

ABSTRACT
The Forkhead box O (FoxO) transcription factors are activated, and necessary for the muscle atrophy, in several pathophysiological conditions, including muscle disuse and cancer cachexia. However, the mechanisms that lead to FoxO activation are not well defined. Recent data from our laboratory and others indicate that the activity of FoxO is repressed under basal conditions via reversible lysine acetylation, which becomes compromised during catabolic conditions. Therefore, we aimed to determine how histone deacetylase (HDAC) proteins contribute to activation of FoxO and induction of the muscle atrophy program. Through the use of various pharmacological inhibitors to block HDAC activity, we demonstrate that class I HDACs are key regulators of FoxO and the muscle-atrophy program during both nutrient deprivation and skeletal muscle disuse. Furthermore, we demonstrate, through the use of wild-type and dominant-negative HDAC1 expression plasmids, that HDAC1 is sufficient to activate FoxO and induce muscle fiber atrophy in vivo and is necessary for the atrophy of muscle fibers that is associated with muscle disuse. The ability of HDAC1 to cause muscle atrophy required its deacetylase activity and was linked to the induction of several atrophy genes by HDAC1, including atrogin-1, which required deacetylation of FoxO3a. Moreover, pharmacological inhibition of class I HDACs during muscle disuse, using MS-275, significantly attenuated both disuse muscle fiber atrophy and contractile dysfunction. Together, these data solidify the importance of class I HDACs in the muscle atrophy program and indicate that class I HDAC inhibitors are feasible countermeasures to impede muscle atrophy and weakness.
KEY WORDS: Histone deacetylase, Acetylation, Deacetylation, Muscle disuse, Nutrient deprivation, FoxO
INTRODUCTION
Skeletal muscle atrophy and weakness accompany several pathophysiological conditions, including muscle disuse (D'Antona et al., 2003), aging (Gosselin et al., 1994; Larsson et al., 1997a; Larsson et al., 1997b; Lowe et al., 2001; Thompson and Brown, 1999), cancer (Roberts et al., 2013a; Roberts et al., 2013b) and chronic heart failure (Evans et al., 1995; Greutmann et al., 2011). The loss of skeletal muscle mass and impaired function during these conditions contribute to reduced physical performance and quality of life, prolonged hospital stays and enhanced mortality (Evans, 2010). Unfortunately, effective countermeasures to impede the loss of muscle mass and function during these, often complex and overlapping, conditions are limited, emphasizing the importance of research aimed at understanding the cellular mechanisms of muscle atrophy and dysfunction. Although the underlying cause of atrophy and weakness are unique to each condition, a common transcriptional program of increased atrophy gene (atrogene) expression occurs in multiple models of muscle atrophy (Lecker et al., 2004; Sacheck et al., 2007). Furthermore, the upstream transcription factors that induce these transcriptional changes also appear to be usually involved during conditions of muscle atrophy. For example, the Forkhead box O (FoxO) transcription factors are activated in multiple models of muscle atrophy, and are both sufficient and required for muscle atrophy (Sandri et al., 2004). Indeed, FoxO is necessary for the typical gene expression changes and muscle fiber atrophy associated with skeletal muscle disuse (Reed et al., 2011; Senf et al., 2010), cancer cachexia (Reed et al., 2012) and sepsis (Reed et al., 2012) in vivo, as well as during treatment with dexamethasone (Sandri et al., 2004) and deprivation of nutrients to skeletal myotubes (Raffaello et al., 2010). Given this importance of FoxO in the atrophy program, identifying mechanisms which regulate activation of FoxO in skeletal muscle has tremendous potential for the development of therapeutics to preserve muscle mass and function across a wide-range of distinct, and coinciding, atrophy conditions.
We and others have recently demonstrated that the cellular localization and activity of the FoxO transcription factors in skeletal muscle are regulated by acetylation (Bertaggia et al., 2012; Senf et al., 2011). We found that FoxO interacts with, and is acetylated by, the histone acetyltransferase (HAT) protein complex p300–CBP. We have also found that reducing HAT activity in skeletal muscle was sufficient to induce FoxO transcriptional activity, whereas increasing the activity of HAT prevented nuclear localization, transcriptional activity and target-gene transcription of FoxO in response to nutrient deprivation in C2C12 skeletal myotubes, and in whole muscle in response to muscle disuse in vivo (Senf et al., 2011). Work from Bertaggia et al. has further demonstrated, through mutation of six FoxO3a lysine acetylation sites, that acetylation of FoxO3a, indeed, represses the transcriptional activity and promotes cytosolic localization of FoxO3a (Bertaggia et al., 2012). The authors also demonstrate that 3 days following denervation, the ratio of acetylated to total FoxO3a is acutely decreased in skeletal muscle, which contributes to FoxO3a-dependent transcription of atrophy genes. Thereafter, a progressive increase in acetylation of FoxO3a is observed and this was attributed as a protective mechanism to promote FoxO3a cytosolic redistribution in an effort to turn off the atrophy program. These findings collectively indicate that decreased acetylation of FoxO3a in skeletal muscle is an important early mechanism controlling the ability of FoxO3a to drive the atrophy program.
Post-translational modification of proteins through acetylation occurs via the enzymatic activity of HATs, whereas the removal of acetylated residues occurs through the opposing actions of histone deacetylases (HDACs). In skeletal muscle, HATs and HDACs are most well known for their regulation of muscle development and differentiation through the regulation of histone acetylation, which leads to modification of chromatin and transcriptional activation or repression (McKinsey et al., 2001). More recently, the class II HDACs HDAC4 and HDAC5 have been shown to promote neurogenic atrophy through their transcriptional repression of Dach2, which normally acts to repress myogenin-dependent induction of atrophy-related genes (Moresi et al., 2010). However, as previously mentioned, in addition to regulating gene transcription through histone acetylation, the catalytic activity of HATs and HDACs also regulates gene expression through altering the acetylation status and function of transcription factors, such as FoxO. However, limited information currently exists on the specific HDACs which regulate the acetylation status of FoxO in skeletal muscle during normal conditions and those which contribute to decreases in FoxO acetylation and activation during catabolic conditions.
We aimed to determine whether the deacetylase activity of specific HDAC proteins contributes to the activation of FoxO and induction of the muscle atrophy program. Specifically, we determined the role of HDACs on FoxO activity and atrophy associated with nutrient deprivation and skeletal muscle disuse. To do this, we first used the global HDAC inhibitor Trichostatin A (TSA) to inhibit class I and class II HDACs in skeletal muscle cells and whole muscle, in vivo, to determine whether HDACs contribute to FoxO activation and the atrophy program in response to nutrient deprivation. We subsequently determined whether class I or class II HDACs preferentially regulate FoxO activation, and then carried these findings over to the more physiologically relevant model of skeletal muscle disuse. Using a class I HDAC inhibitor, and expression plasmids for specific class I HDACs, we demonstrate the requirement of class I HDACs for FoxO activation, transcription of atrophy genes, skeletal muscle atrophy and contractile dysfunction during muscle disuse. Furthermore, our findings pinpoint the class I HDAC, HDAC1, as a novel regulator of FoxO signaling in skeletal muscle that is both sufficient and required for skeletal muscle atrophy.
RESULTS
FoxO nuclear localization and activation in response to nutrient deprivation is mediated by HDAC activity
To determine whether the transcriptional activity of FoxO in skeletal muscle is regulated by class I and II HDACs, we treated skeletal myotubes that had been differentiated for 3 days and transfected with a FoxO-responsive reporter plasmid with TSA, which inhibits both class I and II HDACs. Myotubes were treated with TSA (or vehicle) under control conditions and during nutrient deprivation, which we and others have previously shown increases the nuclear localization and transcriptional activity of FoxO (Mammucari et al., 2007; Senf et al., 2011). As shown in Fig. 1A, TSA strongly repressed FoxO reporter activity in myotubes under normal conditions, as well as after 18 hours of nutrient deprivation. These data indicate that class I and/or class II HDACs maintain basal levels of FoxO activity in skeletal muscle cells and facilitate FoxO activation in response to nutrient deprivation. Another mechanism to increase FoxO activity is to reduce the basal activity of Akt, which normally phosphorylates and causes FoxO transcription factors to be retained in the cytosol. Therefore, we transfected skeletal myoblasts with a FoxO-responsive reporter plasmid, plus a dominant-negative Akt expression plasmid (or empty vector), to reduce endogenous Akt activity and increase FoxO activity. Following 3 days of differentiation, we treated myotubes with TSA (or vehicle) for 24 hours to determine if TSA could reverse the dominant-negative effect. Overexpression of Akt induced an increase in the FoxO reporter. As shown in Fig. 1B, dominant-negative Akt induced the FoxO-reporter by 50%, which was reversed in the presence of TSA. Therefore, this demonstrates that treatment with TSA can block activation of FoxO, even when signaling through Akt is suppressed, and furthermore, suggests that TSA-mediated repression of FoxO is not dependent on Akt signaling.
