

Abstract
As the distribution of Candida species and their susceptibility to antifungal agents have changed, a new means of accurately and rapidly identifying these species is necessary for the successful early resolution of infection and the subsequent reduction of morbidity and mortality. The current work aimed to evaluate ribosomal RNA gene sequencing for the identification of medically relevant Candida species in comparison with a standard phenotypic method. Eighteen reference strains (RSs), 69 phenotypically identified isolates and 20 inconclusively identified isolates were examined. Internal transcribed spaces (ITSs) and D1/D2 of the 26S ribosomal RNA gene regions were used as targets for sequencing. Additionally, the sequences of the ITS regions were used to establish evolutionary relationships. The sequencing of the ITS regions was successful for 88% (94/107) of the RS and isolates, whereas 100% of the remaining 12% (13/107) of the samples were successfully analysed by sequencing the D1/D2 region. Similarly, genotypic analysis identified all of the RS and isolates, including the 20 isolates that were not phenotypically identified. Phenotypic analysis, however, misidentified 10% (7/69) of the isolates. Phylogenetic analysis allowed the confirmation of the relationships between evolutionarily close species. Currently, the use of genotypic methods is necessary for the correct identification of Candida species.
Keywords: Candida, ribosomal DNA, DNA sequence analysis
Candida species are fungal pathogens that can cause a wide range of superficial and deep mycoses, collectively known as candidiasis, that are commonly observed in immunocompromised patients. Candidaemia is the most clinically important Candida infection, both because is the most frequent yeast infection in hospitalised patients and because this infection results in significant mortality. The incidence has risen over the past decades as the number of immunocompromised patients has increased (Pfaller & Diekema 2002, Falagas et al. 2010) and currently, candidaemia is the fourth most common nosocomial bloodstream infection in the United States of America (USA) (Pfaller et al. 1998, Edmond et al. 1999, Wisplinghoff et al. 2004).
Although Candida albicans remains the most frequently isolated species in human infection, more than 50% of candidaemias are due to infection with other Candida species (Price et al. 1994, Nguyen et al. 1996, Abi-Said et al. 1997, Trick et al. 2002, Hajjeh et al. 2004, Wisplinghoff et al. 2004). For example, in Argentina, C. albicans (38.4%), Candida parapsilosis (26%), Candida tropicalis (15.4%) and Candida glabrata (4.3%) are frequently isolated species (Cordoba et al. 2011).
The proper identification of Candida species is increasingly necessary, not only because the distribution of Candida species has changed, but also because these species differ in their susceptibility to antifungal agents (Pfaller & Diekema 2002, Ellepola & Morrison 2005). Accurate and rapid identification can facilitate the successful early resolution of infections and the subsequent reduction of morbidity and mortality (Pincus et al. 2007). The classical identification of fungi has been based on the morphological and physiological features of the sexual and/or asexual state. Because most medically important yeast species lack a sexual state and/or distinctive asexual morphological features, the correct identification of these species is often difficult and inconclusive when based solely on physiological traits. These morphological and physiological characteristics are often unstable, variable and subjective (Latouche et al. 1997). Furthermore, genetically diverse yeast species can yield similar phenotypic profiles, resulting in poor discrimination between unrelated yeast species (Sullivan et al. 1996). In this context, genotypic identification may be preferable as this method is faster and more accurate (Pincus et al. 2007).
The ribosomal RNA (rDNA) gene complex is largely used as a target in many polymerase chain reaction (PCR)-based assays because the complex is present in all microorganisms, occurs as tandem repeats of as many as 100-200 copies and contains highly conserved domains separated by variable domains, thus enabling the design of universal PCR primers for fungi (White et al. 1990, Kurtzman & Fell 1998, Iwen et al. 2002, Pincus et al. 2007). Moreover, nucleotide-sequence heterogeneity within this complex may be used to phylogenetically classify microorganisms, including yeast species (Kurtzman & Robnett 1998, Fell et al. 2000, Iwen et al. 2002).
The taxonomy of yeast and other fungal species has profoundly changed since the advent of DNA sequencing for the classification of microorganisms. Single gene sequences, such as those from the D1/D2 region of 26S rDNA or from the internal transcribed space (ITS) regions, are commonly used to identify yeast species (Kurtzman 2006, 2010). Recently, the sequence of the ITS region has been proposed to be a primary fungal barcode marker by the Consortium for the Barcode of Life (Schoch et al. 2012).
At present, the rDNA sequences of nearly all clinically relevant yeast species are available in public databases, such as GenBank (ncbi.nlm.nih.gov/genbank/) or the Centraalbureau voor Schimmelcultures (CBS) Yeast Database (cbs.knaw.nl), making it possible to genetically identify one unknown yeast isolate by comparing its rDNA sequences with those sequences in the database.
The aim of the current study was to compare a classical phenotypic method with rDNA sequencing for the identification of medically relevant Candida species. ITS sequences were also used to evaluate the genetic relationships between the samples analysed.
