

Abstract
Pathophysiological responses to peripheral nerve injury include alterations in the activity, intrinsic membrane properties and excitability of spinal neurons. The intrinsic excitability of α-motoneurons is controlled in part by the expression, regulation, and distribution of membrane-bound ion channels. Ion channels, such as Kv2.1 and SK, which underlie delayed rectifier potassium currents and afterhyperpolarization respectively, are localized in high-density clusters at specific postsynaptic sites (Deardorff et al., 2013; Muennich and Fyffe, 2004). Previous work has indicated that Kv2.1 channel clustering and kinetics are regulated by a variety of stimuli including ischemia, hypoxia, neuromodulator action and increased activity. Regulation occurs via channel dephosphorylation leading to both declustering and alterations in channel kinetics, thus normalizing activity (Misonou et al., 2004; Misonou et al., 2005; Misonou et al., 2008; Mohapatra et al., 2009; Park et al., 2006). Here we demonstrate using immunohistochemistry that peripheral nerve injury is also sufficient to alter the surface distribution of Kv2.1 channels on motoneurons. The dynamic changes in channel localization include a rapid progressive decline in cluster size, beginning immediately after axotomy, reaching maximum within one week. With reinnervation, the organization and size of Kv2.1 clusters do not fully recover. However, in the absence of reinnervation Kv2.1 cluster sizes fully recover. Moreover, unilateral peripheral nerve injury evokes parallel, but smaller, effects bilaterally. These results suggest that homeostatic regulation of motoneuron Kv2.1 membrane distribution after axon injury is largely independent of axon reinnervation.
Keywords: Kv2.1, Peripheral Nerve Injury, Voltage-gated ion channels, Motoneuron
1. Introduction
Injured peripheral nerves can successfully reinnervate peripheral targets, but complete functional recovery seldom occurs (Alvarez et al., 2011; Bullinger et al., 2011; Cope and Clark, 1993; Cope et al., 1994; Haftel et al., 2005; Prather et al., 2011). Reorganization of central circuits accounts, at least partially, for abnormal sensorimotor integration and stretch areflexia following peripheral nerve injury (Alvarez et al., 2011; Bullinger et al., 2011). Furthermore, injury induced changes to α-motoneuron (MN) intrinsic properties and cellular excitability are well documented (Bichler et al., 2007; Foehring et al., 1986a; Gustafsson and Pinter, 1984; Kuno et al., 1974a; Kuno et al., 1974b; Nakanishi et al., 2005) and may also contribute to dysfunctional or maladaptive signaling in intact spinal circuitry.
Neuronal biophysical properties are controlled in part by the expression patterns and subcellular distribution of highly regulated ion channels (Cerda and Trimmer, 2010; Deardorff et al., 2013; Duflocq et al., 2011; Lai and Jan, 2006; Magee, 2000; Misonou, 2010). It is now recognized that certain voltage-gated ion channels are not uniformly distributed in the surface membrane of α-MNs but exhibit polarized distributions and/or are concentrated in discrete microdomains (Bui et al., 2006; Carlin et al., 2000; Deardorff et al., 2013; Deng and Fyffe, 2004; Elbasiouny et al., 2005; Muennich and Fyffe, 2004). Ion channel localization and density in the surface membrane can be dynamically regulated at a number of levels including gene expression, protein trafficking and membrane insertion, posttranslational modifications, and interactions with macromolecular complexes or lipid rafts. Additionally, different channel subunits can interact to form heteromeric channels with altered channel properties (Benton et al., 2003; Kihira et al., 2010; Monaghan et al., 2004; Strassmaier et al., 2005). Moreover, channel subunits may associate with and be regulated by accessory subunits, neurotransmitter or neuromodulator receptors, scaffolding proteins and postsynaptic density proteins (e.g. PSD-95), and other signaling molecules. Yet few studies have examined channel regulation in α-MNs following peripheral nerve injury.
Delayed rectifier Kv2.1 channels regulate the excitability of a wide range of vertebrate neurons. Kv2.1 channels are typically sequestered, or ‘clustered’, via unique C-terminal phosphorylation sites into highly regulated microdomains with other ion channels, neurotransmitter receptors, and signaling molecules (Deardorff et al., 2013; Deng and Fyffe, 2004; Muennich and Fyffe, 2004; Wilson et al., 2004). Sequestration creates energetically and kinetically favorable mechanism for neurons to initiate rapid and robust responses to changes in synaptic inputs and cellular activity (Misonou et al., 2004; Misonou et al., 2005; Misonou et al., 2008; Misonou, 2010; Park et al., 2006). In α-MNs, the largest Kv2.1 clusters are located at C-bouton postsynaptic sites, where they comprise a unique cellular domain with SK2/3 channels, subsurface cisternae, and mAChR2 receptors (Deardorff et al., 2013; Muennich and Fyffe, 2004). Smaller Kv2.1 clusters are located at excitatory S-type synapses and may also be apposed to extrasynaptic neuroglia processes (Muennich and Fyffe, 2004).
Kv2.1 channel properties and membrane clustering are phosphorylation dependent (Misonou et al., 2004; Murakoshi et al., 1997; Park et al., 2006; Zhang et al., 2008). In the high-density clusters, Kv2.1 channels may be low or non-conducting, and it is possible that the channel proteins exhibit additional non-conducting functions (Fox et al., 2013; O’Connell et al., 2010). A variety of stimuli (ex: ischemia, hypoxia, and synaptic activity) cause Ca2+-calcineurin dependent channel dephosphorylation. (Misonou et al., 2004; Misonou et al., 2005). Dephosphorylated Kv2.1 undergoes a large (≈25 mV) hyperpolarizing shift in voltage dependence, increasing the amplitude of Kv2.1 delayed rectifier currents (Misonou et al., 2004; Mohapatra et al., 2009) and prolonging the duration of the interspike interval (Mohapatra et al., 2009; Surmeier and Foehring, 2004). Dephosphorylated Kv2.1 channels also spatially migrate from the initially clustered state (Misonou et al., 2004). In hippocampal neurons the modulation of Kv2.1 currents by this mechanism suppresses neuronal firing frequency following prolonged depolarization and increased synaptic activity (Mohapatra et al., 2009), strongly indicating a role for Kv2.1 in homeostatic regulation of cellular excitability.
The aim of this study was to use quantitative immunohistochemical techniques to investigate the membrane organization and spatial distribution of Kv2.1 immunoreactivity (IR) in rat medial and lateral gastrocnemius α-motoneurons (MNs) following tibial nerve injury. We first tested the effects of glutamate induced excitation in in vitro spinal cord slices to demonstrate the redistribution and reduced clustering of membrane Kv2.1 channels in spinal α-motoneurons, similar to the membrane redistribution seen following increase in activity in other neurons in vitro. We then tested two types of in vivo peripheral nerve injury in the rat including a tibial crush injury, in which peripheral reinnervation was permitted, and a tibial ligation injury, in which peripheral reinnervation was prevented. Both in vivo injuries resulted in declustering of Kv2.1 suggesting that the regulation of Kv2.1 may contribute to modifications in motoneuron intrinsic excitability following peripheral nerve injury.
