Mor) (n = 8; p = 0.00023 and 0.00040 for respective paired comparisons)]. Similarly, α3L158CK183Cβ2 was sensitive to DTT treatment, with IDTT/Icontrol = 1.10 ± 0.01 for 125 μM ACh and IDTT/Icontrol = 1.19 ± 0.03 for 125 μM ACh + 10 μM Mor challenges, respectively (n = 3; p = 0.026 and 0.0015 for the paired comparisons).
We also tested the reversibility of disulfide formation. After currents were decreased as expected by treatment with H2O2 only, the currents of α3L158CA179Cβ2 were restored to control levels, with IDTT/Icontrol (ACh) = 0.99 ± 0.04 and IDTT/Icontrol (ACh + Mor) = 1.06 ± 0.06 (n = 6). Similarly, the original amplitudes of evoked currents were achieved by DTT treatment of α3L158CK183Cβ2 receptors post-oxidation, with IDTT/Icontrol (ACh) = 0.97 ± 0.03 and IDTT/Icontrol (ACh + Mor) = 1.02 ± 0.02 (n = 8). Thus, these further studies of the α3L158CA179Cβ2 and α3L158CK183Cβ2 double-cysteine receptors demonstrate the importance to receptor activity of residues in this region of α3. While the oxidation (decrease in currents) and reduction (increase in currents) effects are consistent for the three double-cysteine pairs we studied, the magnitude of the effects differs depending on conditions, suggesting that the residues do not contribute equally to receptor activity.
4. Discussion
4.1 Disulfide Bond Formation Limits Receptor Activity
Residues L158 and G181 of the α subunit are predicted in the homology model to be 3.3 Å apart (closest heavy atom distance). Similarly, A179 and K183, which point toward L158 due to the β9 strand configuration, are predicted to be 8.5 and 5.9 Å, respectively, from L158. However, the relative lack of homology in this region and variation in F-loop length across nAChR subunits means these calculated distances are approximate at best. That the residues A179-K183 are all within the optimal range for disulfide bond formation (4–8 Å between cysteine β carbons; Bass et al., 2007) with L158, coupled with the moderate effects of oxidation, might explain the lack of a clear pattern of reactivity for the three double-cysteine pairs. (See also Section 4.3.)
Our results are consistent with oxidation promoting disulfide bond formation between the pairs of cysteines engineered into the α subunit. Oxidation with H2O2 and reduction with DTT (or TCEP) were mutually reversible: each consistently decreased or increased currents, respectively, regardless of the order of treatment (Figures 3 and 6). Furthermore, probing these sites with thiol-specific MTS reagents showed effects consistent with oxidation and reduction of disulfide bonds (Figure 5). We found evidence for spontaneous disulfide formation and, surprisingly, apparent lability of these disulfide bonds (Figures 3 and 4). Spontaneous disulfide formation has also been observed in disulfide trapping studies of the GABAA receptor (e.g., Horenstein et al., 2001; Jansen and Akabas, 2006). The bonding promoted by oxidation may hold these residues, and consequently this region of the protein, in a strained position which is then relieved by breaking the disulfide bond (Goldenberg et al., 1993; Kuwajima et al., 1990). The time-dependent reversal of oxidation effects (Figure 4) may be particular to this region of the receptor, because double-cysteines we have studied in other locations in nAChRs do not show such reversal and are even resistant to reduction (unpublished observations). Oxidation of cysteine sulfhydryls to other forms such as sulfinic or sulfenic moieties is formally possible (Bass et al., 2006), but the selectivity of reduction of disulfide bonds by DTT and TCEP (e.g., Burns et al., 1991) makes our interpretation that the effects are due to disulfide chemistry the most plausible.
Morantel potentiation of α3β2 nAChRs is due to increased channel efficacy (Wu et al., 2008). The proportional decrease in currents evoked by ACh and ACh + Mor under some conditions (Figures 3 and 6) is consistent with a decrease in efficacy for the receptor produced by oxidation, in agreement with previous results for chemical modification of cysteine-mutant nAChRs (Seo et al., 2009). Thus, limiting movement across the β8/F loop–β9 cleft with a disulfide bond decreases channel function. Nonetheless, oxidation of α3L158CG181Cβ2 with H2O2 alone resulted in a smaller decrease in currents than when the treatment included ACh, Mor or ACh + Mor (Figure 2), suggesting that these positions are closer or more reactive in the open or desensitized states of the receptor. In other words, channel activation favors disulfide bond formation, but a disulfide bond does not favor the open state. Due to slow solution exchange for oocyte recordings, we cannot measure changes in desensitization properties in our experiments, but effects of disulfide bond formation on desensitized states remains a formal possibility (e.g., Reeves et al., 2005). This finding strongly suggests relative movement between positions 158 and 181 during conformational inter-conversions. In addition, that the disulfide bond was maintained by repeated channel activation and lost after a period of inactivity (Figure 4) further supports this conclusion.