MATERIALS AND METHODS
Reference strains (RSs) and isolates - Eighteen RSs, which our laboratory commonly uses for identification and/or susceptibility testing, and 89 additional isolates (Supplementary data) were included in the study. The RS were obtained from the American Type Culture Collection, USA, the culture collection of the Carlos III Institute of Health, Spain, and the culture collection of the Department of Mycology of the National Institute of Infectious Diseases Dr Carlos G Malbrán, Argentina. Based on their phenotypic identification, isolates were selected from those received at the yeast identification laboratory from January 2009-December 2010. A total of 69 isolates of Candida species frequently isolated from patient in Argentina were included in the study (Rodero et al. 2005, Cordoba et al. 2011), as well as isolates with known resistance to common antifungal drugs. Also included were 20 isolates with ambiguous or inconclusive phenotypic identification, hereafter referred to as "not identified".
Phenotypic identification - The phenotypic identification was performed using standard methods (Kurtzman & Fell 1998), including an assessment of growth on 19 carbon and two nitrogen sources by the auxanographic method, the fermentation of six carbohydrates, growth at 35ºC and 37ºC, urea hydrolysis and morphological features. Discrimination between C. albicans and Candida dubliniensis was achieved using the agar tobacco test (Bosco-Borgeat et al. 2011).
DNA extraction - DNA extraction was performed according to the method reported by Möller et al. (1992), modified as previously described (Bosco-Borgeat et al. 2011). The DNA was preserved at -20ºC until use.
ITS amplification - The ITS1 (5?-TCCGTAGGTGAA-CCTGCGG-3?) and ITS4 (5?-TCCTCCGCTTATTGAT-ATGC-3?) primers were used (White et al. 1990). The reactions were performed in a volume of 100 µL containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2 mM Mg2Cl2, 5.2% DMSO, 0.2 mM each of dATP, dCTP, dGTP and dTTP (Fermentas International, Inc), 0.1 µM each of the primers ITS1 and ITS4, 1 U Taq DNA polymerase (Invitrogen-Life Technologies, Brazil) and 30 ng of DNA. All of the amplifications were performed in an iCycler (Bio-Rad Laboratories, Inc) using the following parameters: 95ºC for 7 min, followed by 40 cycles at 95ºC for 1 min, 54ºC for 2 min, 72ºC for 1 min and a final extension at 72ºC for 10 min.
Amplification of D1/D2 region of 26S rDNA - The NL1 (5?-GCATATCAATAAGCGGAGGAAAAG-3?) and NL4 (5?-GGTCCGTGTTTCAAGACGG-3?) primers were used (White et al. 1990). The reactions were performed in a volume of 100 µL containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.5 mM Mg2Cl, 5% DMSO, 0.2 mM each of dATP, dCTP, dGTP and dTTP, 0.1 µM each of the primers NL1 and NL4, 1 U Taq DNA polymerase and 30 ng of DNA. All of the amplifications were performed in an iCycler using the following parameters: 95ºC for 7 min, followed by 40 cycles at 95ºC for 1 min, 53ºC for 2 min, 72ºC for 1 min and a final extension at 72ºC for 10 min.
Agarose gel electrophoresis - The PCR products were electrophoresed on 1.5% agarose gels in 40 mM Tris-Acetate and 1 mM EDTA buffer (1X TAE) for 1 h at 100 V, stained with ethidium bromide (10 µg/mL) and then visualised under ultraviolet (UV) light and photo-documented using an LAS 3000 version 2.1 (Fuji Photo Film Co, Ltd). A GeneRuler 100 bp DNA Ladder (Fermentas Internationa, Inc) was used.
Purification of PCR products - The PCR products were purified using a PureLink PCR Purification Kit (Invitrogen). The products were then electrophoresed on 1.5% agarose gels in 1X TAE for 1 h at 100 V, stained with ethidium bromide (10 µg/mL) and visualised under UV light. A ready-to-use MassRuler Express DNA Ladder, LR Reverse (Fermentas), was used.
DNA sequencing and editing - The PCR products were sequenced in the forward and reverse directions using the initial amplification primers and an automated DNA sequencer (Genetic Analyzer 3500, Applied Biosystems). The sequences were edited and the consensus sequences were obtained using BioEdit version 7.0.0 (Hall 1999). All of the sequences were deposited in the GenBank database; the GenBank accessions are listed in Supplementary data.
Genotypic identification by sequence similarity - Sequence similarity was obtained using either the BLASTN tool of the National Center for Biotechnology Information (NCBI) website (ncbi.nlm.nih.gov/BLAST/) (Library of Medicine, Bethesda, MD , USA) or the pairwise sequence alignment tool of the Fungal Biodiversity Centre on the CBS website (cbs.knaw.nl/collections/BioloMICSSequences.aspx) (The Netherlands).
The identity of each isolate was determined based on the sequence similarity of the ITS regions, specifically using those results with > 97% similarity (Nilsson et al. 2008) and 99% coverage. When the similarity of the sequences of the ITS regions was ≤ 97%, the D1/D2 region of the 26S rDNA was sequenced using those results with 99% similarity (Kurtzman 2006) and 99% coverage.