Fig. 1.

Inhibition of class I and II HDACs by TSA blocks induction of the muscle atrophy program. (A) FoxO-dependent luciferase reporter (3×FHRE) activity was normalized to Renilla reniformis luciferase from 3-day-differentiated skeletal myotubes treated with TSA or vehicle during 18 hours of nutrient deprivation or control conditions (Con). (B) FoxO-dependent luciferase reporter activity was normalized to Renilla luciferase from 4-day-differentiated skeletal myotubes transfected as myoblasts with a dominant-negative (d.n.) Akt expression plasmid (or empty vector), and treated with TSA or vehicle 24 hours before harvest. (C–G) 3-day-differentiated myotubes expressing ectopic FoxO3a–DsRed or FoxO1–GFP were deprived of nutrients for 6 hours in the presence of TSA (or vehicle). Cellular localization of the proteins was subsequently determined using fluorescence microscopy following fixation and incubation with DAPI to label cell nuclei. The mean fluorescence of FoxO3a–DsRed and FoxO1–GFP in nuclear and cytoplasmic compartments was calculated for each condition and is expressed as a ratio to indicate the relative localization (C) Representative images from each condition are shown in D–G. The panels in E and G are enlarged images of the areas indicated by white boxes in the corresponding images in D and F, respectively. In F, images taken from control or nutrient-deprived skeletal myotubes are in alignment with those of D. (H) The relative mRNA levels of the FoxO target genes, atrogin-1, MuRF1, Gadd45a, p21, Lc3 and 4e-bp1 in 3-day-differentiated myotubes following 6 hours of nutrient deprivation (or control conditions) in the presence of TSA or vehicle. ND, nutrient deprived. All data represent n = 3 and are reported as means ± s.e.m., normalized to the absolute control group. *P<0.05 (compared with absolute control group). †P<0.05 (compared with vehicle within respective treatment group).
We further determined whether inhibition of HDACs through TSA regulates nuclear localization of FoxO. Skeletal myoblasts were transfected with plasmids expressing FoxO3a tagged with red fluorescent protein (FoxO3a–DsRed) or FoxO1 tagged with green fluorescent protein (FoxO1–GFP) and, following 3 days of differentiation, myotubes were deprived of nutrients in the presence of TSA or vehicle. The localization of ectopic FoxO3a–DsRed and FoxO1–GFP were visualized through fluorescence microscopy, and the ratio of nuclear to cytoplasmic fluorescence was calculated (Fig. 1C). As depicted in the representative images, FoxO3a–DsRed (Fig. 1D,E) and FoxO1–GFP (Fig. 1F,G) were localized predominately to the cytoplasm during control conditions but showed increased localization to the nucleus in response to nutrient deprivation, which is confirmed by co-fluorescence with DAPI-stained nuclei. By contrast, inhibition of class I and II HDACs, through treatment with TSA, prevented the increase in both FoxO3a–DsRed and FoxO1–GFP nuclear localization in response to nutrient deprivation.
To further determine whether inhibition of class I and II HDACs also prevents the increased gene expression of atrophy-related and FoxO target genes during nutrient deprivation, we performed quantitative (q)RT-PCR on 3-day-differentiated myotubes following 6 hours of nutrient deprivation (or control conditions) in the presence or absence of TSA. As shown in Fig. 1H, TSA repressed the increase in the FoxO target genes atrogin-1/MAFbx (Fbxo32), MuRF1 (Trim63) and Lc3 (Map1lc3b), which play a role in degradation, as well as Gadd45a and p21 (Cdkn1a), which are involved in growth arrest. Taken together, these data indicate that class I and II HDACs regulate the nuclear localization and transcriptional activation of FoxO in response to nutrient deprivation, and furthermore, are necessary for the increased transcription of several atrophy-related target genes.
Inhibition of class I and class II HDACs during nutrient deprivation in vivo prevents skeletal muscle fiber atrophy
We next sought to carry over our findings to nutrient deprivation, in vivo. We therefore determined whether TSA could prevent the muscle fiber atrophy associated with nutrient deprivation in mice. Mice were injected intraperitoneally with either vehicle (sterile 1× PBS) or TSA, and were then assigned to a control group (fed) or a nutrient-deprivation group. Following three days of nutrient deprivation, muscles from both groups were harvested. To ensure TSA was, indeed, altering protein acetylation in muscle, we examined the effect of TSA on the acetylation of a known class I HDAC target, histone H3, and a known class II HDAC target, α-tubulin. As shown in Fig. 2A, muscle treated with TSA showed an increase in acetylated histone H3 and α-tubulin. To determine the effect of TSA on muscle fiber cross-sectional area (CSA), cross-sections of skeletal muscle, taken from plantaris muscles, were incubated in wheatgerm agglutinin to outline fiber membranes and the average muscle fiber CSA was calculated for each group. Representative images of muscle cross-sections from each group are shown in Fig. 2B. In response to 3 days of nutrient deprivation, skeletal muscle fiber CSA decreased by 27% in vehicle-treated mice, which was completely prevented in nutrient-deprived mice treated with TSA (Fig. 2C). Therefore, these data demonstrate that class I and/or class II HDACs are necessary for muscle fiber atrophy in response to nutrient deprivation, in vivo. Furthermore, these data provide additional physiological significance to our findings in skeletal muscle cells, which indicate that HDACs regulate the atrophy-associated transcription factor FoxO and promote transcription of atrophy genes in response to nutrient deprivation.
Fig. 2.
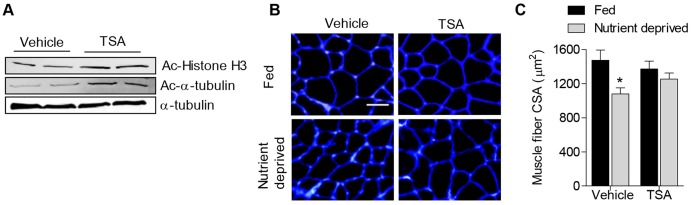
TSA treatment prevents skeletal muscle atrophy in vivo. Mice were treated with TSA or vehicle (sterile PBS) for 3 days during normal (fed) conditions or during nutrient deprivation. (A) Representative western blots for acetylated (Ac) histone H3, acetylated-α-tubulin or α-tubulin as a control, from vehicle- or TSA-treated muscles. (B) Representative muscle cross-sections incubated with wheatgerm agglutinin to visualize muscle fiber membranes (blue). (C) Average plantaris muscle fiber CSA from all groups. Bars represent mean ± s.e.m. for six muscles per group. *P<0.05 (compared with absolute control group).
Class I HDACs preferentially regulate FoxO activation during nutrient deprivation
Because TSA inhibits class I and class II HDACs, which each comprise several distinct HDAC family members, the possible HDAC(s) that could regulate the FoxO transcription factors are numerous. We therefore sought to narrow down the potential HDAC proteins that could regulate FoxO, through determining whether class I or class II HDACs preferentially regulate FoxO activity. To investigate this, we treated skeletal muscle myotubes that had been transfected with a FoxO reporter, with MC-1568 (class II HDAC inhibitor) or MS-275 (class I HDAC inhibitor) under control conditions or 6 hours of nutrient deprivation. As shown in Fig. 3A, treatment with MC-1568 or MS-275 reduced basal levels of FoxO reporter activity, although MS-275 reduced basal FoxO reporter activity to a greater magnitude. By contrast, during nutrient deprivation, inhibition of class II HDACs via MC-1568 reduced FoxO reporter activity only marginally, whereas inhibition of class I HDACs via MS-275 completely prevented FoxO reporter activation in response to nutrient deprivation. These data indicate that both class I and class II HDACs regulate basal levels of FoxO activity in skeletal muscle cells; however, class I HDACs are necessary for FoxO activation in response to a catabolic stimulus.
Fig. 3.