4.2 Movement of the β9 Strand Is Important for Modulation
Our results revealing the importance of residues in the region of the β8/F loop and β9 strands are substantiated by several studies of other Cys-loop receptors. The β9 and β10 strands are the “roots” of the C loop, a component of the canonical agonist binding site; movement in the C loop underlies agonist activation (e.g., Jadey and Auerbach, 2012, and references therein). The β9 position 181 is very well-conserved as glycine or alanine (small volume side chains) in all but one of the rat nAChR subunits, and position 183 is very well-conserved as a positively-charged lysine or arginine (Figure 1B). In contrast, positions 158 and 179 are fairly conserved in neuronal forms, but vary in side chain volume and polarity across all nAChR subunits. All told, we expected these residues to have important functional roles on the basis of sequence conservation.
Only a few studies of the role of the outer surface of the extracellular domain of nAChRs have been reported. α7 receptors are modulated by divalent cations from a site at the N-terminus of the β9 strand. Using chemical modification of substituted cysteines, Rosenberg and colleagues concluded that this modulatory site communicates with the canonical agonist binding site via movement of the β9 strand (Lyford et al., 2003). They also demonstrated that the outer β sheet (strands β7, β9 and β10) contributes to channel activation and allosteric modulation of α7, possibly through a torsional motion of the β sheet, connecting the agonist site above to the transmembrane domain below (McLaughlin et al., 2007).
Investigations of the interactions of several β9 strand residues with their neighbors in GABAA receptors are particularly relevant to our study. In the β2 subunit, which contributes the C loop to the canonical agonist site, an electrostatic interaction between residues on adjoining strands is critical for GABA activation and pentobarbital allosteric modulation (Venkatachalan and Czajkowski, 2008). In addition, cross-linking these positions via substituted cysteines diminished GABA currents to different degrees depending on the presence or absence of GABA (Venkatachalan and Czajkowski, 2008). In the γ2 subunit, which contributes (−) interface residues to the benzodiazepine allosteric site, two double-cysteine mutant pairs spanning the cleft formed by the β8/F loop and β9 strands displayed lower GABA EC50s and significantly diminished flurazepam potentiation (Hanson and Czajkowski, 2011). Interestingly, the disulfide bonds for these two pairs, deduced to be present due to lack of MTS reactivity in the mutant receptors, formed spontaneously upon protein expression and were resistant to reduction with DTT (Hanson and Czajkowski, 2011). Together, these findings demonstrate that the region in and around strand β9 contributes to activation and allosteric modulation of GABAA channels, and suggest that this is a general property of the Cys-loop receptors.
Our results likewise support the conclusion that the β8/F loop–β9 region of the α3 nAChR subunit functions in channel activation. Disulfide bonds for three cysteine pairs, created across the cleft formed by these two parallel stretches (Figure 1), decrease both agonist activation and allosteric modulation (Figure 6), by decreasing channel efficacy. Disulfide bond formation depends on whether the receptors are ligand-bound (Figure 2) and the bonds are apparently strained in the closed state (Figure 4), suggesting that these strands move with respect to each other during channel activation. Although sequence homology between the α3 nAChR and γ2 GABAA subunits in this region is low, α3G181 aligns with γ2F203; this residue and γ2L206 are positions that disrupt GABA activation and benzodiazepine potentiation when cross-linked (Hanson and Czajkowski, 2011). The changes to ACh activation and Mor modulation characteristics in our single and double mutants (Table 1) further indicate the importance of the residues in receptor function. The lack of Mor potentiation for α3K183Cβ2 and α3L158CK183Cβ2 is particularly interesting because K183 is predicted to be 4.8 Å from residue D155; this pair might have a functionally important electrostatic interaction such as that found in the GABAA β2 subunit (Venkatachalan and Czajkowski, 2008). Taken together, our evidence, bolstered by previous findings, indicates that the residues we studied, particularly those in strand β9, mediate allosteric modulation of α3β2 nAChRs.