Phylogenetic analyses - All of the sequences of the ITS regions were aligned using the CLUSTALW program (Thompson et al. 1994) and a phylogenetic tree was constructed using MEGA version 4.0.2 software (Tamura et al. 2007). The neighbour-joining algorithm and the number-of-differences model were implemented. All of the gaps were excluded from the analysis and branch support was ascertained using 2,000 bootstrap replicates.
RESULTS
Genotypic identification by sequence similarity - Supplementary data lists all of the identification results. The PCR using the primers ITS1 and ITS4 successfully amplified the ITS regions of all of the RS and isolates (data not shown). However, of the total 18 RS and 89 isolates, three RS and nine isolates yielded illegible sequences for their ITS regions for three replicates. Of the 80 isolates with legible sequences, 78 isolates had ? 97% similarity with the sequences deposited in the public databases. For the remaining two isolates and all three RS and nine isolates with illegible sequences for their ITS regions, sequencing of the D1/D2 region was performed. All of these RS and isolates yielded legible sequences and were 99% similar to the sequences deposited in the public databases.
In total, 15 RS and 78 isolates were genotypically identified by sequencing the ITS regions alone. The remaining three RS and 11 isolates required sequencing of the D1/D2 region for identification. Two special cases, listed below, are comprised by the cases described in this paragraph.
Isolate 113940, which was not identified phenotypically, exhibited 94% similarity to Candida magnoliae SL040806 (AM408497.1, GenBank) using the BLASTN program and 99.7% similarity to Candida sorbosivorans CBS10293 (CBS website) for the ITS sequences. However, this isolate had 99% similarity to C. sorbosivorans CBS2250 (AY521567.1, GenBank) and 98% similarity to C. magnoliae ESAB9 (AJ749827.1, GenBank) using the BLASTN program and 100% similarity to C. sorbosivorans CBS8768, CBS8824 and CBS10296 and 98.8% similarity to C. magnoliae CBS2800 (CBS website) for the D1/D2 sequence. This isolate was thus identified as C. sorbosivorans.
Isolate 103840, which was not identified phenotypically, had 94% and 99% similarity to Candida pseudorugosa XH1164 (DQ234792.1 and DQ234791.1, GenBank) for the ITS and D1/D2 regions, respectively. This isolate was identified as a Candida species closely related to C. pseudorugosa.
Comparison of phenotypic and genotypic identification - Table shows the concordance values obtained for the phenotypic and genotypic identifications. By comparing these methods, all of the RS and isolates phenotypically identified as C. albicans , C. dubliniensis ,> C. tropicalis , C. glabrata , Candida haemulonii , Candida kefyr , Candida krusei , Candida lusitaniae , Candida pelliculosa and Candida guilliermondii var. membranifaciens demonstrated 100% concordance with their genotypic identifications. In contrast, the phenotypic identification did not agree with the genotypic identification in seven cases: four C. guilliermondii were misidentified as Candida famata (isolates 113934-37), isolate 113891 ( C. haemulonii var. vulnera ) was misidentified as C. guilliermondii , isolate 113933 (Candida fermentati) was misidentified as C. guilliermondii and isolate 113916 ( Candida orthopsilosis ) was misidentified as C. parapsilosis .
TABLE. Concordance between phenotypic and genotypic identification of Candida spp isolates.

a: number of microorganism from the culture collection of the Department of Mycology of the National Institute of Infectious Diseases Dr Carlos G Malbrán, Argentina; ND: not determined; IS: illegible sequence.
All of the isolates with ambiguous or inconclusive phenotypic identification (that is, not identified) were genotypically identified by sequencing one or both of the target regions.
Phylogenetic analyses - The phylogenetic tree for the ITS sequences (Figure) exhibits different clusters that are composed of isolates belonging to the same species. One exception was the C. fermentati isolate (113933), which clustered in the C. guilliermondii group. These species have only a three-nucleotide difference in their ITS sequences (data not shown). A low-level sequence divergence was observed among the isolates in each cluster, although intraspecies diversity was observed for C. famata , C. haemulonii and C. lusitaniae . An analysis of the C. haemulonii cluster showed that C. haemulonii var. vulnera (isolate 113891) was included in a separate branch. When analysing the C. lusitaniae cluster, the level of sequence diversity may be compared with the interspecies divergence observed between C. albicans and C. dubliniensis or between C. parapsilosis and C. orthopsilosis , for example (Figure).
Neighbour joining tree based on the internal transcribed space regions sequences showing the phylogenetic relationship among Candida species and isolates. Bootstrap percentages from 2,000 replicates are shown in each node. Scale bar indicates number of differences. The teleomorph name correspondent to each anamorph Candida species is included between brackets.