2. Results
2.1 Redistribution of Kv2.1-IR results from increased in vitro activity in α-motoneurons
Previous work with Kv2.1 in both primary hippocampal cells and HEK293 cultures has shown a lateral translocation of Kv2.1 from clusters to a more uniform distribution (‘declustering’) in the membrane following glutamate treatment (Misonou et al., 2004; Misonou et al., 2005; Mohapatra et al., 2009; Murakoshi et al., 1997). Here we treated lumbar spinal cord slices with 10 μM glutamate for 10 minutes to determine if Kv2.1 clusters behave similarly in αMNs. En face Kv2.1-IR macrocluster (diameters > 1μm) areas from 20 αMNs (6 rats, 2 litters) were measured from control and glutamate-treated groups. Treatment with glutamate resulted in a significant 47% reduction in Kv2.1 macrocluster areas on lumbar αMNs in less than 10 minutes (figure 2). The reduction of macrocluster area is consistent with the previous work in other cell types that Kv2.1-IR is redistributed following increased activity.
Figure 2.
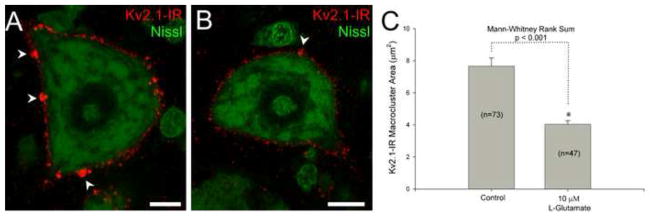
Kv2.1-IR cluster areas decrease in lumbar α-motoneurons following increased in vitro activity. Bath application of either artificial cerebral spinal fluid (ACSF) (panel A) or 10 μM glutamate (panel B) was applied to lumbar spinal cord slices for 10 minutes. Arrowheads point to the large Kv2.1-IR macroclusters. (A) Micrograph representation of single optical confocal section of tissue slice incubated in ACSF. Scale bar is 10 μm. (B) Micrograph representation of single optical confocal section of tissue slice incubated in artificial cerebral spinal fluid with 10 μM glutamate. Scale bar is 10 μm. (C) Quantitative analysis showing significant decrease of Kv2.1-IR macrocluster areas following glutamate treatment. Number of clusters sampled is displayed as “n” from 20 total cells of each treatment group. Data is presented as mean ± SEM.
2.2 Tibial nerve crush results in a significant decrease of both Kv2.1-IR macrocluster area and Kv2.1-IR macrocluster number
The goal of experiments reported in this section was to determine the effects of PNI on Kv2.1-IR macroclusters in rats by implementing a tibial nerve crush injury that permits proper reinnervation of peripheral targets. Spinal MNs with axons in the tibial nerve, specifically those that innervate the medial and lateral gastrocnemius muscles (MG/LG), were visualized with retrograde tracers injected intramuscularly prior to the injury. MNs positive for the retrograde tracers were imaged with confocal microscopy and quantitatively analyzed (figure 1). Initial observations revealed a statistically significant bilateral effect of unilateral nerve damage on Kv2.1-IR immunoreactivity in α-MNs after nerve crush or ligation (see section 2.5 below), although the reduction of Kv2.1 cluster sizes in contralateral MNs was always much less than on the side ipsilateral to the nerve damage. No effects were detected following sham operations. Therefore for accuracy, statistical analyses and graphs compare injury data to both sides of sham animals. However, to eliminate image variability resulting from artifacts or inconsistencies in tissue processing, confocal images included for illustrative purposes compare immunoreactivity from injured MG/LG MNs to contralateral, injury-spared MG/LG MNs within the same tissue slice (figures 3 & 4). It should be recognized that at the time points chosen for the ‘side-by-side’ confocal images presented here (8 days post injury) there was not a significant contralateral effect; however, because there may be a contralateral effect, the illustrated data potentially underestimates the ‘absolute’ degree of change in cluster size that occurs in an injured neuron.
Figure 1.

Intramuscular injections of retrograde tracer were used to identify motoneurons innervating the medial and lateral gastrocnemius (MG/LG). Only MG/LG motoneurons were selected for quantitative analysis in this study. Yellow arrow points to a population of motoneuron somas in the lateral lamina IX in the ventral horn of spinal cord segment L5 containing cholera toxin subunit B (CTB) conjugated to 488 fluorophore (green), one of the retrograde tracers used in this study. Nissl-IR (White) was used as a general neuronal stain. Retrogradely labeled cells selected for quantitative analysis were located in appropriate position within the motoneuron pool consistent with previous reports identifying location of MG/LG motoneuron somas (Romanes, 1951). For every injured αMN a contralateral control αMN was also selected from the appropriate position within lamina IX (see sec 4.4 methods) Scale bar is 200 μm.
Figure 3.

Kv2.1-IR macrocluster areas decrease on soma of medial and lateral gastrocnemius α-motoneurons following tibial nerve crush injury. To eliminate image variability resulting from artifacts or inconsistencies in tissue processing cells shown in panels A and B are from the same tissue section and imaged with fixed parameters. However, due to observed contralateral effects (see section 2.5) the illustrated data potentially underestimates the ‘absolute’ degree of change in cluster size that occurs in an injured neuron.(A) Micrograph of a confocal stack (32 × 1μ z-steps) of a contralateral motoneuron showing representative Kv2.1-IR clustering. Scale bar is 20 μm. (B) Micrograph of a confocal stack (32 × 1μ z-steps) of an 8 days post tibial nerve crushed motoneuron showing representative Kv2.1-IR clustering with reduced areas compared to the spared motoneuron. Scale bar is 20 μm. (C) Quantitative analysis of reduced Kv2.1-IR soma macrocluster areas on lumbar α-motoneurons 8 days following tibial nerve crush. (D) Quantitative analysis of reduced numbers of Kv2.1-IR soma macroclusters on lumbar α-motoneurons 8 days following tibial nerve crush. (E) Kv2.1 macrocluster nearest neighbor distance increases following 8 days post tibial nerve crush. In panels C, D, &E significance (p<0.05) is indicated with asterisk and determined with Pairwise T-Test and data is presented as mean ± SEM.
Figure 4.
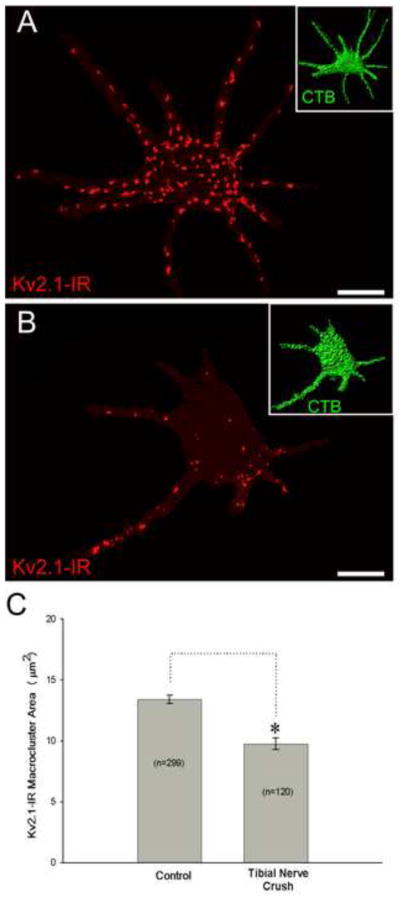
Kv2.1-IR Macrocluster areas decrease on proximal dendrites of medial and lateral gastrocnemius α-motoneurons following tibial nerve crush. Both cells shown in these panels are from the same spinal cord tissue slice and are scaled equally. Scale bar is 30 μm. Inserts in the top right of panel A and B shows the CTB retrograde fills from gastrocnemius muscle that was used to reconstruct the cell somas and proximal dendrites. (A) Micrograph showing representative 3-D reconstruction, from confocal microscopy image stacks (57 × 0.5 μm z-steps), of a contralateral motoneuron. (B) Micrograph showing representative 3-D reconstruction, from confocal microscopy image stacks (41 × 0.5 μm z-steps), of a motoneuron 8 days following nerve crush. (C) Quantitative data from 10 control and 10 injured motoneurons showing Kv2.1-IR macrocluster areas significantly decrease on proximal dendrites following both 8 days post tibial nerve crush. Data is presented as mean ± SEM. Significance is set to p<0.05.