4.3 A Possible Pathway between Heterotropic Allosteric Ligand Sites
The Mor allosteric binding site, which we previously delineated to be in the β2(+)/α3(−) interface (Cesa et al., 2012; Seo et al., 2009), is 29 Å from the ACh binding site (Figure 1). As such, these sites communicate over a distance on the same order as the ~50 Å separating the agonist binding site and channel pore (Unwin, 2005). Because the α3 subunit contributes to both ligand sites, we hypothesize that movement within the α3 subunit is the primary pathway of this allosteric communication.
The juncture between the outer (β9-10-7) and inner (β1-2-6-5/5′) β sheets, including the irregular structure between strands β8 and β9 (“loop 9”), suggested a sort of “fault line” along which the two sheets may move. Thus, we chose the series of α3 residues A179, G181 and K183 paired with L158 to test for a systematic pattern of reactivity for the substituted cysteines that might indicate a direction of movement. While we conclude that these positions are at least involved in channel activation processes, our data unfortunately do not allow us to discriminate individual roles of these positions, except perhaps that the L158C-G181C pair, predicted to be the closest residues, differs from the other two.
Mutations of these residues interrupt the communication between the Mor and ACh sites. Decreases in ACh- and ACh/Mor-evoked currents depended on oxidation conditions for the α3L158CG181Cβ2 receptor (Figures 2, 5 and 6), in agreement with similar state-dependence for disulfide trapping of GABAA receptors (Horenstein et al., 2001; Jansen and Akabas, 2006; Venkatachalan and Czajkowski, 2008). Similarly, the mutations themselves impacted the modulation of the receptors. For example, while the α3K183C and α3L158CK183C mutations resulted in slightly greater ACh potency relative to wild type α3, Mor potentiation was nearly completely abolished for these two (Table 1). Thus, perturbations in this region of α3 appear to have differential effects on ACh and Mor activities, from which we infer a disruption of the transfer of Mor-binding information.
In this light, our finding that the oxidation treatment including ACh alone, Mor alone and ACh + Mor decreased currents equivalently (Figure 2) suggests that Mor binding alone moves the receptor into something like an active state, despite eliciting very little current on its own (data not shown; see Seo et al., 2009). If so, it may be possible to discriminate movements elicited by the different agonist conditions of Mor alone, ACh alone and ACh + Mor, but would require double-cysteine mutants with greater overall sensitivity to oxidation. In continuing work, we are studying other locations in the neuronal nAChR as putative determinants of allosteric modulation and thus likely regions of intra-molecular movement.
Highlights.
Double cysteine substitution and disulfide trapping perturb neuronal nAChR function
Oxidation and reduction of mutant receptors alter efficacy
Effects depend on residue position in α3 subunit and functional state of receptor
Communication between modulator and agonist sites possibly disrupted
Acknowledgments
This paper is dedicated to W.W. Cleland. We thank Mark King and Chao-Wei Hung for early pilot experiments, and Drs. Peter Chivers, Ron Raines and Jeffery Jonkman for helpful discussions. We also thank the reviewers of the original manuscript for helpful suggestions. This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [grant 1R15 NS070760-01 (to MML)], the Howard Hughes Medical Institute [Undergraduate Science Education grant 52006298 (to Grinnell College)], and by funding from Grinnell College.