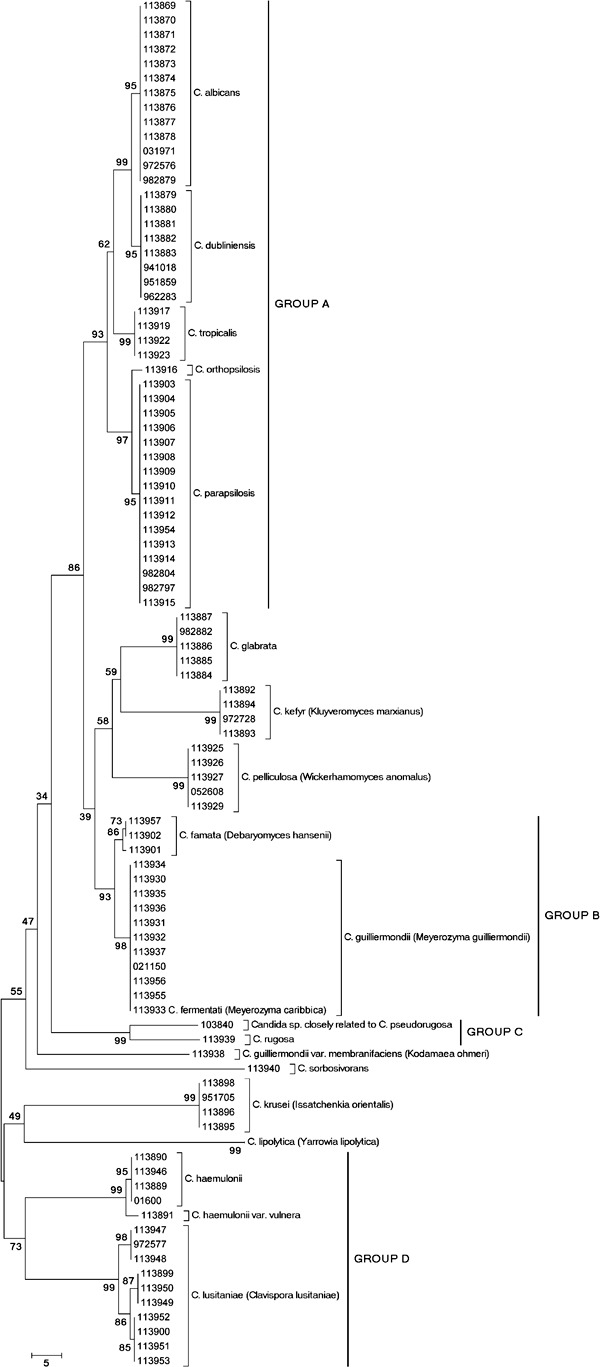
Certain species formed well-supported clades (bootstrap > 70) (Group A: C. albicans , C. dubliniensis , C. tropicalis , C. parapsilosis and C. orthopsilosis ; Group B: C. famata , C. guilliermondii and C. fermentati ; Group C: C. pseudorugosa and Candida rugosa ; Group F: C. haemulonii and C. lusitaniae ).
DISCUSSION
The advent of DNA sequencing has yielded many new tools for fungal identification that are used by taxonomists and non-taxonomists alike. However, certain factors should be considered when performing genotypic identification using rDNA sequencing. First, rDNA consists of tandem repeats, with as many as 100-200 copies (Kurtzman & Fell 1998). Individual copies typically evolve nearly in unison, meaning that each gene copy shares the same set of mutations with the other copies. This uniformity arises from sequence homogenisation mechanisms that are collectively referred to as concerted evolution (Alvarez & Wendel 2003). However, these mechanisms may be out of pace with variation-generating processes. It thus cannot be assumed that only one sequence type exists (Alvarez & Wendel 2003). In our study, 12 specimens yielded an illegible ITS chromatogram, presumably because these specimens contained more than one type of ITS sequence. In such cases, sequencing of the D1/D2 region was performed, resulting in a legible sequence of that domain, likely because the 26S rDNA has evolved slowly and is more conserved than the ITS regions.
Second, it is often assumed that fungal intraspecies variability in the ITS regions is generally low and represented by a percentage interval of 0-3% (Ciardo et al. 2006, Nilsson et al. 2008). In a large study of the sequences of the ITS regions available in international sequence databases, Nilsson et al. (2008) determined that the canonical 3% threshold value for intraspecies variation is surprisingly accurate for fungi, but this threshold is nevertheless refuted by multiple examples from all of the fungal phyla. However, the authors calculated that the weighted average of the intraspecies ITS variability of the kingdom Fungi is 2.51 ± 4.57% (1.96 ± 3.73% for Ascomycota), demonstrating the apparent futility of identifying a single unifying yet stringent fungus-wide cut-off value to demarcate ITS intraspecies variability from interspecies variability. In connection with the D1/D2 region of the 26S rDNA sequence, Kurtzman (2006) showed that species strains exhibit no more than zero-three nucleotide differences (0-0.5%) in this domain and strains showing six or more noncontiguous substitutions (1%) are typically considered separate species. However, certain species exhibit an intraspecies variability of > 1% (Lachance et al. 2003, Tavanti et al. 2005, Vaughan-Martini et al. 2005, Kurtzman 2006). In the current study, isolate 103840 showed ≤ 97% similarity to the sequences of the ITS regions in the public databases. Isolate 103840 was first reported following a case of bloodstream infection and was identified as a Candida species closely related to C. pseudorugosa (Taverna et al. 2012). The present study indicates that this isolate may belong to a species that is more polymorphic than other species or may be distinct from previously identified strains.