HDAC1 deacetylase activity is sufficient to cause skeletal muscle atrophy. (A,B) FoxO-dependent luciferase reporter activity (A) was normalized to Renilla reniformis luciferase from 3-day-differentiated skeletal myotubes treated with vehicle, MC-1568 (class II HDAC inhibitor) or MS-275 (class I HDAC inhibitor) for 6 hours under control conditions or during nutrient deprivation and (B) from soleus muscles of rats transfected with either GFP, WT or dominant-negative HDAC1, HDAC2 or HDAC3 expression plasmids. (C) Overexpression of HDAC2 and HDAC3 was confirmed by western blot. (D–F) Soleus muscles were transfected with GFP, WT HDAC1–GFP or dominant-negative HDAC1–GFP expression plasmids and harvested 7 days after transfection to visualize muscle fiber CSA (D), measure the mean fiber CSA (E), and quantify the relative mRNA levels of atrogin-1, MuRF1, Ctsl and Lc3 normalized to 18S (F). (G) Relative luciferase activity from soleus muscles transfected with a luciferase reporter plasmid driven by the promoter of atrogin-1, plus expression plasmids for GFP or WT HDAC1, plus empty vector (EV), WT FoxO3a or FoxO3a 6KQ. (H) The ability of HDAC1 to interact with, and regulate the acetylation of, endogenous FoxO1 and FoxO3a was determined in soleus muscles injected with GFP, WT HDAC1–GFP or dominant-negative HDAC1–GFP expression plasmids. Equal amounts of protein extract were incubated with either an antibody against acetyl-lysine to immunoprecipitate (IP) total acetylated proteins (Ac-k), an antibody against HDAC1 to IP HDAC1 protein complexes, or IgG as a negative control. Precipitated proteins were subjected to SDS-PAGE and immunoblotted for either FoxO3a or FoxO1. Experiments were independently repeated three times. Western blots were also performed on equal amounts of whole-muscle lysates from the same samples using antibodies for total FoxO1 and FoxO3a or phosphorylated FoxO1 or FoxO3a (P-FoxO1 and P-FoxO3a, respectively). (I) Representative muscle cross-sections and (J) mean muscle fiber CSA of soleus muscles co-transfected with expression plasmids for DsRed plus GFP, DsRed plus WT HDAC1–GFP, dominant-negative FoxO plus GFP or dominant-negative FoxO plus WT HDAC1–GFP. Scale bars: 50 µm. All data represent the mean ± s.e.m. for six muscles per group. *P<0.05 (compared with absolute control group). †P<0.05 (compared with control within respective treatment group). aP<0.05 (compared with EV plus WT HDAC1). d.n., dominant negative.
HDAC1 is sufficient to increase FoxO transcriptional activity
Although MS-275 is a class I HDAC inhibitor, it does not inhibit HDAC8 (Hu et al., 2003). We therefore screened the remaining class I HDACs (HDAC1, HDAC2 and HDAC3) to determine which of these proteins regulate the activity of FoxO. To do this we injected and electroporated whole rat soleus muscles in vivo with a FoxO-dependent luciferase reporter plasmid plus an empty vector, or expression plasmids for wild-type (WT) or dominant-negative HDAC1, 2 or 3. Despite successful overexpression of the HDAC2 and HDAC3 constructs (Fig. 3C), neither regulated FoxO activity. However, WT HDAC1 was sufficient to increase FoxO transcriptional activity ∼3-fold, which required its deacetylase activity (Fig. 3B). Because HDAC1 increases FoxO activity, and MS-275, which preferentially inhibits HDAC1 (IC50 = 300 nM) (Hu et al., 2003), prevents FoxO activation, together, these findings demonstrate that HDAC1 regulates FoxO signaling in skeletal muscle.
HDAC1 is sufficient to induce muscle fiber atrophy, in vivo
Given our finding that overexpression of HDAC1 is sufficient to increase FoxO activity, and that FoxO is sufficient to cause skeletal muscle fiber atrophy (Sandri et al., 2004), we hypothesized that HDAC1 might be sufficient to cause skeletal muscle fiber atrophy. In order to test this hypothesis, we injected and electrotransferred rat soleus muscles with expression plasmids for GFP only, or GFP constructs also expressing WT HDAC1 or dominant-negative HDAC1 and harvested muscles 7 days later for CSA or gene expression analyses. As shown in the representative cross-sections in Fig. 3D, fibers expressing WT HDAC1–GFP were visually smaller than fibers expressing GFP alone or dominant-negative HDAC1–GFP. Quantification of the average CSA of the transfected fibers revealed that fibers expressing WT HDAC1–GFP were 42% smaller than fibers expressing GFP alone (Fig. 3F), demonstrating that HDAC1 is sufficient to induce muscle fiber atrophy in the absence of any physiological stimulus. Furthermore, as the CSA of fibers expressing dominant-negative HDAC1–GFP was not different from those expressing GFP, this further demonstrates that HDAC1 causes muscle fiber atrophy through its deacetylase activity.
In order to determine whether the HDAC1-mediated increase in FoxO activity and muscle fiber atrophy is associated with the transcriptional activation of known atrophy-related FoxO target genes, we further measured the mRNA levels of atrogin-1, MuRF1, Ctsl (cathepsin L) and Lc3, which are elevated in skeletal muscle in response to multiple catabolic conditions and are involved in protein degradation (Mammucari et al., 2007; Sandri et al., 2004). Overexpression of WT HDAC1 was sufficient to induce the gene expression of atrogin-1 (60%), MuRF1 (45%), Ctsl (25%) and Lc3 (25%), which required its deacetylase activity, as dominant-negative HDAC1 did not similarly increase the mRNA levels of these genes (Fig. 3F). Importantly, the transcriptional activity of the FoxO transcription factors can be regulated via direct acetylation of lysine residues, which has been recently demonstrated as a regulatory mechanism to inhibit FoxO in skeletal muscle (Bertaggia et al., 2012; Senf et al., 2011). Indeed, the ability of a FoxO3a protein that cannot be phosphorylated by AKT (FoxO3a-TM) to induce an atrogin-1 promoter construct, and cause skeletal muscle fiber atrophy, was recently shown by Bertaggia et al. to be reversed via mutation of six lysine residue acetylation sites within the FoxO3a-TM construct to mimic the acetylated form (FoxO3a-TM-6KQ). Therefore, we next sought to determine whether HDAC1-induced transcription of atrogin-1 was mediated through deacetylation of FoxO3a at these specific lysine residues. To do this, we injected and electrotransferred muscles with a reporter plasmid for the promoter of atrogin-1 plus expression plasmids for WT HDAC1 (or GFP), plus empty vector, WT FoxO3a or FoxO3a-6KQ (which cannot be regulated via deacetylation at the indicated residues). If WT HDAC1 does interact with, and deacetylate, endogenous FoxO3a to increase the transcription of atrogin-1, the presence of FoxO3a-6KQ should interfere with the ability of WT HDAC1 to induce the atrogin-1 promoter. As shown in Fig. 3G, both WT HDAC1 and WT FoxO3a are sufficient to increase the atrogin-1 reporter, and show an additive effect when coexpressed. However, similar to the findings of Bertaggia et al., the FoxO3a-6KQ mutant is not sufficient to induce the atrogin-1 promoter (Bertaggia et al., 2012). Furthermore, the FoxO3a-6KQ mutant significantly interferes with the ability of HDAC1 to increase activation of the atrogin-1 promoter, providing strong evidence that HDAC1 regulates atrogin-1 through deacetylation of FoxO3a at these specific lysine residues.
Given this finding, we next determined whether we could detect changes in the acetylation of endogenous FoxO3a in muscles transfected with WT or dominant-negative HDAC1 expression plasmids. Total acetylated proteins were immunoprecipitated from muscles using an antibody against acetyl-lysine and subsequently immunoblotted for FoxO3a. As shown in Fig. 3H, muscles injected with dominant-negative HDAC1 demonstrated an increase in acetylated FoxO3a, which indicates that FoxO3a is, indeed, a target of HDAC1 deacetylase activity. In order to determine whether HDAC1 directly complexes with FoxO3a, we also immunoprecipitated HDAC1 from muscles, subsequently immunoblotted for FoxO3a, and found that these proteins co-precipitate. We repeated these experiments using an antibody against FoxO1 and found that HDAC1 also complexes with FoxO1 and is a target for the deacetylation activity of HDAC1 (Fig. 3H). Because we and others have previously demonstrated that increases in FoxO1 and FoxO3a acetylation are associated with increases in their phosphorylation status (Bertaggia et al., 2012; Matsuzaki et al., 2005; Senf et al., 2011), we further measured the effect of HDAC1 on the phosphorylation of endogenous FoxO1 and FoxO3a. WT HDAC1 had no observable effect on the basal levels of FoxO phosphorylation, whereas dominant-negative HDAC1 modestly increased the levels of both phospho-FoxO1 and phospho-FoxO3a.
Although WT HDAC1 did not decrease the basal levels of FoxO phosphorylation or acetylation, WT HDAC1 was nonetheless sufficient to increase FoxO activity and induce muscle atrophy. Therefore, we next determined whether HDAC1-induced muscle fiber atrophy required activation of FoxO. To do this we co-injected WT HDAC1–GFP (or GFP) with DsRed or a dominant-negative FoxO–DsRed expression plasmid to inhibit FoxO-dependent transcription, and harvested muscles seven days later for analysis of CSA. Expression of dominant-negative FoxO–DsRed localized to the nucleus and induced significant muscle hypertrophy, both of which we have demonstrated previously (Reed et al., 2012; Reed et al., 2011), whereas WT HDAC1–GFP induced significant atrophy of myofibers (Fig. 3D,I). Interestingly, coexpression of dominant-negative FoxO–DsRed and WT HDAC1–GFP together did not significantly alter the size of muscle fibers (Fig. 3I,J). This finding indicates that HDAC1-induced muscle atrophy is counteracted by the hypertrophic and/or anti-atrophic effects of dominant-negative FoxO, and suggests that endogenous FoxO could mediate the atrophy that is induced by HDAC1. However, because HDAC1 also prevented the hypertrophy induced by dominant-negative FoxO, this further suggests that HDAC1 likely regulates the size of myofibers through additional pathways, independent of FoxO.