Kv2.1-IR macroclusters (diameters > 1μm) visualized en face were selected and measured 8 days following tibial nerve crush injury (figure 3). Kv2.1-IR macrocluster cross-sectional areas are on average reduced by about 50% on the injured αMNs (4.75 μm2 ± 2.36 SD, n=937 clusters, 76 αMNs, 5 rats) compared to sham surgery control αMNs (9.29μm2 ± 4.82 SD, n=543 clusters, 50 αMNs, 3 rats) and the contralateral spared αMNs (9.35μm2 ± 3.76 SD, n=1123 clusters, 77 αMNs, 5 rats) (figure 3C). The Kv2.1 cluster areas on the contralateral sides were not found to be significantly different from sham surgery control animals at this time point (see section 2.5 below).
In addition to the reduction in size, the crush injury also induces a significant decrease in the number of Kv2.1-IR macroclusters on the soma of medial and lateral gastrocnemius MNs, with a maximum 55% reduction in number (injured mean=24.4 ± 9.68 SD, n=44 αMNs, 5 rats) 8 days after the injury compared to sham surgery controls (sham surgery control mean=54.6 ± 15.91 SD, n=36 αMNs, 3 rats). There were no significant differences in macrocluster number on the contralateral side (mean=50.8 ± 13.89 SD, n=53 αMNs, 5 rats), compared to sham surgery controls (figure 3D).
Furthermore, the linear distance between individual Kv2.1-IR macroclusters were measured in a nearest neighbor analysis, in 10 control and injured αMNs 8 days post injury. There are significant increases in distances between nearest neighbor Kv2.1 macroclusters following tibial nerve crush (figure 3E). Altogether, these data are consistent with fewer macroclusters with reduced areas.
Kv2.1-IR macroclusters located on proximal dendrites (up to approximately 150 μm from soma) were measured through 3D reconstructions to determine if channel clusters on dendrites also were affected by PNI. Kv2.1-IR macroclusters located on the proximal dendrites are 27% reduced in area (sham surgery control 13.42μm2 ± 6.054 SD, n=299 clusters, 20 αMNs, 2 rats; injured 9.764μm2 ± 5.091 SD, n=120 clusters, 10 αMNs, 2 rats) 8 days following tibial nerve crush (figure 4). Although dendritic Kv2.1-IR macroclusters (some of which are at least as large as clusters on the soma) are affected by PNI, they exhibit smaller on average percentage reduction of area than do macroclusters located on the soma membrane itself.
2.3 Tibial nerve crush results in a redistribution of Kv2.1-IR in α-motoneuron membrane
In cultures of hippocampal cells, use of Kv2.1 surface membrane-impermeant biotinylation assays following glutamate application revealed that the amount of membrane-localized Kv2.1 did not change even though the cluster areas qualitatively appeared reduced (Misonou et al., 2004). These results indicated a lateral translocation and redistribution of Kv2.1 in the membrane following the glutamate treatment rather than Kv2.1 internalization, and we wished to determine if this is the case in αMNs after injury. Indeed, in our experiments, when the axotomized MN membrane is examined in single optical confocal sections and small stacks of en face membrane, the small distinct punctae, or microclusters, typical of Kv2.1-IR distributions in uninjured αMNs (figure 5A&C) appear to be spread into a more diffuse field (figure 5B&D). Our previous studies of Kv2.1 immunoreactivity demonstrates the ability of our methods to quantitatively measure very small sized patches of immunoreactivity (Muennich and Fyffe, 2004). However, following injury the diffuse Kv2.1-IR in the membrane consists largely of punctae so small that accurate area measurements are precluded. However, we were able to assess quantitatively the relative levels of immunoreactivity in areas devoid of macroclusters but containing microclusters (measurable diameters <1.0 μm) and/or even smaller punctae. Analysis of regions of interest in en face membrane were selected in 10 uninjured and 10 injured MNs 8 days post tibial nerve crush. The minimum and average intensities measured in these regions were conserved following injury, with only the maximum intensity significantly decreased (figure 5E). However, there are no significant changes in the integrated optical density (density normalized to area sampled) of these same regions suggesting that the density of fluorescent pixels is conserved within the regions of interest (figure 5F). Finally we evaluated the same membrane regions for the fraction of pixels that deviate more than 10% from the average intensity and found there is a significant 88% reduction in the degree of heterogeneity of the Kv2.1-IR signal after injury (figure 5G). These results suggest that the Kv2.1-IR is less organized into microclusters, but still present in the membrane in a diffuse redistributed (or declustered) field of average intensity.
Figure 5.

Kv2.1 microcluster immunoreactivity becomes more diffuse on soma of medial and lateral gastrocnemius α-motoneurons following tibial nerve crush injury. To eliminate image variability resulting from artifacts or inconsistencies in tissue processing cells shown in panels A and B are single optical confocal sections from the same tissue section and imaged with fixed parameters. However, due to observed contralateral effects (see section 2.5) the illustrated data potentially underestimates the ‘absolute’ degree of change in cluster size that occurs in an injured neuron. Both panels A and B are single optical confocal sections through the center of the cell from the same tissue section and imaged with fixed parameters. Full confocal stacks of these two cells are shown in figure 3. (A1) Micrograph of single optical section of a contralateral motoneuron showing representative Kv2.1-IR clustering. (A2) Inset is zoomed region of the membrane, indicated by dotted box, with arrowheads pointing to distinct Kv2.-IR microcluster punctae in the membrane. Scale bar is 20 μm. (B1) Micrograph of single optical section of an 8 days post tibial nerve crushed motoneuron showing representative diffuse Kv2.1 microcluster immunoreactivity compared to the spared motoneuron. (B2) Inset is zoomed region of the membrane, indicated by dotted box. Note diffuse Kv2.1 microcluster immunoreactivity in the membrane compared to distinct punctae shown by arrowheads in A2. Scale bar is 20 μm. (C&D) Micrographs of small confocal stacks (10 × 1.0 μm Z-steps) of small patches of en face membrane regions. The distinct microcluster punctae in the spared motoneurons (panels C) appear more diffuse 8 days after tibial nerve crush (panels D). Scale bars are 10 μm. (E) Quantitative analysis of Kv2.1-IR microcluster intensity of lumbar α-motoneuron somas of spared and 8 day post tibial nerve crush. (F) Quantitative analysis of Kv2.1 microcluster integrated optical density of immunoreactive pixels on lumbar α-motoneuron somas of spared and 8-day post tibial nerve crush. (G) Quantitative analysis of Kv2.1-IR microcluster heterogeneity, the fraction of pixels that deviate more than 10% from the average intensity, on lumbar α-motoneuron somas of control and 8 day post crush. Data is presented as mean ± SEM. Significance (p<0.05) as indicated with asterisk was determined with t-test.