Third, several public databases and bioinformatics tools currently greatly aid genotypic identification by sequencing. GenBank, the most popular of the public databases, contains numerous sequences, including those of fungal rDNA. The drawbacks of GenBank include the presence of certain sequencing and nomenclature errors and infrequent nomenclature updates and expert-based reclassifications of mislabelled sequences. In this context, other databases comprising well-characterised sequences are preferable. For example, the CBS Yeast Database contains results for roughly 6,500 strains available from the CBS collection, as well as descriptions of up to 900 yeast species (Pincus et al. 2007). However, GenBank contains sequences of novel species from around the world. As a consequence, in certain cases, it may be useful to consult both databases. In our study, the identification of isolate 103940 as C. sorbosivorans necessitated such a dual analysis. Notably, although isolate 103940 C. sorbosivorans was obtained from a blood sample, this species had never been reported as a human pathogen. Unfortunately, we do not have follow-up information on the case, so nothing further can be reported.
Based on our comparative analysis of methods of phenotypic and genotypic identification, seven isolates were found to have been misidentified by phenotypic identification. These isolates corresponded to species that share a phenotypic profile with other species or that are cryptic species (Tavanti et al. 2005, Vaughan-Martini et al. 2005, Desnos-Ollivier et al. 2008, Cendejas-Bueno et al. 2012). Additionally, a phylogenetic analysis revealed a close relationship between the misidentified species, consistent with previous studies (Tavanti et al. 2005, Vaughan-Martini et al. 2005, Cendejas-Bueno et al. 2012, Taverna et al. 2012). The phylogenetic tree also indicated four well-supported groups. Group A comprised the species most frequently isolated in humans, all of which have no known teleomorph, but occur within the Lodderomyces clade, as proposed by others (Kurtzman & Suzuki 2010). Group B was composed of members of the Debaryomyces and Meyerozyma genera that are difficult to discriminate by phenotypic methods (Nishikawa et al. 1999, Vaughan-Martini et al. 2005, Desnos-Ollivier et al. 2008, Castanheira et al. 2012). Group C was composed of a potentially new Candida species closely related to C. pseudorugosa and C. rugosa. Finally, Group D was formed of members of the C. haemulonii complex and C. lusitaniae. The high level of sequence diversity within the C. haemulonii complex has been previously studied, leading to reclassification (Cendejas-Bueno et al. 2012). In contrast, the high level of sequence diversity of C. lusitaniae is in agreement with the unusually polymorphic sequences of the D1/D2 region in these strains (Lachance et al. 2003). Further studies focussing on a reclassification of the C. lusitaniae complex should be pursued.
The concept of a barcode marker for fungi has been discussed in recent years. The ITS regions have demonstrated the highest probability of successful identification for the broadest range of fungi, with the most clearly defined barcode gap between interspecies and intraspecies variation and high ITS PCR amplification success (Schoch et al. 2012). The possibility of a two-marker barcoding system for fungi, as previously adopted for plants, is often discussed among mycologists and particularly those researching ascomycetous yeasts who prefer a system combining ITS and 26S rDNA sequences. Additionally, the concept of one fungus having one name, whether the fungus exhibits sexual reproduction, instead of dual fungal nomenclature has been integrated into the new International Code of Nomenclature for algae, fungi and plants (Taylor 2011).
In the present study, a phylogenetic analysis was performed based on a single marker. However, single-gene analyses do not yield sufficient information to resolve the phylogenies. Well-resolved phylogenies often include not only rDNA genes, but also protein-coding genes (Kurtzman & Robnett 2003, Rokas et al. 2003, Diezmann et al. 2004, Suh et al. 2006, Tsui et al. 2008, Schoch et al. 2012). Other molecular studies have demonstrated that sequencing multiple genes or portions of genes and analysing the resultant data by phylogenetic methods is a robust strategy for identifying fungal species. This strategy is known as Genealogical Concordance Phylogenetic Species Recognition (Taylor et al. 2000). However, such a methodology is expensive and requires phylogenetic expertise, which may be limiting factors in clinical laboratories in which rapid identification is required (Balajee et al. 2009).
In conclusion, genotypic identification allowed the accurate identification of species frequently misidentified by phenotypic methods, cryptic species and potential new species. Yet, phenotypic data should not be disregarded and yeast identification should encompass a comprehensive analysis of both phenotypic and genotypic data.
Appendix
TABLE. Reference strains and isolates, phenotypic and genotypic identification and GenBank accessions.