Abbreviations
- nAChR
nicotinic acetylcholine receptor
- Mor
morantel
- MTS
methanesthiosulfonate
- TCEP
tris(2-carboxyethyl)phosphine
- GABAA
γ-aminobutyric acid A (receptor)
- OR2
oocyte Ringer’s
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Rayes D, Bouzat C. Molecular determinants of pyrantel selectivity in nicotinic receptors. Mol Pharmacol. 2006;70:1307–18. doi: 10.1124/mol.106.026336. [DOI] [PubMed] [Google Scholar]
- Bass RB, Butler SL, Chervitz SA, Gloor SL, Falke JJ. Use of site-directed cysteine and disulfide chemistry to probe protein structure and dynamics: Applications to soluble and transmembrane receptors of bacterial chemotaxis. Methods Enzymol. 2007;423:25–51. doi: 10.1016/S0076-6879(07)23002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Cooper E, Valera S, Rungger D, Ballivet M. Electrophysiology of neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes following nuclear injection of genes or cDNAs. Methods Neurosci. 1991;4:174–193. [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Boulter J, Connolly J, Deneris E, Goldman D, Heinemann S, Patrick J. Functional expression of two neuronal nicotinic acetylcholine receptors from cDNA clones identifies a gene family. Proc Natl Acad Sci USA. 1987;84:7763–7767. doi: 10.1073/pnas.84.21.7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Burns JA, Butler JC, Moran J, Whitesides GM. Selective reduction of disulfides by tris(2-carboxyethyl)phosphine. J Org Chem. 1991;56:2648–2650. [Google Scholar]
- Cesa LC, Higgins CA, Sando SR, Kuo DW, Levandoski MM. Specificity determinants of allosteric modulation in the neuronal nicotinic acetylcholine receptor: A fine line between inhibition and potentiation. Mol Pharmacol. 2012;81:239–249. doi: 10.1124/mol.111.076059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damle VN, Karlin A. Effects of agonists and antagonists on reactivity of the binding site disulfide in acetylcholine receptor from Torpedo californica. Biochem. 1980;19:3924–3932. doi: 10.1021/bi00558a006. [DOI] [PubMed] [Google Scholar]
- Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 Å resolution. Nature neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- Goldenberg DP, Bekeart LS, Laheru DA, Zhou JD. Probing the determinants of disulfide stability in native pancreatic trypsin inhibitor. Biochem. 1993;32:2835–2844. doi: 10.1021/bi00062a015. [DOI] [PubMed] [Google Scholar]
- Grossberg GT, Schmitt FA, Meng X, Tekin S, Olin J. Effects of transdermal rivastigmine on ADAS-cog items in mild-to-moderate Alzheimer’s disease. Am J Alzheimers Dis Other Dementias. 2010;25:627–633. doi: 10.1177/1533317510385808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Czajkowski C. Disulphide trapping of the GABAA receptor reveals the importance of the coupling interface in the action of benzodiazepines. Br J Pharmacol. 2011;162:673–687. doi: 10.1111/j.1476-5381.2010.01073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsøe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T. Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci. 2011;31:10759–10766. doi: 10.1523/JNEUROSCI.1509-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar SN, Dunlop J. Neuronal nicotinic acetylcholine receptors - targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr Top Med Chem. 2010;10:144–152. doi: 10.2174/156802610790410983. [DOI] [PubMed] [Google Scholar]
- Hilf RJC, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Hilf RJC, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–119. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- Horenstein J, Wagner DA, Czajkwoski C, Akabas MH. Protein mobility and GABA-induced conformational changes in GABAA receptor pore-lining M2 segment. Nature neurosci. 2001;4:477–485. doi: 10.1038/87425. [DOI] [PubMed] [Google Scholar]
- Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol Therapeutics. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Jadey S, Auerbach A. An integrated catch-and-hold mechanism activates nicotinic acetylcholine receptors. J Gen Physiol. 2010;140:17–28. doi: 10.1085/jgp.201210801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Akabas MH. State-dependent cross-linking of the M2 and M3 segments: Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J Neurosci. 2006;26:4492–4499. doi: 10.1523/JNEUROSCI.0224-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–45. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- Kuwajima K, Ikeguchi M, Sugawara T, Hiraoka Y, Sugai S. Kinetics of disulfide bond reduction in α-lactalbumin by dithiothreitol and molecular basis of superreactivity of the cys6-cys 120 disulfide bond. Biochem. 1990;29:8240–8249. doi: 10.1021/bi00488a007. [DOI] [PubMed] [Google Scholar]
- Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM, Chen L. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nature neurosci. 2011;14:1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig J, Hoffle-Maas A, Samochocki M, Luttmann E, Albuquerque EX, Fels G, Maelicke A. Localization by site-directed mutagenesis of a galantamine binding site on α7 nicotinic acetylcholine receptor extracellular domain. J Rec Signal Trans Res. 2010;30:469–483. doi: 10.3109/10799893.2010.505239. [DOI] [PubMed] [Google Scholar]
- Lyford LK, Sproul AD, Eddins D, McLaughlin JT, Rosenberg RL. Agonist-induced conformational changes in the extracellular domain of α7 nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:650–658. doi: 10.1124/mol.64.3.650. [DOI] [PubMed] [Google Scholar]
- Maelicke A, Albuquerque EX. Allosteric modulation of nicotinic acetylcholine receptors as a treatment strategy for Alzheimer’s disease. Eur J Pharmacol. 2000;393:165–70. doi: 10.1016/s0014-2999(00)00093-5. [DOI] [PubMed] [Google Scholar]
- Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC, Moroni M, Bermudez I. Additional acetylcholine (ACh) binding site at α4/α4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J Biol Chem. 2011;286:31043–31054. doi: 10.1074/jbc.M111.262014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JT, Fu J, Sproul AD, Rosenberg RL. Role of the outer β-sheet in divalent cation modulation of α7 nicotinic receptors. Mol Pharmacol. 2006;70:16–22. doi: 10.1124/mol.106.023259. [DOI] [PubMed] [Google Scholar]
- Mills EJ, Wu P, Lockhart I, Thorlund K, Puhan M, Ebbert JO. Comparisons of high-dose and combination nicotine replacement therapy, varenicline, and bupropion for smoking cessation: A systematic review and multiple treatment meta-analysis. Ann Med. 2012;44:588–597. doi: 10.3109/07853890.2012.705016. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Sine SM. An intersubunit trigger of channel gating in the muscle nicotinic receptor. J Neurosci. 2007;27:4110–4119. doi: 10.1523/JNEUROSCI.0025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459:451–455. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortells MO, Arias HR. Neuronal networks of nicotine addiction. Int J Biochem Cell Biol. 2010;42:1931–1935. doi: 10.1016/j.biocel.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Pandya A, Yakel JL. Allosteric modulators of the α4β2 subtype of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2011;82:952–958. doi: 10.1016/j.bcp.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Hernandez CM, Dineley KT. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem Pharmacol. 2011;82:931–942. doi: 10.1016/j.bcp.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3:1–12. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P, Auerbach A. Acetylcholine receptor gating: movement in the α-subunit extracellular domain. J Gen Physiol. 2007;130:569–579. doi: 10.1085/jgp.200709858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayes D, De Rosa MJ, Sine SM, Bouzat C. Number and locations of agonist binding sites required to activate homomeric Cys-loop receptors. J Neurosci. 2009;29:6022–6032. doi: 10.1523/JNEUROSCI.0627-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves DC, Jansen M, Bali M, Lemster T, Akabas MH. A Role for the β1–β2 loop in the gating of 5-HT3 receptors. J Neurosci. 2005;25:9358–9366. doi: 10.1523/JNEUROSCI.1045-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallette J, Bohler S, Benoit P, Soudant M, Pons S, Le Novere N, Changeux JP, Corringer PJ. An extracellular protein microdomain controls up-regulation of neuronal nicotinic acetylcholine receptors by nicotine. J Biol Chem. 2004;279:18767–18775. doi: 10.1074/jbc.M308260200. [DOI] [PubMed] [Google Scholar]
- Seo S, Henry JT, Lewis AH, Wang N, Levandoski MM. The positive allosteric modulator morantel binds at noncanonical subunit interfaces of neuronal nicotinic acetylcholine receptors. J Neurosci. 2009;29:8734–8742. doi: 10.1523/JNEUROSCI.1859-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Venkatachalan SP, Czajkowski C. A conserved salt bridge critical for GABAA receptor function and loop C dynamics. Proc Natl Acad Sci USA. 2008;105:13604–13609. doi: 10.1073/pnas.0801854105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Toghraee R, Papke D, Cheng XL, McCammon JA, Ravaioli U, Sine SM. Single-channel current through nicotinic receptor produced by closure of binding site C-loop. Biophysical J. 2009;96:3582–3590. doi: 10.1016/j.bpj.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DK, Wang J, Papke RL. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: Advantages and limitations. Biochem Pharmacol. 2011;82:915–930. doi: 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TY, Smith CM, Sine SM, Levandoski MM. Morantel allosterically enhances channel gating of neuronal nicotinic acetylcholine α3β2 receptors. Mol Pharmacol. 2008;74:466–475. doi: 10.1124/mol.107.044388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman J, Gill SJ. Binding and Linkage: Functional Chemistry of Biological Macromolecules. University Science Books; Mill Valley, CA: 1990. [Google Scholar]