HDAC1 is required for muscle fiber atrophy, in vivo
Importantly, our studies thus far have focused on the ability of HDAC1 to induce the muscle-atrophy program in the absence of a physiological stimulus of atrophy. Therefore, we next sought to determine whether the deacetylase activity of HDAC1 mediates physiological muscle atrophy that is induced by muscle disuse. To test this, we injected GFP or dominant-negative HDAC1–GFP into rat solei and cast-immobilized muscles for 7 days before analyses of CSA. As shown in the representative muscle cross-sections of immobilized muscles in Fig. 4A, GFP-positive fibers were not visibly different from nontransfected fibers. However, fibers positive for dominant-negative HDAC1–GFP were visibly larger than the surrounding nontransfected fibers in immobilized muscle. Measurement of the mean (±s.e.m.) fiber CSA in immobilized muscles demonstrates that fibers expressing dominant-negative HDAC1–GFP (2048±154 µm2) are significantly larger (74%) than GFP-expressing fibers (1178±16 µm2) (Fig. 4B). When calculated as a percentage of fiber CSA from muscles of weight-bearing mice (mobile mice that support their own bodyweight), immobilization caused a 55% decrease in fiber size in GFP-transfected fibers that was attenuated by 60% in fibers expressing dominant-negative HDAC1–GFP. As dominant-negative HDAC1–GFP did not affect fiber CSA under weight-bearing conditions, these data provide strong evidence that HDAC1 is necessary for the progression of muscle atrophy resulting from muscle disuse, and that this is mediated through the deacetylase activity of HDAC1.
Fig. 4.

HDAC1 deacetylase activity is required for skeletal muscle atrophy. (A) Representative muscle cross-sections and (B) mean fiber CSA from 7-day-immobilized soleus muscles injected with GFP or dominant-negative (d.n.) HDAC1–GFP. The dashed line represents the average CSA of weight-bearing GFP-transfected fibers. Scale bar: 50 µm. (C) Relative luciferase activity driven by a FoxO-dependent reporter plasmid and (D) relative mRNA levels of atrogin-1, MuRF1, Ctsl and Lc3 normalized to 18S from the soleus of 3-day-immobilized rats transfected with GFP, WT or dominant-negative HDAC1–GFP expression plasmids. All data from immobilized muscles are normalized to data collected from the absolute control group (weight-bearing, GFP) to reflect the raw data. All data represent mean ± s.e.m. for six muscles per group. *P<0.05 (compared with absolute control group). †P<0.05 (compared with control within respective treatment group). (E) Total protein levels of FoxO1 and FoxO3a, or phosphorylated FoxO1 and FoxO3a (P-FoxO1 and P-FoxO3a, respectively) from 7-day-immobilized (Imm) soleus muscles injected with GFP, WT or dominant-negative HDAC1–GFP was measured by western blot. (F) The levels of endogenous HDAC1 in whole-muscle lysate, or cytosolic or nuclear fractions in weight-bearing or immobilized (Imm) muscles were measured using western blot. Western blots for histone H1 and Sod1 on nuclear or cytosolic fractions are included, demonstrating successful separation of the nuclear and cytosolic fractions.
We next sought to determine whether HDAC1 was necessary for the activation of FoxO and the increased transcription of atrophy-related FoxO target genes during muscle disuse. To do this, we transfected rat solei with either expression plasmids for WT HDAC1–GFP, dominant-negative HDAC1–GFP or GFP, with a subset of rats also co-transfected with a FoxO-responsive luciferase reporter plasmid. Rats were subsequently assigned to weight-bearing or cast-immobilized conditions for 3 days to induce muscle disuse, after which muscles were harvested for measurement of luciferase activity or mRNA analysis. Measurement of FoxO-dependent luciferase activity in immobilized muscles revealed that WT HDAC1 potentiated the immobilization-induced increase in the activity of FoxO, whereas dominant-negative HDAC1 attenuated the increase in FoxO activity (Fig. 4C). Thus, this finding demonstrates that the deacetylase activity of HDAC1 is required for the normal increase in FoxO activity in response to muscle disuse.
As shown in Fig. 4D, as expected, cast-immobilization significantly increased the levels of atrogin-1, MuRF1, Ctsl and Lc3 mRNA. Similar to our findings during conditions where mice were mobile, when normalized to the control group (weight-bearing, GFP), overexpression of WT HDAC1 during immobilization further increased the mRNA levels of these atrophy genes, which required the deacetylase activity of HDAC1. Moreover, expression of dominant-negative HDAC1 during immobilization repressed the immobilization-induced increase in these atrophy genes. As dominant-negative HDAC1 did not affect the expression of atrophy genes in weight-bearing muscle (Fig. 3F), these data indicate that HDAC1 is necessary for induction of atrophy genes in response to muscle disuse and that this requires the deacetylase activity of HDAC1. Thus, based on these collective findings, HDAC1 could mediate muscle-disuse-mediated atrophy through deacetylating and activating FoxO to induce atrophy gene transcription. Importantly, because a reduction in phosphorylation of FoxO is a widely used marker of FoxO activation during atrophy conditions, we further measured the effect of the HDAC constructs on the phosphorylation of endogenous FoxO during muscle disuse. As shown in Fig. 4E, overexpression of WT HDAC1 reduced the phosphorylation of both FoxO1 and FoxO3a, whereas overexpression of dominant-negative HDAC1 strongly increased their phosphorylation. Thus, HDAC1 might contribute to the muscle atrophy phenotype during muscle disuse through both deacetylating FoxO and reducing the sensitivity of FoxO to phosphorylation. Moreover, based on additional data analyzing the total abundance, and cellular localization, of endogenous HDAC1, HDAC1 could shuttle out of the nucleus to exert its effect on FoxO within the cytoplasm. Indeed, although total protein levels of HDAC1 were unchanged, the relative abundance of HDAC1 in the nuclear fraction decreased; however, HDAC1 increased in the cytosol (Fig. 4F). Thus, HDAC1-mediated deacetylation of FoxO in the cytosol might be an important signal that leads to decreased phosphorylation and nuclear localization of FoxO. However, this relocalization of HDAC1 to the cytosol during disuse could also lead to increased deacetylation of other HDAC1 substrates located in the cytosol, which could also contribute to the muscle-atrophy phenotype.
Inhibition of class I HDACs during skeletal muscle disuse prevents contractile dysfunction and reduces the extent of fiber atrophy
Because we found that HDAC1 deacetylase activity was an important regulator of the muscle-atrophy program associated with muscle disuse, we next sought to determine whether disuse-mediated muscle atrophy, and the associated muscle weakness, could be prevented by treatment with MS-275. As mentioned above, MS-275 is a class I HDAC inhibitor that exerts strong preference towards HDAC1 (Hu et al., 2003). Mice were therefore injected intraperitoneally with either vehicle or MS-275, and were assigned to a control (weight-bearing) group or immobilized group. Mice continued to receive daily injections of MS-275 or vehicle and, after 10 days of immobilization, soleus muscles were harvested from both groups. To confirm MS-275 was altering the acetylation of proteins in muscle, we examined the effect of MS-275 on the acetylation of a known class I HDAC target, histone H3, and a known class II HDAC target, α-tubulin. Unlike TSA, which increased the acetylation of both histone H3 and α-tubulin (Fig. 2A), MS-275 only increased the acetylation of histone H3 (Fig. 5A). To determine the effect of MS-275 on soleus muscle fiber atrophy, sections were incubated in wheatgerm agglutinin, to outline fiber membranes, and the average muscle fiber CSA was calculated for each group. Representative images of soleus muscle cross-sections from each group are shown in Fig. 5B. Following 10 days of cast-immobilization, soleus muscle fiber CSA decreased 41% in vehicle-treated mice, which was attenuated by 39% in immobilized mice that had been treated with MS-275 (Fig. 5C).
Fig. 5.