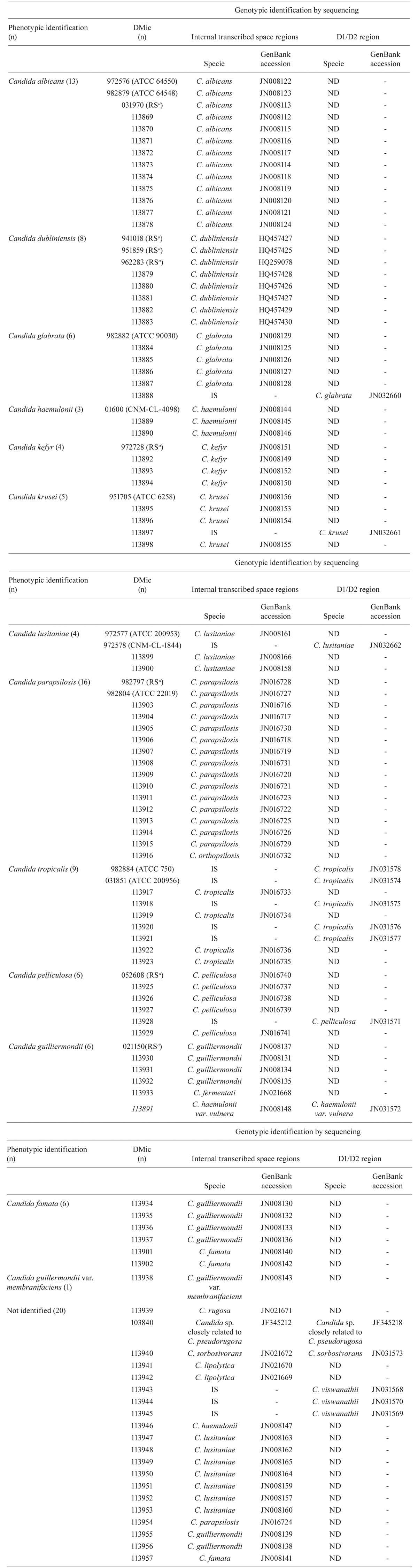
a: reference strain (RS) of the culture collection of the Department of Mycology of the National Institute of Infectious Diseases Dr Carlos G Malbrán, Argentina (DMic); ND: not determined; IS: illegible sequence.
REFERENCES
- Abi-Said D, Anaissie E, Uzun O, Raad I, Pinzcowski H, Vartivarian S. The epidemiology of hematogenous candidiasis caused by different Candida species. Clin Infect Dis. 1997;24:1122–1128. doi: 10.1086/513663. [DOI] [PubMed] [Google Scholar]
- Alvarez I, Wendel JF. Ribosomal ITS sequences and plant phylogenetic inference. Mol Phylogenet Evol. 2003;29:417–434. doi: 10.1016/s1055-7903(03)00208-2. [DOI] [PubMed] [Google Scholar]
- Balajee SA, Borman AM, Brandt ME, Cano J, Cuenca-Estrella M, Dannaoui E, Guarro J, Haase G, Kibbler CC, Meyer W, O´Donnell K, Petti CA, Rodriguez-Tudela JL, Sutton D, Velegraki A, Wickes BL. Sequence-based identification of Aspergillus, Fusarium and Mucorales species in the clinical mycology laboratory: where are we and where should we go from here? J Clin Microbiol. 2009;47:877–884. doi: 10.1128/JCM.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco-Borgeat ME, Taverna CG, Cordoba S, Isla MG, Murisengo OA, Szusz W, Vivot W, Davel G. Prevalence of Candida dubliniensis fungemia in Argentina: identification by a novel multiplex PCR and comparison of different phenotypic methods. Mycopathologia. 2011;172:407–414. doi: 10.1007/s11046-011-9450-6. [DOI] [PubMed] [Google Scholar]
- Castanheira M, Woosley LN, Diekema DJ, Jones RN, Pfaller MA. Candida guilliermondii and other species of Candida misidentified as Candida famata: assessment by the Vitek 2, DNA-Sequencing Analysis and MALDI-TOF MS in two global antifungal surveillance programs. J Clin Microbiol. 2012;51:117–124. doi: 10.1128/JCM.01686-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cendejas-Bueno E, Kolecka A, Alastruey-Izquierdo A, Theelen B, Groenewald M, Kostrzewa M, Cuenca-Estrella M, Gómez-López A, Boekhout T. Reclassification of the Candida haemulonii complex, C. duobushaemulonii sp. nov. (C. haemulonii group II) and C. haemulonii var. vulnera var. nov.: two multiresistant human pathogenic yeasts. J Clin Microbiol. 2012;50:3641–3651. doi: 10.1128/JCM.02248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardo DE, Schar G, Bottger EC, Altwegg M, Bosshard PP. Internal transcribed spacer sequencing versus biochemical profiling for identification of medically important yeasts. J Clin Microbiol. 2006;44:77–84. doi: 10.1128/JCM.44.1.77-84.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba S, Vivot W, Bosco-Borgeat ME, Taverna C, Szusz W, Murisengo O, Isla G, Davel G. Species distribution and susceptibility profile of yeast isolated from blood cultures: results of a multicenter active laboratory-based surveillance study in Argentina. Rev Argent Microbiol. 2011;43:176–185. doi: 10.1590/S0325-75412011000300003. [DOI] [PubMed] [Google Scholar]
- Desnos-Ollivier M, Ragon M, Robert V, Raoux D, Gantier JC, Dromer F. Debaryomyces hansenii (Candida famata), a rare human fungal pathogen often misidentified as Pichia guilliermondii (Candida guilliermondii) J Clin Microbiol. 2008;46:3237–3242. doi: 10.1128/JCM.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezmann S, Cox CJ, Schönian G, Vilgalys RJ, Mitchell TG. Phylogeny and evolution of medical species of Candida and related taxa: a multigenic analysis. J Clin Microbiol. 2004;42:5624–5635. doi: 10.1128/JCM.42.12.5624-5635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmond MB, Wallace SE, McClish DK, Pfaller MA, Jones RN, Wenzel RP. Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin Infect Dis. 1999;29:239–244. doi: 10.1086/520192. [DOI] [PubMed] [Google Scholar]
- Ellepola AN, Morrison CJ. Laboratory diagnosis of invasive candidiasis. J Microbiol. 2005;43(Suppl):65–84. [PubMed] [Google Scholar]