2.4 Kv2.1 macroclusters are not completely restored after peripheral reinnervation
Properties shaping intrinsic excitability following peripheral nerve injury generally return to normal ranges upon proper reinnervation of the peripheral target (Bichler et al., 2007; Foehring et al., 1986a; Foehring et al., 1986b; Kuno et al., 1974a). To determine if Kv2.1 ‘reclusters’ upon reinnervation, en face Kv2.1-IR macroclusters (diameters >1μm) were selected and measured for cross sectional area at 20 minutes, 3 days, 8 days, 2 weeks, 4 weeks and 6 months following tibial nerve crush injury (figure 6, table 1). At the latter two of these time points it is likely that reinnervation of target muscle is underway and/or has occurred. Significant differences in Kv2.1-IR macrocluster area exist between the injured αMNs and the sham surgery control αMNs as early as 20 minutes following injury (20% reduction), progressing to a maximum 51% reduction at 3 days post-injury. Subsequently, there is a gradual increase in cluster size, eventually attaining about 87% of normal average cluster size 6 months after injury (figure 6A).
Figure 6.

Time course of Kv2.1-IR declustering of gastrocnemius motoneuron somas following tibial nerve crush at key time points (20 minutes, 3 days, 8 days, 2 weeks, 4 weeks, and 6 months). Kv2.1-IR cluster sizes are still significantly smaller 6 months following injury with reinnervation. All comparisons are made to sham surgery control animals analyzed at 3 day, 8 day and 4 weeks following surgery. No significant differences were found between or within time points for the ipsilateral and contralateral sides in sham surgery control animals. Data is presented as mean ± SEM. Significance (p<0.05) was determined with pairwise ANOVA. (A) Significant decrease (indicated by single asterisk) in Kv2.1-IR areas on injured motoneurons appear as early as 20 minutes following injury and are significantly reduced at all time points following injury. There are also small, yet significant (indicated by double asterisk), decreases on the injury spared contralateral side of the rat spinal cord at 20min, 3 days, 2 weeks and 4 weeks post injury. (B) The number of Kv2.1-IR macroclusters on motoneurons somas decrease significantly (indicated with asterisk) 3 days following tibial nerve crush and are significantly reduced at all following time points.
Table 1.
Kv2.1-IR macrocluster means and sample sizes for tibial nerve crush injury
| TIBIAL NERVE CRUSH INJURY
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Sham | 20 min | 3 Day | 8 Day | 2 Week | 4 Week | 6 Month | ||
| Injury (ipsilateral) | Kv2.1-IR Mean Macrocluster Area (μm2) | 9.29 ± 4.82 | 7.47 ± 3.23 | 4.54 ± 2.32 | 4.74 ± 2.36 | 5.14 ± 3.01 | 6.48 ± 2.61 | 8.10 ± 3.41 |
| Clusters, Cells, Animals sampled | n=543, 50, 3 | n= 915, 77, 3 | n= 563, 25, 2 | n= 937, 76, 5 | n= 792, 59, 4 | n= 744, 56, 3 | n= 920, 57, 2 | |
|
| ||||||||
| Contralateral Controls | Kv2.1-IR Mean Macrocluster Area (μm2) | 9.24 ± 5.17 | 8.81 ± 4.03 | 7.28 ± 3.62 | 9.35 ± 3.76 | 8.65 ± 3.59 | 7.81 ± 3.54 | 9.01 ± 3.64 |
| Clusters, Cells, Animals sampled | n=504, 49, 3 | n= 920, 75, 3 | n= 519, 24, 2 | n= 1123, 77, 5 | n= 747, 47, 4 | n= 1116, 51, 3 | n= 1204, 62, 2 | |
±SD
To determine if the number of Kv2.1-IR macroclusters is restored after reinnervation, Kv2.1-IR macroclusters (diameters >1μm) were counted on MN somas at 20 minutes, 3 days, 8 days, 2 weeks, 4 weeks and 6 months following tibial nerve crush (figure 6B). Significant differences in the number of Kv2.1-IR macroclusters exist between the injured αMNs and sham surgery control with approximately 31% reduction 3 days following nerve crush and progressing to a maximum 54% reduction at 8 days following crush and returning to about 85% of normal 6 months after injury (figure 6B).
In a separate set of experiments, over the same time points post injury, we sought to determine the effects on Kv2.1-IR clustering when reinnervation was prevented through tibial nerve ligation (see methods). Following ligation, there is a gradual, yet ultimately significant declustering of Kv2.1-IR macrocluster areas. Significance is reached 3 days following this type of injury, when there is a 25% reduction in Kv2.1-IR macrocluster area, and a maximal 34% reduction by 4 weeks after the tibial nerve ligation when compared to sham surgery controls. Interestingly, by 6 months, the average Kv2.1-IR macrocluster areas are no longer significantly different from the sham controls (figure 7A&B, table 2). Similar to the tibial nerve crush injury, there is a significant decrease in the number of Kv2.1-IR macroclusters, but this too occurs over a slower time course, with a maximum 40% reduction 2 weeks after ligation (figure 7B).
Figure 7.
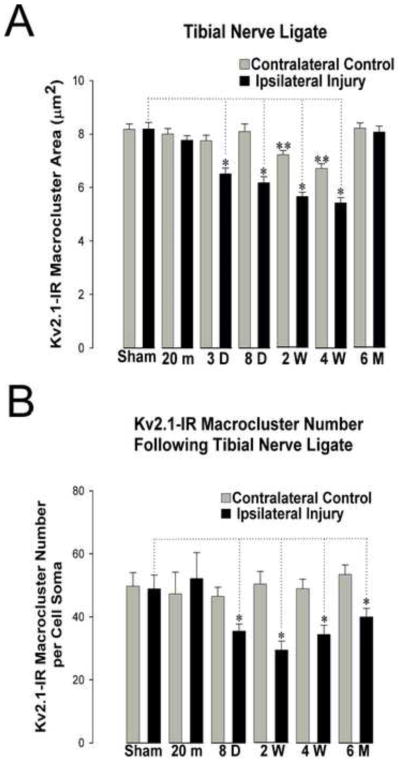
Time course of Kv2.1-IR declustering of gastrocnemius motoneuron somas following tibial nerve ligation at key time points (20 minutes, 3 days, 8 days, 2 weeks, 4 weeks, and 6 months). Kv2.1-IR clusters are reduced in number but return to pre-injury size 6 months following injury without reinnervation. All comparisons are made to sham surgery control animals analyzed at 3 day, 8 day and 4 weeks following surgery. No significant differences were found between or within time points for the ipsilateral and contralateral sides in sham surgery control animals. Data is presented as mean ± SEM. Significance (p<0.05) was determined with pairwise ANOVA. (A) Kv2.1 areas are significantly reduced (indicated by single asterisk) at 3 days, 8 days, 2 weeks, and 4 weeks following injury. There are small, yet significant (indicated by double asterisk), decreases on the injury spared contralateral side of the rat spinal cord at 2 weeks and 4 weeks post injury. (B) The number of Kv2.1-IR macroclusters decrease significantly (indicated with asterisk) 3 days following tibial nerve crush and are significantly reduced at all following time points.
Table 2.