MS-275 prevents contractile dysfunction and attenuates muscle fiber atrophy associated with muscle disuse. Mice were treated with MS-275 or vehicle for 10 days under normal weight-bearing conditions or 10 days of muscle disuse, induced by cast-immobilization of the hind limbs. (A) Representative western blots are shown for acetylated histone H3 (Ac-histone H3), acetylated-α-tubulin (Ac-α-tubulin) or α-tubulin (as loading control) from vehicle- or MS-275-treated muscles. (B) Representative soleus muscle cross-sections were incubated with wheatgerm agglutinin to visualize muscle fiber membranes (blue). Scale bar: 50 µm. (C) The average muscle fiber CSA from each group was quantified. (D–G) The force-generating capacity of soleus muscles from each group was measured, including (D) absolute force–frequency relationship, (E) maximal absolute force, (F) specific force–frequency relationship (force normalized to muscle weight), and (G) maximal specific force. (H) The relative protein content of myosin heavy chain (MHC) was determined using SDS-PAGE. All data represent the mean ± s.e.m. for six muscles per group. *P<0.05 (compared with the weight-bearing group treated with vehicle). †P<0.05 (compared with the vehicle-treated control within respective treatment group). Imm, immobilized.
To determine whether MS-275 could also protect against the muscle weakness induced by immobilization, we subsequently measured force production by soleus muscle, in vitro, in a subset of mice. Following 10 days of immobilization, absolute force in the soleus muscle was decreased 55–61% across all stimulation frequencies ≥50 Hz, demonstrating both submaximal and maximal force deficits in response to muscle disuse (Fig. 5D). However, solei from immobilized mice that had been treated with MS-275 showed a 31–35% attenuation of the force deficits observed in both submaximal and maximal absolute force across all stimulation frequencies ≥80 Hz (Fig. 5D,E). As production of skeletal-muscle force is a function of both muscle mass and the intrinsic contractile properties of the muscle, we subsequently normalized force to muscle weight and plotted the specific force–frequency relationship. In vehicle-treated mice, a 25–31% decrease in submaximal and maximal specific force was apparent across all stimulation frequencies ≥80 Hz, indicating significant contractile dysfunction. However, this decrease in specific force was completely prevented in mice treated with MS-275 (Fig. 5F,G). Reductions in muscle force that are evident following normalization to muscle mass indicate impairments in contractile function. Therefore, our finding that MS-275 completely prevented the decrease in specific force in 10-day-immobilized muscles suggests that class I HDACs contribute to contractile dysfunction during disuse. There are several potential mechanisms that might contribute to contractile dysfunction during muscle disuse, including (but not limited to) shifts in myosin isoforms (Caiozzo et al., 1998; Caiozzo et al., 1996; Campione et al., 1993; Fitts et al., 2000), alterations in Ca2+ release and sensitivity (Fraysse et al., 2003), and the preferential degradation of myosin heavy chain (MHC) (Derde et al., 2012; Ochala et al., 2011), which is mediated through the FoxO target gene MuRF1 (Clarke et al., 2007). Because we found that HDAC1 was necessary for both activation of FoxO and the expression of MuRF1, and MS-275 preferentially inhibits HDAC1, we hypothesized that the preservation of specific force might be related to the sparing of MHC. Thus, we isolated myofibrillar proteins from gastrocnemius muscles of control and 10-day-immobilized mice treated with MS-275 or vehicle and measured the relative levels of MHC and actin from equal amounts of protein lysate. As shown in Fig. 5H, cast-immobilization resulted in a significant reduction in the relative abundance of MHC, which was prevented in immobilized mice that had been treated with MS-275. Although the levels of actin showed a slight decrease in content in response to immobilization, this difference was not statistically significant and was unchanged by treatment with MS-275. Given that the ratio of myosin to actin can dictate contractile function, the sparing of myosin by MS-275 during immobilization could explain, in part, the protection from contractile dysfunction. In summary, these findings collectively demonstrate that class I HDACs are crucial regulators of the muscle-atrophy program and contribute to both muscle fiber atrophy and contractile dysfunction during disuse.
DISCUSSION
The results of this study demonstrate that class I HDACs, and specifically HDAC1, are necessary for the muscle atrophy and contractile dysfunction associated with skeletal muscle disuse. We show that HDAC1-dependent atrophy during disuse requires its deacetylase activity, and is mediated, in part, through its activation of FoxO and the expression of several atrophy-related genes. Moreover, overexpression of HDAC1 in skeletal muscle was also sufficient to cause significant muscle atrophy in the absence of any physiological atrophy stimulus. Together, these findings solidify the importance of HDAC1 in the regulation of the muscle atrophy program, and indicate that therapeutics targeting HDAC1 could be feasible countermeasures to impede muscle atrophy. Furthermore, because inhibition of class I HDACs during muscle disuse also rescued the decrease in the specific force of skeletal muscle, these data also suggest that targeting class I HDACs could preserve muscle function, not only through sparing of muscle fiber size, but also through additional mechanisms that directly regulate contractile function.
The class I HDAC proteins include HDACs 1, 2, 3 and 8. The class I HDAC inhibitor used in the current study, MS-275, inhibits the catalytic activity of HDAC1, 2 and 3, but has the greatest inhibitory effect on HDAC1 (Dokmanovic et al., 2007; Hu et al., 2003; Kennedy et al., 2013). From our experiments using both MS-275, and HDAC1, HDAC2 and HDAC3 expression plasmids, our findings pinpoint HDAC1 as a primary regulator of FoxO in skeletal muscle and as a key regulator of the atrophy program. However, as HDAC1 and HDAC2 are often found in complex together, HDAC1 might work in conjunction with HDAC2 to regulate the activity of FoxO. HDAC1 and HDAC2 are generally thought of as global transcriptional repressors owing to their role in the deacetylation of histones, which limits accessibility to gene promoters. However, gene array analyses of skeletal muscle from HDAC1 and HDAC2 double-knockout mice show only modest changes in global gene expression when compared to muscles from control mice (Moresi et al., 2012). Based on this finding, the authors of that study concluded that the functions of HDAC1 and HDAC2 in skeletal muscle are, likely, more specific than global transcriptional repression. In fact, they found that HDAC1 and HDAC2 were necessary for the maintenance of normal skeletal muscle structure and function. This finding was linked to HDAC1- and HDAC2-dependent induction of several genes associated with autophagy, including Atg5, Gabarapl1, Lc3 and p62 (Sqstm1), and the regulation of autophagic flux. Flux through autophagy is required for cellular homeostasis during normal conditions; however, increased autophagic flux during catabolic conditions contributes to the muscle-atrophy process (Mammucari et al., 2007; Masiero and Sandri, 2010). Although we did not focus on autophagy in the current manuscript, our findings that HDAC1 is both sufficient and required for physiological muscle atrophy could be related to its role in the induction of autophagy. In relation to this, FoxO3a also induces autophagy and muscle atrophy (Mammucari et al., 2007; Zhao et al., 2007), and we found that HDAC1 is both sufficient and required for FoxO activation. Thus, it seems plausible that the induction of atrophy by HDAC1 could involve FoxO-dependent induction of autophagy. In support of this we found that HDAC1 was both sufficient and required for the induction of Lc3, which is a known FoxO target gene involved in autophagy. However, HDAC1 was also necessary for the increased gene expression of other FoxO target genes involved in the ubiquitin proteasome pathway (atrogin-1 and MuRF1) and in the inhibition of protein synthesis (4e-bp1, also known as Eif4ebp1). Therefore, HDAC1 could promote muscle atrophy through increasing FoxO-dependent transcription of target genes involved in several different pathways that lead to increased protein turnover.
Recent data have demonstrated that decreased acetylation of FoxO3a during atrophy conditions is a crucial mechanism that activates FoxO3a-dependent transcription and its ability to induce muscle fiber atrophy (Bertaggia et al., 2012; Senf et al., 2011). However, until now, the specific proteins regulating FoxO3a deacetylation in skeletal muscle were unknown. Our findings indicate that HDAC1 directly deacetylates FoxO and is necessary for activation of FoxO in response to disuse of skeletal muscle. Interestingly, because we found that endogenous HDAC1 relocalizes from the nucleus to the cytosol in response to muscle disuse, we hypothesize that HDAC1 could deacetylate FoxO in the cytosolic compartment to facilitate the nuclear localization, and transcriptional activation, of FoxO.
Although this is the first evidence to support class I HDACs as activators of FoxO in skeletal muscle and in the induction of muscle atrophy, class I HDACs have previously been identified as therapeutic targets for muscular dystrophy (Colussi et al., 2008; Consalvi et al., 2011; Minetti et al., 2006). Class I HDACs associate with MyoD and repress MyoD-dependent transcription of target genes involved in satellite-cell-mediated myofiber growth and regeneration (Puri et al., 2001), which is the rationale for the use of HDAC inhibitors in muscle dystrophy. Minetti et al. demonstrated that, in mdx mice, inhibition of class I HDACs through MS-275 reduced muscle fibrosis and cellular infiltrate, increased muscle fiber CSA and enhanced the time to exhaustion during an exercise performance test (Minetti et al., 2006). These findings were associated with the induction of follistatin, which is a MyoD-target gene that promotes myoblast fusion and hypernucleation of myofibers through its negative regulation of myostatin. Interestingly, myostatin is elevated in some models of disuse muscle atrophy, although the importance of myostatin for disuse atrophy is controversial, with evidence to support (Murphy et al., 2011) and refute (Hamrick et al., 2007) its involvement. Therefore, although we did not measure follistatin levels in the current study, increased transcription of follistatin and subsequent repression of myostatin signaling following inhibition of class I HDACs could also be involved in the attenuation of disuse muscle fiber atrophy and weakness in the current study.