- Falagas ME, Roussos N, Vardakas KZ. Relative frequency of albicans and the various non-albicans Candida spp among candidemia isolates from inpatients in various parts of the world: a systematic review. Int J Infect Dis. 2010;14:e954–966. doi: 10.1016/j.ijid.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Fell JW, Boekhout T, Fonseca A, Scorzetti G, Statzell-Tallman A. Biodiversity and systematics of basidiomycetous yeasts as determined by large-subunit rDNA D1/D2 domain sequence analysis. Int J Syst Evol Microbiol. 2000;50:1351–1371. doi: 10.1099/00207713-50-3-1351. [DOI] [PubMed] [Google Scholar]
- Hajjeh RA, Sofair AN, Harrison LH, Lyon GM, Arthington-Skaggs BA, Mirza SA, Phelan M, Morgan J, Lee-Yang W, Ciblak MA, Benjamin LE, Sanza LT, Huie S, Yeo SF, Brandt ME, Warnock DW. Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population-based active surveillance program. J Clin Microbiol. 2004;42:1519–1527. doi: 10.1128/JCM.42.4.1519-1527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdita user-friendly biological sequence aligment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Iwen PC, Hinrichs SH, Rupp ME. Utilization of the internal transcribed spacer regions as molecular targets to detect and identify human fungal pathogens. Med Mycol. 2002;40:87–109. doi: 10.1080/mmy.40.1.87.109. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP. Yeast species recognition from gene sequence analyses and other molecular methods. Mycoscience. 2006;47:65–71. [Google Scholar]
- Kurtzman CP. Description of new yeast species - is one strain enough? The Bulletin of BISMiS. 2010;1:17–24. [Google Scholar]
- Kurtzman CP, Fell JW. The yeast, a taxonomic study. 4th ed. Elsevier; New York: 1998. 1054 pp [Google Scholar]
- Kurtzman CP, Robnett CJ. Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek. 1998;73:331–371. doi: 10.1023/a:1001761008817. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP, Robnett CJ. Phylogenetic relationships among yeasts of the "Saccharomyces complex" determined from multigene sequence analyses. FEMS Yeast Res. 2003;3:417–432. doi: 10.1016/S1567-1356(03)00012-6. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP, Suzuki M. Phylogenetic analysis of ascomycete yeasts that form coenzyme Q-9 and the proposal of the new genera Babjeviella, Meyerozyma, Millerozyma, Priceomyces and Scheffersomyces . Mycoscience. 2010;51:2–14. [Google Scholar]
- Lachance MA, Daniel HM, Meyer W, Prasad GS, Gautam SP, Boundy-Mills K. The D1/D2 domain of the large-subunit rDNA of the yeast species Clavispora lusitaniae is unusually polymorphic. FEMS Yeast Res. 2003;4:253–258. doi: 10.1016/S1567-1356(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Latouche GN, Daniel HM, Lee OC, Mitchell TG, Sorrell TC, Meyer W. Comparison of use of phenotypic and genotypic characteristics for identification of species of the anamorph genus Candida and related teleomorph yeast species. J Clin Microbiol. 1997;35:3171–3180. doi: 10.1128/jcm.35.12.3171-3180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller EM, Bahnweg G, Sanderman H, Geiger HH. A simple and efficient protocol for isolation of high molecular weigh DNA from filamentous fungi, fruit bodies and infected plant tissues. Nucleic Acids Res. 1992;20:6115–6116. doi: 10.1093/nar/20.22.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MH, Peacock JE, Jr, Morris AJ, Tanner DC, Nguyen ML, Snydman DR, Wagener MM, Rinaldi MG, Yu VL. The changing face of candidemia: emergence of non-Candida albicans species and antifungal resistance. Am J Med. 1996;100:617–623. doi: 10.1016/s0002-9343(95)00010-0. [DOI] [PubMed] [Google Scholar]
- Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson KH. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform Online. 2008;4:193–201. doi: 10.4137/ebo.s653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa A, Sugita T, Shinoda T. Rapid identification of Debaryomyces hansenii/Candida famata by polymerase chain reaction. Med Mycol. 1999;37:101–104. [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ. Role of sentinel surveillance of candidemia: trends in species distribution and antifungal susceptibility. J Clin Microbiol. 2002;40:3551–3557. doi: 10.1128/JCM.40.10.3551-3557.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Jones RN, Doern GV, Sader HS, Hollis RJ, Messer SA. International surveillance of bloodstream infections due to Candida species: frequency of occurrence and antifungal susceptibilities of isolates collected in 1997 in the United States, Canada and South America for the SENTRY Program. The SENTRY Participant Group. J Clin Microbiol. 1998;36:1886–1889. doi: 10.1128/jcm.36.7.1886-1889.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus DH, Orenga S, Chatellier S. Yeast identification - past, present and future methods. Med Mycol. 2007;45:97–121. doi: 10.1080/13693780601059936. [DOI] [PubMed] [Google Scholar]