Kv2.1-IR macrocluster means and sample sizes for tibial nerve ligate injury
| TIBIAL NERVE LIGATION INJURY
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Sham | 20 min | 3 Day | 8 Day | 2 Week | 4 Week | 6 Month | ||
| Injury (ipsilateral) | Kv2.1-IR Mean Area (μm2) | 8.19 ± 5.50 | 7.77 ± 4.13 | 6.51 ± 4.59 | 6.18 ± 4.06 | 5.66 ± 3.95 | 5.43 ± 4.18 | 8.07 ± 5.11 |
| Clusters, Cells, Animals sampled | n=521, 64, 2 | n=624, 74, 2 | n=453, 50, 2 | n=319, 33, 2 | n=617, 83, 2 | n=466, 68, 3 | n=522, 73, 3 | |
|
| ||||||||
| Contralateral Controls | Kv2.1-IR Mean Area (μm2) | 8.18 ± 4.84 | 8.00 ± 4.75 | 7.75 ± 4.65 | 8.10 ± 4.89 | 7.23 ± 4.37 | 6.71 ± 3.98 | 8.22 ± 4.86 |
| Clusters, Cells, Animals sampled | n=589, 58, 2 | n=537, 54, 2 | n=523, 61, 2 | n=301, 30, 2 | n=766, 72, 2 | n=482, 65, 3 | n=682, 74, 3 | |
±SD
Using the same 3-D reconstruction techniques preciously described for the tibial nerve crush injury, we calculated the Kv2.1 macrocluster areas on proximal dendrites of MG/LG αMNs following tibial nerve ligation. For these analyses we selected 4 weeks post ligation due to the maximum effect of ligation on somatic αMN Kv2.1-IR macroclusters. On proximal dendrites, there is a significant 23% reduction in soma Kv2.1-IR macrocluster areas (sham surgery control 13.42μm2 ± 6.054 SD, n=299 clusters, 20 αMNs, 2 rats; injured 10.347μm2 ± 6.388 SD, n=125 clusters, 10 αMNs, 2 rats). No significant differences of Kv2.1 macrocluster areas were ever found within the population of the control MNs that were 3-D reconstructed.
2.5 Bilateral Kv2.1-IR declustering occurs following PNI
As mentioned in section 2.2, we found that Kv2.1-IR redistribution occurs bilaterally following PNI. Kv2.1-IR macroclusters on the αMN cell body also decrease on the uninjured contralateral side following PNI (figure 6A). The contralateral effect in the tibial nerve crush results in a significant maximum 22% reduction at 3 days post injury compared to sham surgery controls, and there is complete recovery of cluster size by 8 days. However, this recovery appears to be transient, because a second significant reduction (16%) occurs at 28 days. While this biphasic contralateral effect is present and significant, there was, however, a greater decrease in Kv2.1-IR macrocluster area on the corresponding ipsilateral side at all time points. Like the tibial nerve crush, there is also a small yet significant contralateral effect (18% maximum reduction in Kv2.1-IR macrocluster area) on the contralateral side 28 days following ligation (figure 7A). While not significant, there is a trend resembling the biphasic response observed in the tibial nerve crush injury, with an initial transient reduction (5%) in Kv2.1 cluster size 3 days after the ligation injury.
2.6 No Changes in Motoneuron Cell Diameter following Peripheral Nerve Injury
Axotomy can cause morphological changes in neurons such as cell shrinkage, dissolution of nissl bodies, and acentric localization of the nucleus, all referred to as chromatolysis. Axotomy, and associated chromatolysis, was used to identify central motor cell columns in the cat before the development of retrograde tracers (Romanes, 1951). However, unlike our PNIs, repeated acute injury to the peripheral nerves was necessary for generating the degree of chromatolysis necessary for identifying the motor pools. Here, we sought to determine if the changes in Kv2.1-IR cluster areas are a result of changes in MN sizes, first by determining if any correlations exist between Kv2.1 cluster sizes and MN diameter, and secondly to determine if there are detectable cell diameter changes following axotomy. In sham surgery control MNs, there was no correlation between Kv2.1-IR cluster sizes and the mean cell diameter of the MG/LG MNs (n=520 clusters, 80 cells, 2 rats) (supplemental figure 1A). Furthermore, there were no significant changes in the mean cell diameter 8 days after the tibial nerve crush and 4 weeks after tibial nerve ligation when compared to contralateral controls (supplemental figure 1B). It is therefore unlikely the observed changes in Kv2.1-IR macrocluster areas following axotomy are a result of changes in MN size.
3. Discussion
Members of the voltage-gated potassium channel family are essential for numerous neuronal functions, and have been implicated in a wide variety of responses ranging from neuroprotection to apoptosis. Kv2.1 channels underlie delayed rectifyer currents in many central neurons, including MNs where they are expressed in high-density clusters (Blaine and Ribera, 2001; Muennich and Fyffe, 2004). The striking membrane organization of Kv2.1 channels makes them a valuable model for study of channel regulation under conditions of altered activity including injury. In hippocampal cells, Kv2.1 undergoes dynamic membrane declustering following increased activity both in vivo (ischemia, hypoxia and seizure) and in vitro (glutamate application) (Misonou et al., 2004; Misonou et al., 2005). Furthermore, an increase in Kv2.1 clustering has been observed in some model systems, triggered by physiological stimuli such as repetitive firing, or removal of pathological stimuli (Aras et al., 2009; Cerda and Trimmer, 2011; Misonou et al., 2004; Misonou et al., 2006; Mulholland et al., 2008; Zhang et al., 2008). Here we show, for the first time, glutamate induced Kv2.1 cluster dispersal in lumbar spinal MNs, in vitro. This in vitro cluster dispersal occurs on a time scale (<10 minutes) similar to that previously reported for hippocampal and HEK293 cells. Moreover, we demonstrate in vivo axotomy as a novel stimulus causing significant rapid declustering of Kv2.1, as early as 20 minutes, representing the earliest accessible time point following surgical axotomy. Similar to ischemia, hypoxia, and glutamate application, axotomy induced Kv2.1 declustering is likely a product of altered activity.
Peripheral nerve injury is complex because it results in axotomy of both MNs and sensory afferents known to modulate MN function; however, given its clinical relevance, it remains a valuable model from which to extract new information on the capacity of central neurons to respond to and recover from injury. Although the results of axotomy and reinnervation studies depend on a large number of factors (e.g. lesion location, age, degree of reinnervation, etc.), several consistent outcomes have been reported by a number of laboratories (Eccles et al., 1958; Kuno and Llinas, 1970a; Kuno and Llinas, 1970b; Kuno et al., 1974a; Kuno et al., 1974b; Kuno, 1976). Within several days following PNI there are many changes in MN intrinsic properties, such as reduction in rheobase, modulation of AHP, increase in input resistance, and slowing of axonal conduction velocity proximal to the injury (Bichler et al., 2007; Gustafsson and Pinter, 1984; Kuno et al., 1974b; Nakanishi et al., 2005). Interestingly, most membrane properties appear to return to pre-axotomy states upon reinnervation of the peripheral targets (Bichler et al., 2007; Kuno et al., 1974a; Nakanishi et al., 2005). The altered membrane properties following axotomy likely reflect the cumulative alterations to multiple channel types, including Kv2.1, that contribute to a coordinated cellular response.
In this study, we sought to determine what happens to Kv2.1-IR distribution in the absence or presence of reinnervation by the injured neurons, using a tibial nerve crush injury that permits reinnervation of the peripheral target (Fournier and Strittmatter, 2002; Nguyen et al., 2002) and a ligation injury that prevents reinnervation. As a consequence of initial axon injury in each model we expected the time course of manifestation of injury responses to be similar at least until the time of reinnervation that occurs about 4–6 weeks after crush injury (Alvarez et al., 2011; Bichler et al., 2007). However, long before reinnervation occurs, we observed injury type-specific changes. Most surprisingly, the magnitude of effects on Kv2.1 immunoreactivity was apparently greater following nerve crush than following nerve cut and ligation. Paradoxically, after reinnervation occurs, normal Kv2.1 cluster sizes are not fully restored; however, cluster sizes do recover when reinnervation is prevented.