In conclusion, our data pinpoints HDAC1 as a primary regulator of FoxO in skeletal muscle that is both sufficient and required for skeletal muscle atrophy. Importantly, our findings also demonstrate that, during muscle disuse, class I HDACs are necessary for not only fiber atrophy and the associated muscle weakness, but that they also contribute to additional cellular processes that cause contractile dysfunction independently of the loss of muscle mass. These findings collectively indicate that class I HDAC inhibitors are feasible countermeasures to inhibit muscle atrophy and weakness that might be effective in multiple conditions of muscle atrophy.
MATERIALS AND METHODS
Animals
Sprague-Dawley male rats weighing ∼200 g, and C57BL/6 mice weighing ∼20 g, were purchased from Charles River Laboratories (Wilmington, MA). Animals were maintained in a temperature-controlled environment with a 12-hour light and dark cycle, and provided a standard diet and water ad libitum. The University of Florida Institutional Animal Care and Use Committee approved all animal procedures.
Animal models
The hind limbs of rats were bilaterally cast-immobilized, 4 days after plasmid injection, for either 3 or 7 days, to induce muscle disuse as has been detailed previously (Senf et al., 2008). For experiments using MS-275, mice were treated daily with intraperitoneal injections of either MS-275 (Selleckchem, Houston, TX, 5 mg/kg of body weight) or vehicle [1% dimethyl sulfoxide (DMSO)] beginning 1 day before the 10-day period of weight-bearing conditions or bilateral cast-immobilization of the hind limbs. For nutrient-deprivation experiments, mice in the fed or nutrient-deprived groups were treated daily with TSA (Sigma-Aldrich, St Louis, MO, 0.6 mg/kg of body weight) or vehicle (sterile 1× PBS) via intraperitoneal injection beginning 1 day before the 3-day period of control (fed) conditions or withholding of food (nutrient deprived).
Cell culture experiments
C2C12 mouse skeletal muscle myoblasts were purchased from American Type Culture Collection (Manassas, VA). Cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and antibiotics. Myoblasts were passaged at 70% confluence and were seeded onto 0.1% gelatin-coated tissue-culture plates. Transient transfection was performed with FuGENE® HD Transfection Reagent (Promega, Madison, WI) when cells were at 80% confluence. At 24 hours following transfection, differentiation into myotubes was induced by replacing the growth medium with differentiation medium containing 2% horse serum and antibiotics in high-glucose DMEM (Cellgro, Manassas, VA). Medium was replenished every 2 days. Cells were maintained at 37°C under 5% CO2 and 95% air. 3- to 5-day-differentiated myotubes were used for experiments. For treatment with HDAC inhibitors, 3-day-differentiated myotubes were either treated with vehicle (1% DMSO), 50 nM of TSA (Sigma-Aldrich), 5 µM of MC1568 or 5 µM of MS-275 (Selleckchem) for 18 hours (reporter experiments) or 6 hours (gene expression and localization experiments) in differentiation medium or Hank's Balanced Salt Solution (nutrient deprivation) as described previously (Senf et al., 2011). The relative localization of ectopic FoxO3a–DsRed and FoxO1–GFP was determined by calculating the average fluorescence (mean gray value) in DAPI-stained (nuclear) regions versus non-DAPI-stained (cytosolic) areas of transfected myofibers using ImageJ software and calculating the ratio of nuclear to cytosolic mean fluorescence.
Plasmids
The FoxO reporter plasmid, containing three Forkhead-responsive elements (3×FHRE) was obtained from Addgene (plasmid 1789), deposited by Michael Greenberg (Harvard University, Boston, MA), has been described previously (Brunet et al., 1999). The WT HDAC1 expression plasmid was obtained from Addgene (plasmid 11054), deposited by Ramesh Shivdasani (Harvard University, Boston, MA), has been described previously (Tou et al., 2004). The dominant-negative HDAC1 expression plasmid was a gift from Yi Qiu (University of Florida, Gainesville, FL) and has been described previously (Qiu et al., 2011). The dominant-negative HDAC1 protein contains a histidine to alanine replacement at amino acid residue 141 which renders the enzyme catalytically inactive, yet retains its ability to interact with binding partners and targets of its deacetylase activity (Hassig et al., 1998; Mal et al., 2001). This deacetylase mutant is commonly referred to as a dominant-negative protein owing to its ability to block endogenous HDAC1-mediated events, which is presumed to occur through outcompeting endogenous HDAC1 for binding in protein complexes (Ito et al., 2002; Lei et al., 2010; Mal et al., 2001). The WT and dominant-negative HDAC2 and HDAC3 expression plasmids were gifts from Ed Seto (Moffit Cancer Center, Tampa, FL) and have been described previously (Juan et al., 2000; Qiu et al., 2011). Mutagenesis was used to restore the Akt phosphorylation sites in the FoxO3a-TM-6KQ construct (provided by Marco Sandri, Venetian Institute of Molecular Medicine, Padova, Italy) to generate the FoxO3a-6KQ plasmid. The FoxO3a-TM-6KQ construct has been described previously (Bertaggia et al., 2012). The dominant-negative Akt, atrogin-1-GL2 (reporter) and dominant-negative FoxO–DsRed constructs have also been used and described previously (Reed et al., 2011; Senf et al., 2008; Senf et al., 2011). pRL-TK-Renilla luciferase control reporter plasmid was purchased from Promega (Madison, WI). Plasmid DNA was amplified and isolated from bacterial cultures using Endotoxin-Free Maxi or Mega Prep Kits (Qiagen, Valencia, CA) and resuspended in sterile filtered PBS for transfections in vivo, or Tris-EDTA buffer for transfections in culture, as described previously (Senf and Judge, 2012).
In vivo plasmid delivery
Rats were acutely anesthetized and a small incision was made on the lateral side of the lower leg to expose the soleus muscle. Each soleus was injected with 50 µl of sterile 1× PBS containing 10 µg of expression plasmid and/or 40 µg of reporter plasmid, followed by electroporation at 75 V/cm using an electric pulse generator (Electro Square Porator ECM 830: BTX, Hawthorne, NY) as described previously (Senf et al., 2008).
In vitro muscle contractile properties
The methods and solutions used for the measurements of soleus muscle function have been described previously (Ferreira et al., 2010; Roberts et al., 2013b). Upon muscle removal, one end of the soleus was tied to a Dual-Mode Muscle Lever System (300C-LR, Aurora Scientific, Aurora, Canada) and the other end was secured to a glass rod using 4.0 silk sutures. Following 20 minutes of thermoequilibration, the soleus was placed at optimal length and force–frequency measurements began. The soleus was stimulated with a supramaximal current (600–800 mA) with pulses of 0.25 mseconds delivered through a stimulator (701C, Aurora Scientific) and a train duration of 500 mseconds. All data were recorded and analyzed using commercial software (Dynamic Muscle Control and Analysis Software, Aurora Scientific).
Histochemistry and CSA analyses
To measure the muscle fiber cross-sectional area (CSA), 10-µm sections were taken from the midbelly of the soleus muscle using a Microm HM 550 cryostat (Microm International, Walldorf, Germany). Sections were incubated with Alexa-Fluor-350-conjugated wheatgerm agglutinin (Invitrogen) for 2 hours and subsequently washed in PBS. Areas containing transfected fibers in muscle cross-sections were visualized, and images captured, using a Leica DM5000B microscope (Leica Microsystems, Wetzlar, Germany) and the Leica Application Suite, version 3.5.0 software. This software was also used to trace and measure muscle fiber CSA.
Reporter assays
For reporter experiments, tissue was harvested in Passive Lysis Buffer (Promega) and a Modulus Single Tube Multimode reader (Promega) was used to determine luciferase activity. For in vitro experiments, luciferase activity was determined by normalizing firefly luciferase activity to pRL-TK-Renilla luciferase activity using a dual-luciferase reporter assay (Promega) (Senf et al., 2008).