- Price MF, LaRocco MT, Gentry LO. Fluconazole susceptibilities of Candida species and distribution of species recovered from blood cultures over a 5-year period. Antimicrob Agents Chemother. 1994;38:1422–1424. doi: 10.1128/aac.38.6.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero L, Davel G, Soria M, Vivot W, Cordoba S, Canteros CE, Saporiti A. Multicenter study of fungemia due to yeasts in Argentina. Rev Argent Microbiol. 2005;37:189–195. [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi . Proc Natl Acad Sci USA. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SO, Blackwell M, Kurtzman CP, Lachance MA. Phylogenetics of Saccharomycetales, the ascomycete yeasts. Mycologia. 2006;98:1006–1017. doi: 10.3852/mycologia.98.6.1006. [DOI] [PubMed] [Google Scholar]
- Sullivan DJ, Henman MC, Moran GP, O'Neill LC, Bennett DE, Shanley DB, Coleman DC. Molecular genetic approaches to identification, epidemiology and taxonomy of non-albicans Candida species. J Med Microbiol. 1996;44:399–408. doi: 10.1099/00222615-44-6-399. [DOI] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tavanti A, Davidson AD, Gow NA, Maiden MC, Odds FC. Candida orthopsilosis and Candida metapsilosis spp nov. to replace Candida parapsilosis groups II and III. J Clin Microbiol. 2005;43:284–292. doi: 10.1128/JCM.43.1.284-292.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna CG, Córdoba S, Isla G, Fernández N, García S, Mazza M, Murisengo OA, Vivot W, Szusz W, Davel G, Tiraboschi IN, Bosco-Borgeat ME. First case report of bloodstream infection due to a Candida species closely related to the novel species Candida pseudorugosa . J Clin Microbiol. 2012;50:2165–2169. doi: 10.1128/JCM.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW. One fungus = one name: DNA and fungal nomenclature twenty years after PCR. IMA Fungus. 2011;2:113–120. doi: 10.5598/imafungus.2011.02.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. Phylogenetic species recognition and species concepts in Fungi . Fungal Genet Biol. 2000;31:21–32. doi: 10.1006/fgbi.2000.1228. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTALWimproving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trick WE, Fridkin SK, Edwards JR, Hajjeh RA, Gaynes RP. Secular trend of hospital-acquired candidemia among intensive care unit patients in the United States during 1989-1999. Clin Infect Dis. 2002;35:627–630. doi: 10.1086/342300. [DOI] [PubMed] [Google Scholar]
- Tsui CK, Daniel HM, Robert V, Meyer W. Re-examining the phylogeny of clinically relevant Candida species and allied genera based on multigene analyses. FEMS Yeast Res. 2008;8:651–659. doi: 10.1111/j.1567-1364.2007.00342.x. [DOI] [PubMed] [Google Scholar]
- Vaughan-Martini A, Kurtzman CP, Meyer SA, O'Neill EB. Two new species in the Pichia guilliermondii clade: Pichia caribbica sp. nov., the ascosporic state of Candida fermentati and Candida carpophila comb. nov. FEMS Yeast Res. 2005;5:463–469. doi: 10.1016/j.femsyr.2004.10.008. [DOI] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor JW. Innis MA, Gelfand DH, Sninsky JJ, White TJ. PCR protocols: a guide to methods and applications. Academic Press; New York: 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics; pp. 315–322. [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE. Reference strains and isolates, phenotypic and genotypic identification and GenBank accessions.
a: reference strain (RS) of the culture collection of the Department of Mycology of the National Institute of Infectious Diseases Dr Carlos G Malbrán, Argentina (DMic); ND: not determined; IS: illegible sequence.