Other investigators have reported injury-type specific changes (Bullinger et al., 2011; He et al., 1996; Nielsch et al., 1987; Prather et al., 2011; Waite and Cragg, 1982). However, even when muscle reinnervation occurs, the initial form of injury (crush or cut) provokes different outcomes. In the cat, when muscles reinnervated by crushed nerves are stretched, MNs yield a more forceful reflex contraction despite the decrease in monosynaptic stretch evoked EPSPs (Prather et al., 2011). Contrasting results were observed in both rat and cat in which nerves were cut and the stretch reflex is absent following reinnervation (Alvarez et al., 2010; Bullinger et al., 2011; Cope and Clark, 1993). The range of pathophysiological and structural changes now observed following crush or cut injuries suggests that additional investigation into the differences between cut and crush nerve injury are warranted.
One significant structural change that occurs after peripheral nerve injury is the loss of synaptic inputs on MNs (synaptic stripping) (Alvarez et al., 2011; Blinzinger and Kreutzberg, 1968; Brannstrom and Kellerth, 1998; Brannstrom and Kellerth, 1999; Gonzalez-Forero et al., 2004; Hughes et al., 2004; Sumner, 1975; Tiraihi and Rezaie, 2004). Moreover, there is a permanent loss of vesicular glutamate transporter isoform 1 (VGLUT1) positive primary sensory afferent synapses following nerve cut and a transient reduction of synaptic coverage from cholinergic C-boutons MNs that is partially restored upon peripheral reinnervation (Alvarez et al., 2010; Alvarez et al., 2011; Bullinger et al., 2011; Sumner, 1975). Because ‘macroclusters’ of Kv2.1-IR are selectively apposed to C-boutons (Muennich and Fyffe, 2004), it is interesting that the time course of effects on these clusters mirrors the VAChT-IR stripping in nerve cut injuries. The role that synaptic stripping has in the regulation and dysregulation of Kv2.1 clusters is currently unknown, and any speculation here is complicated by the paucity of data on synaptic stripping following nerve crush injuries.
3.1 Kv2.1 and Homeostatic Plasticity
Kv2.1 has increasingly been described as a target for mechanisms of homeostatic plasticity (Cudmore and Turrigiano, 2004; Kihira et al., 2010; Misonou et al., 2004; Misonou et al., 2008; Nataraj et al., 2010; Surmeier and Foehring, 2004). Neuronal homeostatic plasticity reflects the ability of a neuron to respond to changes in activity and maintain an optimal level of output. Multiple homeostatic mechanisms have been described, invoking both pre- and post-synaptic changes (Burrone and Murthy, 2003; Murphy, 2003; Schulz et al., 2006; Surmeier and Foehring, 2004; Walmsley et al., 2006). These include ‘synaptic scaling’, reducing synaptic strength at presynaptic inputs in response to increased postsynaptic activity (Turrigiano et al., 1998). Regulation of postsynaptic machinery through either translational/transcriptional regulation or post-translational modifications may play a major role in homeostatic responses. Activation of immediate early genes (Leah et al., 1991; Sheng and Greenberg, 1990), which are known to regulate activity dependent expression of certain proteins including potassium channels (Leao et al., 2010), can occur within an hour of PNI (New et al., 1989). However, the slow time course for transcription, translation and trafficking to the membrane would not allow de novo expression or downregulation of ion channels to compensate for immediate changes in activity. Trafficking through the Golgi apparatus alone in cell lines can take up to 20 minutes (Patterson et al., 2008). Here, we saw a significant decrease in the Kv2.1 cluster area within 20 minutes following crush injury. This supports that idea that regulation of Kv2.1 in MNs is likely to be a consequence of post-translational modification, such as changes in redox state or phosphorylation. In hippocampal pyramidal neurons and cell cultures, the lateral translocation of Kv2.1 is homeostatically regulated through calcineurin dependent dephosphorylation (Misonou et al., 2004; Misonou et al., 2005; Murakoshi et al., 1997; Park et al., 2006). This effect is mimicked by injecting phosphatases into the cell and is blocked with calcineurin inhibitors (Misonou et al., 2004; Misonou et al., 2005; Mohapatra et al., 2009). Given the speed at which declustering occurs in the spinal cord after injury (within 20 minutes), regulation through phosphorylation mechanisms appears to be a likely candidate. Regulating MN excitability and activity through modification of post-synaptic voltage-gated ion channels, such as Kv2.1, may be a more energetically favorable mechanism because regulating the firing rate can be isolated to just the cell body and proximal dendrites of the neuron rather than requiring changes in thousands of input synapses (Kihira et al., 2010). The signal that triggers the response is not yet defined, but it is possible that the instantaneous ‘injury discharge’ in sensory and motor axons can change activity sufficiently to evoke the rapid post-translational modifications of channel state. It is also possible that the magnitude and duration of injury discharges following different types of insult may differ and be sufficient to trigger different and long-lasting regulatory changes.
Placing axotomy-induced responses of MNs in the context of homeostasis is difficult because the exact in vivo activity level of MNs, and their state dependent modifications, following PNI and the instantaneous but relatively brief injury discharge is unknown. Moreover, multiple factors, including pre- and postsynaptic changes are likely to coordinate in the injury response to increase or decrease activity. Phosphatases regulate Kv2.1 channels in two critical ways, simultaneously altering both localization and channel activity. Modeling suggests that the disruption of Kv2.1 clusters, with all other factors constant, results in decreased excitability (Mohapatra et al., 2009; Surmeier and Foehring, 2004). However following axotomy, MNs become more excitable. One possible explanation to this paradox is that homeostatic mechanisms may not be strong enough to overcome pathophysiologically induced or sustained hyperexcitability (Misonou et al., 2004; Misonou et al., 2005; Misonou et al., 2008; O’Leary et al., 2010). It has been speculated that some ‘homeostatic’ mechanisms may instead be providing neuroprotection against neuronal damage and death (Misonou et al., 2005; Surmeier and Foehring, 2004). For example, in the brain Kv2.1 is clustered near astroglial processes expressing high levels of glutamate transporter (Misonou et al., 2008). During ischemia, the astrocytes become dysfunctional resulting in an accumulation of extracellular glutamate. The positioning of the Kv2.1 clusters permits a rapid response to the high glutamate levels thus potentially protecting the neuron from a potentially damaging excitotoxic effect (Misonou et al., 2008; Misonou, 2010). In the rat lumbar spinal cord, Kv2.1-IR clusters are selectively localized at specific synapses and at glial sites (Muennich and Fyffe, 2004) and are thus also appropriately positioned for a quick neuroprotective response to excitatory insult.
There are still unanswered questions about what happens to the phosphorylation state of the channels after the declustering of Kv2.1. The restoration of cluster areas could be explained by alterations in the ratio of kinase to phosphatase activity or because of protein turnover. However, protein turnover is not likely to be the sole reason for reclustering. In quantum dot assays in HEK cells, new channels are regularly trafficked to the Kv2.1 clusters (O’Connell et al., 2006). This trafficking is frequent enough that within twelve minutes there is significant GFP recovery of a photobleached Kv2.1 cluster (O’Connell et al., 2006). Our data here shows that there is continued declustering of Kv2.1 clusters for at least 8 days after the injury suggesting that some other mechanism is regulating and maintaining the declustered state. Furthermore, constitutive insertion of de novo proteins to the membrane may also explain why we are never completely devoid of Kv2.1 clusters.
Kv2.1 also has a role in mediating the K+ loss in apoptosis. Increased phosphorylation of S800 in the C-terminal tail of Kv2.1 by p38 Map Kinase leads to enhanced K+ currents through SNARE mediated membrane insertion in cultured cells (Redman et al., 2007; Yao et al., 2009). This site is distinct from the identified calcineurin dependent phosphorylation sites that play a role in homeostatic plasticity (Kihira et al., 2010; Misonou et al., 2004; Park et al., 2006). Here we see evidence that Kv2.1 reversibly declusters, suggesting that the regulation of Kv2.1 after PNI is dynamic and is likely a protective, or homeostatic, response rather than an apoptotic response. This suggests that different pathological conditions can give rise to differential Kv2.1 expression and/or modification mediating multiple physiological effects.