RNA isolation and qRT-PCR
RNA was isolated from skeletal myotubes and skeletal muscle tissue using a TRIzol-based method as described previously (Senf et al., 2011). cDNA was generated from 1 µg of RNA using an Ambion® RETROscript® First Strand Synthesis Kit (Life Technologies, Grand Island, NY) and was used as a template for qRT-PCR, using a 7300 real-time PCR system (Applied Biosystems, Austin, TX). Primers used for qRT-PCR were purchased from Applied Biosystems. Primers used for C2C12 muscle cells were: atrogin-1/MAFbx (Fbxo32, GeneBank NM_026346.2), MuRF1 (Trim63, GeneBank NM_001039048.2), Gadd45a (GeneBank NM_007836.1), p21 (GeneBank NM_001104569.1), Lc3 (Map1lc3a, GeneBank NM_025735.3), 4e-bp1 (Eif4ebp1, GeneBank NM_053857.1) and Mrpl32 (GeneBank NM_029271.2). Primers used for rat muscle tissue: atrogin-1/MAFbx (GeneBank NM_133521.1), MuRF1 (GeneBank NM_080903.1), Gadd45a (GeneBank NM_024127.2), p21 (GeneBank NM_001106498.1), Lc3 (GeneBank NM_012823.1), Ctsl (GeneBank NM_001912.4), and 18S (GeneBank X03205.1). Quantification of gene expression was performed using the relative standard-curve method and all data was normalized to the absolute control group and subsequently normalized to the gene expression of either Mrpl32 (C2C12s) or 18S (whole muscle).
Western blotting and immunoprecipitation assays
Skeletal muscles were homogenized and processed for analysis by western blot, as described previously (Senf et al., 2008). Primary antibodies against acetyl-histone H3 (no. 9677, Cell Signaling Technology, Boston, MA); acetyl-α-tubulin (no. 3971, Cell Signaling Technology); α-tubulin (T6199, Sigma-Aldrich, St Louis, MO); FoxO1 (no. 9454, Cell Signaling Technology); phospho-FoxO1 (Ser256) (no. 9461, Cell Signaling Technology); FoxO3a (SC-11351, Santa Cruz Biotechnology, Santa Cruz, CA); phospho-FoxO3a (Ser253) (no. 9466, Cell Signaling Technology); histone H1 (SC-8030, Santa Cruz Biotechnology); superoxide dismutase 1 (Sod1) (SC-11407, Santa Cruz Biotechnology); HDAC1 (SC-7872, Santa Cruz Biotechnology); HDAC2 (SC-7899); HDAC3 (SC-11417) or acetyl-lysine (no. 05-515, Millipore, Billerica, MA) were used according to the manufacturer's directions. Nuclear and cytosolic fractions were separated as described previously (Senf et al., 2009). Successful fractionation was confirmed by western blot for histone-H1 and Sod1. For the immunoprecipitation assays, 500 µg of protein was incubated overnight with either 4 µg of antibody against acetyl-lysine, HDAC1 or a non-specific IgG control antibody (no. 2729, Cell Signaling Technology) using the Catch and Release v2.0 Reversible Immunoprecipitation System (no. 17-500, Millipore) as used and described previously (Senf et al., 2011). The following day, immunoprecipitated proteins were washed, eluted in denaturing buffer and heated before analysis by western blot.
Contractile protein analyses
For optimal extraction and processing of MHC and actin, muscles were homogenized in a high-salt lysis buffer as described previously (Cosper and Leinwand, 2012; Roberts et al., 2013b). Samples were mixed in Laemmli buffer (Bio-Rad, Hercules, CA) and heated. 0.6 µg of protein was loaded into a 10% polyacrylamide gel (Criterion precast gels; Bio-Rad) and run at 200 V for 50 minutes at 4°C. Coomassie Brilliant Blue (Thermo Fisher Scientific, Waltham, MA) was used to visualize proteins, and an Odyssey Infrared Imaging system (LI-COR, Lincoln, NE) was used to quantify the optical density of MHC. Levels of actin were determined using standard western blot analysis as described previously (Senf et al., 2008). The primary antibody against actin (1∶1000, JLA20; Developmental Studies Hybridoma Bank, Iowa City, IA) was used according to the manufacturer's directions. The Li-Cor Odyssey fluorescence detection system was used to visualize actin following incubation with Alexa-Fluor-680-conjugated secondary antibody (1∶10,000, Invitrogen).
Statistical analyses
All data were analyzed using a two-way analysis of variance followed by Bonferroni post hoc comparisons or, when appropriate, a Student's t-test (GraphPad Software, San Diego, CA). All data are expressed as means±s.e.m. and significance was established at P<0.05.
Acknowledgments
We thank the laboratory of Marco Sandri (Venetian Institute of Molecular Medicine, Padova, Italy) for providing the FoxO3a-TM-6KQ construct and Dan Ryder (University of Florida, Gainesville, FL) for his contribution in construct mutagenesis to generate the FoxO3a-6KQ variant.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
A.R.J. and S.M.S conceived, designed and interpreted the study; A.W.B. conducted animal experiments and performed biochemical and histological analyses, with contributions from B.M.R., A.R.J. and S.M.S.; P.B.S. conducted and analyzed cell culture experiments, with contributions from S.M.S. and A.R.J.; L.F.F. conducted and analyzed muscle function experiments; S.M.S. wrote the manuscript, with contributions from A.W.B. and A.R.J. All authors edited and approved the final manuscript.
Funding
This work was supported by U.S. National Institute of Arthritis and Musculoskeletal and Skin Diseases [grant numbers R01AR060209 to A.R.J., R00HL098453 to L.F.F.]. Deposited in PMC for release after 12 months.
References
- Bertaggia E., Coletto L., Sandri M. (2012). Posttranslational modifications control FoxO3 activity during denervation. Am. J. Physiol. 302, C587–C596 10.1152/ajpcell.00142.2011 [DOI] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 10.1016/S0092--8674(00)80595--4 [DOI] [PubMed] [Google Scholar]
- Caiozzo V. J., Haddad F., Baker M. J., Herrick R. E., Prietto N., Baldwin K. M. (1996). Microgravity-induced transformations of myosin isoforms and contractile properties of skeletal muscle. J. Appl. Physiol. 81, 123–132 [DOI] [PubMed] [Google Scholar]
- Caiozzo V. J., Baker M. J., Baldwin K. M. (1998). Novel transitions in MHC isoforms: separate and combined effects of thyroid hormone and mechanical unloading. J. Appl. Physiol. 85, 2237–2248 [DOI] [PubMed] [Google Scholar]
- Campione M., Ausoni S., Guezennec C. Y., Schiaffino S. (1993). Myosin and troponin changes in rat soleus muscle after hindlimb suspension. J. Appl. Physiol. 74, 1156–1160 [DOI] [PubMed] [Google Scholar]
- Clarke B. A., Drujan D., Willis M. S., Murphy L. O., Corpina R. A., Burova E., Rakhilin S. V., Stitt T. N., Patterson C., Latres E. et al. (2007). The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 6, 376–385 10.1016/j.cmet.2007.09.009 [DOI] [PubMed] [Google Scholar]
- Colussi C., Mozzetta C., Gurtner A., Illi B., Rosati J., Straino S., Ragone G., Pescatori M., Zaccagnini G., Antonini A. et al. (2008). HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 105, 19183–19187 10.1073/pnas.0805514105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consalvi S., Saccone V., Giordani L., Minetti G., Mozzetta C., Puri P. L. (2011). Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol. Med. 17, 457–465 10.2119/molmed.2011.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosper P. F., Leinwand L. A. (2012). Myosin heavy chain is not selectively decreased in murine cancer cachexia. Int. J. Cancer 130, 2722–2727 10.1002/ijc.26298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antona G., Pellegrino M. A., Adami R., Rossi R., Carlizzi C. N., Canepari M., Saltin B., Bottinelli R. (2003). The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J. Physiol. 552, 499–511 10.1113/jphysiol.2003.046276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derde S., Hermans G., Derese I., Güiza F., Hedström Y., Wouters P. J., Bruyninckx F., D'Hoore A., Larsson L., Van den Berghe G. et al. (2012). Muscle atrophy and preferential loss of myosin in prolonged critically ill patients. Crit. Care Med. 40, 79–89 10.1097/CCM.0b013e31822d7c18 [DOI] [PubMed] [Google Scholar]
- Dokmanovic M., Clarke C., Marks P. A. (2007). Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 5, 981–989 10.1158/1541--7786.MCR--07--0324 [DOI] [PubMed] [Google Scholar]
- Evans W. J. (2010). Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 91, 1123S–1127S 10.3945/ajcn.2010.28608A [DOI] [PubMed] [Google Scholar]
- Evans S. A., Watson L., Hawkins M., Cowley A. J., Johnston I. D., Kinnear W. J. (1995). Respiratory muscle strength in chronic heart failure. Thorax 50, 625–628 10.1136/thx.50.6.625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira L. F., Moylan J. S., Gilliam L. A., Smith J. D., Nikolova-Karakashian M., Reid M. B. (2010). Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am. J. Physiol. 299, C552–C560 10.1152/ajpcell.00065.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts R. H., Riley D. R., Widrick J. J. (2000). Physiology of a microgravity environment invited review: microgravity and skeletal muscle. J. Appl. Physiol. 89, 823–839 [DOI] [PubMed] [Google Scholar]
- Fraysse B., Desaphy J. F., Pierno S., De Luca A., Liantonio A., Mitolo C. I., Camerino D. C. (2003). Decrease in resting calcium and calcium entry associated with slow-to-fast transition in unloaded rat soleus muscle. FASEB J. 17, 1916–1918 [DOI] [PubMed] [Google Scholar]