3.2 Contralateral Effect
There are a range of well-documented pathophysiological changes in the spinal cord following unilateral peripheral nerve lesions, including effects on the contralateral side (Koltzenburg et al., 1999). Generally, as is the case with our observations here, the contralateral effects are qualitatively similar to those occurring on the ipsilateral side, but with smaller magnitudes and briefer time courses. Our comparisons of effects in control animals undergoing sham surgery as well as naïve controls, at different time points post surgery revealed that sham surgery does not trigger any changes. It is also unlikely that the contralateral effect is compensatory from the animal using the uninjured leg more, because as in injured motoneurons the decrease in cluster size is already measurable within twenty minutes, before any post-surgical locomotor activity could occur. Thus the contralateral effects must be a product of the peripheral nerve injury itself.
Evidence exists to support both multiple humoral and neuronal mechanisms contribute to a wide range of contralateral effects in rats. It is well established that inflammatory responses contribute to a variety of effects in the central nervous system within the first three days following trauma (Gaudet et al., 2011). It is unknown whether the effect on Kv2.1 expression and localization in contralateral, uninjured, motoneurons results from a systemic inflammatory response or from some other neuronal mechanism (Jancalek et al., 2010; Jancalek et al., 2011; Koltzenburg et al., 1999; Lu and Richardson, 1993; Pachter and Eberstein, 1991). The specific mechanisms underlying the biphasic contralateral response shown here are also unknown. Interestingly, studies of other biophysical properties also detect biphasic responses with peaks at 3 and 28 day time periods following injury (Mizisin and Weerasuriya, 2011; Weerasuriya and Hockman, 1992). Nonetheless, our results further emphasize that caution should be used when using internal bilateral controls to interpret changes in spinal systems.
3.3 Conclusions
Here we show that the dynamic reorganization of Kv2.1 previously shown in other neuronal types also occurs in rat lumbar spinal MNs following increased activity. Moreover, we demonstrate and quantify novel effects on the membrane distribution of Kv2.1 ion channels on rat lumbar spinal MNs following PNI. The dynamic changes in membrane channel localization include a progressive decline in cluster size immediately after axon injury followed by gradual recovery of cluster size over the next several months. Upon motor axon reinnervation of muscle tissue, Kv2.1 clusters do not completely restore to pre-injury sizes, however in the absence of reinnervation, Kv2.1 clusters fully restore. Furthermore, unilateral peripheral nerve injury evokes parallel, but smaller, effects contralaterally in uninjured motoneurons. Altogether these results suggest that in MNs Kv2.1 declustering is likely a homeostatic response to altered activity and its regulation following axon injury is largely independent of muscle reinnervation.
4. Experimental Procedures
All animal procedures were performed according to National Institutes of Health (NIH) guidelines and reviewed by the local Laboratory Animal Use Committee at Wright State University.
4.1 Peripheral Nerve Injuries and Retrograde Labels
Adult female Sprague Dawley rats (250–350 grams) underwent a series of two survival surgeries. In the first surgery the medial and lateral gastrocnemius muscles were injected with a fluorescent tracer to retrogradely label MNs for post-hoc identification in the spinal cord (figure 1). Rats were deeply anesthetized with 5% isoflurane and maintained with 2–3% isoflurane. An Incision was made using sterile technique over the left hindlimb and through biceps femoris muscle to reveal the triceps surae. A total of 40–50 μL of either hydroxystilbamidine (Biotium, Inc., Hayward, CA, USA; 4%), FastBlue (Polysciences, Inc., Warrington, PA, USA; 2%) or Cholera Toxin Subunit B, CTB, (Invitrogen, Carlsbad, CA, USA; 0.5%) was injected into the medial and lateral gastrocnemius muscle in a series of multiple small injections throughout the two muscles. The wound was irrigated and closed in layers. Three days later, the incision was reopened and the tibial nerve was dissected away from surrounding tissues and exposed. The tibial nerve was either crushed, by applying pressure for 10 sec with fine jeweler’s forceps, or ligated, by using microscissors and silk suture to ligate nerve and prevent reinnervation. The wound was irrigated and closed. Surgery was not performed on the right hindlimb to provide uninjured internal controls. Animals received 0.1 ml of 0.3 mg/ml buprenorphine every 12 hours for post-operative pain medication for 48 hours and were monitored closely by professional staff. The sham surgery control animals underwent the same surgical procedures described above except the tibial nerve was only exposed rather than injured. Nerve injured and sham control animals were analyzed at similar post-operative time points
4.2 Immunohistochemistry
All animals were anesthetized with pentobarbital (150 mg/kg, i.p) and transcardially perfused with 4% paraformaldehyde in 0.1M phosphate buffer at pH 7.3. The spinal cords were removed, post fixed for 2 hours and cryoprotected in 15% sucrose in 0.1M phosphate buffer. 50 μm transverse sections were obtained from the L4 and L5 spinal cord segments on a cryostat and immunostained free floating. Kv2.1 immunocytochemistry was performed using mouse anti-Kv2.1 clone D4/11 (catalog number 75-047) at 1:1000 in PBS with 0.1% Triton X pH 7.3 that was developed and/or obtained from the UC Davis/NINDS/NIMH Neuromab facility, supported by NIH grant U24NS0506060 and maintained by the Department of Pharmacology, School of Medicine, University of California, Davis, CA. Immunoreactivity was detected with species specific secondary antibodies conjugated to Cy3 (Jackson Immuno, West Grove, PA, USA). Nissl immunocytochemistry was performed using fluorescein-conjugated nissl (1:100, Molecular Probes, Carlsbad, CA, USA).
4.3 Spinal Cord Slice Preparations
Young Sprague Dawley Rats, P14, (Charles River, Wilmington, MA, USA) were anesthetized with 65mg/ml pentobarbital and decapitated. The spinal cords were immediately dissected in <4°C sucrose modified artificial CSF (26mM NaHCO3, 10mM Glucose, 3mM KCl, 1.25mM NaPO4, 2mM MgCl2, and 218mM Sucrose) with bubbling 95% O2 and 5% CO2. Transverse sections, 300 μm thick, of the L4 – L5 region of the spinal cord were cut on Vibratome Series 1000 Plus (The Vibratome Co., St. Louis, MO, USA). Slices were briefly incubated in 30% PEG (Carp et al., 2008) and transferred to oxygenated aCSF (130 mM NaCl, 26mM NaHCO3, 10mM Glucose, 3mM KCl, 1.25mM NaPO4, 2mM MgCl2, 2mM CaCl2 at 300–310 mOsm) and incubated 1 hour at 37°C to stabilize. Sections were incubated in either 10 μm L-Glutamate (Sigma, St. Louis, MO) diluted in aCSF or aCSF alone for 10 minutes, longer times in glutamate increased cell death. Addition of glutamate to aCSF had minimal effect on osmotic pressure of the solution. Immediately following incubations, the slices were immersion fixed in 4% paraformaldehyde overnight. Tissue slices were sectioned further (50 μm) on cryostat and immunohistochemistry was performed as previously described (refer to immunohistochemistry methods).