- Gosselin L. E., Johnson B. D., Sieck G. C. (1994). Age-related changes in diaphragm muscle contractile properties and myosin heavy chain isoforms. Am. J. Respir. Crit. Care Med. 150, 174–178 10.1164/ajrccm.150.1.8025746 [DOI] [PubMed] [Google Scholar]
- Greutmann M., Le T. L., Tobler D., Biaggi P., Oechslin E. N., Silversides C. K., Granton J. T. (2011). Generalised muscle weakness in young adults with congenital heart disease. Heart 97, 1164–1168 10.1136/hrt.2010.213579 [DOI] [PubMed] [Google Scholar]
- Hamrick M. W., Shi X., Zhang W., Pennington C., Thakore H., Haque M., Kang B., Isales C. M., Fulzele S., Wenger K. H. (2007). Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone 40, 1544–1553 10.1016/j.bone.2007.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig C. A., Tong J. K., Fleischer T. C., Owa T., Grable P. G., Ayer D. E., Schreiber S. L. (1998). A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. USA 95, 3519–3524 10.1073/pnas.95.7.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E., Dul E., Sung C. M., Chen Z., Kirkpatrick R., Zhang G. F., Johanson K., Liu R., Lago A., Hofmann G. et al. (2003). Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 307, 720–728 10.1124/jpet.103.055541 [DOI] [PubMed] [Google Scholar]
- Ito A., Kawaguchi Y., Lai C. H., Kovacs J. J., Higashimoto Y., Appella E., Yao T. P. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236–6245 10.1093/emboj/cdf616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan L. J., Shia W. J., Chen M. H., Yang W. M., Seto E., Lin Y. S., Wu C. W. (2000). Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 275, 20436–20443 10.1074/jbc.M000202200 [DOI] [PubMed] [Google Scholar]
- Kennedy P. J., Feng J., Robison A. J., Maze I., Badimon A., Mouzon E., Chaudhury D., Damez-Werno D. M., Haggarty S. J., Han M. H. et al. (2013). Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat. Neurosci. 16, 434–440 10.1038/nn.3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson L., Li X., Frontera W. R. (1997a). Effects of aging on shortening velocity and myosin isoform composition in single human skeletal muscle cells. Am. J. Physiol. 272, C638–C649 [DOI] [PubMed] [Google Scholar]
- Larsson L., Li X., Yu F., Degens H. (1997b). Age-related changes in contractile properties and expression of myosin isoforms in single skeletal muscle cells. Muscle Nerve Suppl. 20, 74–78 [DOI] [PubMed] [Google Scholar]
- Lecker S. H., Jagoe R. T., Gilbert A., Gomes M., Baracos V., Bailey J., Price S. R., Mitch W. E., Goldberg A. L. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51 10.1096/fj.03--0610com [DOI] [PubMed] [Google Scholar]
- Lei W. W., Zhang K. H., Pan X. C., Wang D. M., Hu Y., Yang Y. N., Song J. G. (2010). Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis. 1, e44 10.1038/cddis.2010.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe D. A., Surek J. T., Thomas D. D., Thompson L. V. (2001). Electron paramagnetic resonance reveals age-related myosin structural changes in rat skeletal muscle fibers. Am. J. Physiol. 280, C540–C547 [DOI] [PubMed] [Google Scholar]
- Mal A., Sturniolo M., Schiltz R. L., Ghosh M. K., Harter M. L. (2001). A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 20, 1739–1753 10.1093/emboj/20.7.1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S. J., Di Lisi R., Sandri C., Zhao J. et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- Masiero E., Sandri M. (2010). Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 6, 307–309 10.4161/auto.6.2.11137 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. (2005). Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 102, 11278–11283 10.1073/pnas.0502738102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Olson E. N. (2001). Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11, 497–504 10.1016/S0959--437X(00)00224--0 [DOI] [PubMed] [Google Scholar]
- Minetti G. C., Colussi C., Adami R., Serra C., Mozzetta C., Parente V., Fortuni S., Straino S., Sampaolesi M., Di Padova M. et al. (2006). Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 12, 1147–1150 10.1038/nm1479 [DOI] [PubMed] [Google Scholar]
- Moresi V., Williams A. H., Meadows E., Flynn J. M., Potthoff M. J., McAnally J., Shelton J. M., Backs J., Klein W. H., Richardson J. A. et al. (2010). Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45 10.1016/j.cell.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresi V., Carrer M., Grueter C. E., Rifki O. F., Shelton J. M., Richardson J. A., Bassel-Duby R., Olson E. N. (2012). Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. USA 109, 1649–1654 10.1073/pnas.1121159109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K. T., Cobani V., Ryall J. G., Ibebunjo C., Lynch G. S. (2011). Acute antibody-directed myostatin inhibition attenuates disuse muscle atrophy and weakness in mice. J. Appl. Physiol. 110, 1065–1072 10.1152/japplphysiol.01183.2010 [DOI] [PubMed] [Google Scholar]
- Ochala J., Gustafson A. M., Diez M. L., Renaud G., Li M., Aare S., Qaisar R., Banduseela V. C., Hedström Y., Tang X. et al. (2011). Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J. Physiol. 589, 2007–2026 10.1113/jphysiol.2010.202044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P. L., Iezzi S., Stiegler P., Chen T. T., Schiltz R. L., Muscat G. E., Giordano A., Kedes L., Wang J. Y., Sartorelli V. (2001). Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8, 885–897 10.1016/S1097--2765(01)00373--2 [DOI] [PubMed] [Google Scholar]
- Qiu Y., Stavreva D. A., Luo Y., Indrawan A., Chang M., Hager G. L. (2011). Dynamic interaction of HDAC1 with a glucocorticoid receptor-regulated gene is modulated by the activity state of the promoter. J. Biol. Chem. 286, 7641–7647 10.1074/jbc.M110.185488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A., Milan G., Masiero E., Carnio S., Lee D., Lanfranchi G., Goldberg A. L., Sandri M. (2010). JunB transcription factor maintains skeletal muscle mass and promotes hypertrophy. J. Cell Biol. 191, 101–113 10.1083/jcb.201001136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S. A., Senf S. M., Cornwell E. W., Kandarian S. C., Judge A. R. (2011). Inhibition of IkappaB kinase alpha (IKKα) or IKKbeta (IKKβ) plus forkhead box O (Foxo) abolishes skeletal muscle atrophy. Biochem. Biophys. Res. Commun. 405, 491–496 10.1016/j.bbrc.2011.01.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S. A., Sandesara P. B., Senf S. M., Judge A. R. (2012). Inhibition of FoxO transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 26, 987–1000 10.1096/fj.11--189977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. M., Ahn B., Smuder A. J., Al-Rajhi M., Gill L. C., Beharry A. W., Powers S. K., Fuller D. D., Ferreira L. F., Judge A. R. (2013a). Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 27, 2600–2610 10.1096/fj.12--222844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. M., Frye G. S., Ahn B., Ferreira L. F., Judge A. R. (2013b). Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochem. Biophys. Res. Commun. 435, 488–492 10.1016/j.bbrc.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacheck J. M., Hyatt J. P., Raffaello A., Jagoe R. T., Roy R. R., Edgerton V. R., Lecker S. H., Goldberg A. L. (2007). Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21, 140–155 10.1096/fj.06--6604com [DOI] [PubMed] [Google Scholar]
- Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., Goldberg A. L. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 10.1016/S0092--8674(04)00400--3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senf S. M., Judge A. R. (2012). Determination of gene promoter activity in skeletal muscles in vivo. Methods Mol. Biol. 798, 461–472 10.1007/978--1--61779--343--1_27 [DOI] [PubMed] [Google Scholar]
- Senf S. M., Dodd S. L., McClung J. M., Judge A. R. (2008). Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 22, 3836–3845 10.1096/fj.08--110163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senf S. M., Dodd S. L., Judge A. R. (2010). FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am. J. Physiol. 298, C38–C45 10.1152/ajpcell.00315.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senf S. M., Sandesara P. B., Reed S. A., Judge A. R. (2011). p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. Am. J. Physiol. 300, C1490–C1501 10.1152/ajpcell.00255.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. V., Brown M. (1999). Age-related changes in contractile properties of single skeletal fibers from the soleus muscle. J. Appl. Physiol. 86, 881–886 [DOI] [PubMed] [Google Scholar]
- Tou L., Liu Q., Shivdasani R. A. (2004). Regulation of mammalian epithelial differentiation and intestine development by class I histone deacetylases. Mol. Cell. Biol. 24, 3132–3139 10.1128/MCB.24.8.3132--3139.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Brault J. J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S. H., Goldberg A. L. (2007). FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483 10.1016/j.cmet.2007.11.004 [DOI] [PubMed] [Google Scholar]