4.4 Confocal Microscopy and Quantitative Analysis
Immunolabeled lumbar αMN images were obtained on a Fluoview FX Olympus (Center Valley, PA, USA) confocal system for tibial nerve crush studies and Fluoview 1000 Olympus (Center Valley, PA, USA) confocal microscope for the tibial nerve ligation studies with a 60x oil immersion objective at 0.5 μm steps at 1.0–2.5 digital zoom (N.A 1.35). For every ipsilateral injured αMN imaged and analyzed, a contralateral injury spared αMN was also selected as an internal bilateral control. Uninjured MNs selected for internal control analysis were of approximate size and position within the motor pools on the contralateral side within each tissue slice. In 4 animals, the uninjured αMNs were retrogradely labeled with different color conjugated fluorophores to differentiate from the injured αMNs. No significant differences were found between these retrogradely labeled uninjured αMNs from the αMNs selected based on size and location (data not shown). Sham surgery control animals that contained the retrograde tracers were used to verify and quantify the ipsilateral and contralateral effects observed following PNI. Image stacks were quantitatively analyzed for Kv2.1-IR cluster areas as previously described (Muennich and Fyffe, 2004) in Image Pro Software (Media Cybernetics, Silver Springs, MD, USA). Cell body Kv2.1-IR macroclusters (diameter >1.0μm), were measured in en face single optical sections on MNs that innervate the medial and lateral gastrocnemius as revealed by retrograde labels (figure 1). All Kv2.1-IR microcluster (diameter <1.0μm) analyses were performed by selecting regions of interest in single optical confocal sections that did not contain macroclusters. Mean cell body diameters were calculated using averaging applications in Image Pro Software based on plasma membrane labeling on single optical sections through the center of the neuron containing the nucleolus. For all analyses, injured MNs were compared to both contralateral internal controls as well as sham surgery control animals. Significance was set at p<0.05 using Pairwise ANOVA and Pairwise T-Test (SigmaStat; Systat Software, Port Richmond, CA, USA).
4.5 Dendritic Analysis and Reconstructions
Our conventional methods for measuring soma Kv2.1 cluster areas were not suitable for measuring cylindrical dendrites due to limited availability of en face dendritic membrane through which accurate 2-D measurements can be obtained. Dendritic Kv2.1 macrocluster areas and numbers were calculated by 3D reconstructions of the confocal image stacks through Imaris Software (Bitplane Scientific Software, Zurich, Switzerland). Prior to being reconstructed, the confocal images where deconvolved on Huygens Essential Software (Scientific Volume Imaging, Hilversum, Netherlands) to reduce noise and z-plane distortion. The deconvolved confocal image stacks were imported into Imaris software where the image is displayed as a 3-dimentional isometric view. Surface rendering and thresholding was used to create a 3-D solid surface of the CTB filled motoneuron and Kv2.1-IR clusters with careful attention to accurately match anatomic dimensions. A masking feature in Imaris software allowed only the Kv2.1 clusters contacting the CTB- filled motoneuron to be selected for analysis. The proximal dendrites and associated Kv2.1-IR clusters were digitally dissected from the soma for analysis. Kv2.1-IR cluster areas from proximal dendrites were exported from the software for statistical analysis. The reconstructed MNs were from 50 μm thick tissue slices, an appropriate thickness for consistently analyzing dendritic lengths up to 150 μm traveling through 3-D space. Kv2.1-IR macrocluster areas located on control MN somas obtained through 3-D reconstructions were not found to be significantly different from the areas measured using our conventional techniques previously described (data not shown).
4.5 Figure composition
Microscope images were prepared by adjusting contrast and brightness in Image Pro Plus Software (Media Cybernetics, Bethesda, MD, USA) and always preserved all the information content of the images. Figures were composed using CorelDraw (v. 12.0). Graphs were composed in SigmaPlot (version 9.0, Systat Software, SPSS Inc, Chicago, IL, USA). Data presented in figures is mean ± SEM. Standard Deviations are listed in tables 1 and 2. Some images were sharpened using a “high gauss” filter in image pro. Quantification was always carried out in original unprocessed images.
Supplementary Material
Highlights.
Kv2.1 ion channel distribution is altered by activity in spinal motoneurons
Peripheral nerve injury causes dynamic redistribution of motoneuron Kv2.1 channels
Unilateral nerve injury impacts uninjured contralateral motoneurons
Kv2.1 channel restoration is independent of proper axon reinnervation
Acknowledgments
This work was supported in part by funds from the NIH grant number 5P01 NS057228.
We thank Mrs. Lori Goss for her invaluable time with animal surgeries and handling, and Raychel Santo for her time and contribution with the cut model analysis and Kv2.1 cluster size correlations.
We thank Dr. David Ladle for use of his Imarus Software that enabled us to perform the dendritic analysis in this body of work.
We thank Dr. Mark Rich for his invaluable time in reviewing this article and for his insightful comments.
We are grateful for the generous contributions in time and resources from Drs. Timothy C. Cope, Francisco Alvarez, and Kathrin Engisch.
We thank Bev Grunden of Wright State University’s Statistical Consulting Center for statistical guidance.
Abbreviations
- MN
Motoneuron
- PNI
Peripheral Nerve Injury
- IR
Immunoreactivity
- CTB
Cholera Toxin Subunit B
- MG
Medial Gastrocnemius Muscle
- LG
Lateral Gastrocnemius Muscle
Footnotes
Author Contributions
REWF conceived experiments and interpreted data. REWF, ASD, and SHR wrote the manuscript. SHR directed all experiments. SHR, KMD, MWG, and RCT performed immunohistochemistry, quantitative microscopic analysis and interpreted data. All experiments and analysis were performed in the laboratory of REWF. All authors critically reviewed the manuscript for accuracy and important intellectual content.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding organizations acknowledged above.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Shannon H. Romer, Email: Shannon.romer@wright.edu.
Kathleen M. Dominguez, Email: Kmdominguez@juno.com.
Marc W. Gelpi, Email: gelpi.2@wright.edu.
Adam S. Deardorff, Email: deardorff.2@wright.edu.
Robert C. Tracy, Email: robert.tracy@osumc.edu.
References
- Alvarez FJ, Bullinger KL, Titus HE, Nardelli P, Cope TC. Permanent reorganization of Ia afferent synapses on motoneurons after peripheral nerve injuries. Ann N Y Acad Sci. 2010;1198:231–41. doi: 10.1111/j.1749-6632.2010.05459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Titus-Mitchell HE, Bullinger KL, Kraszpulski M, Nardelli P, Cope TC. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. I. Loss of VGLUT1/IA synapses on motoneurons. J Neurophysiol. 2011;106:2450–70. doi: 10.1152/jn.01095.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras MA, Saadi RA, Aizenman E. Zn2+ regulates Kv2.1 voltage-dependent gating and localization following ischemia. Eur J Neurosci. 2009;30:2250–7. doi: 10.1111/j.1460-9568.2009.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton DC, Monaghan AS, Hosseini R, Bahia PK, Haylett DG, Moss GW. Small conductance Ca2+-activated K+ channels formed by the expression of rat SK1 and SK2 genes in HEK 293 cells. J Physiol. 2003;553:13–9. doi: 10.1113/jphysiol.2003.054551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichler EK, Carrasco DI, Rich MM, Cope TC, Pinter MJ. Rat motoneuron properties recover following reinnervation in the absence of muscle activity and evoked acetylcholine release. J Physiol. 2007;585:47–56. doi: 10.1113/jphysiol.2007.135541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaine JT, Ribera AB. Kv2 channels form delayed-rectifier potassium channels in situ. J Neurosci. 2001;21:1473–80. doi: 10.1523/JNEUROSCI.21-05-01473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat. 1968;85:145–57. doi: 10.1007/BF00325030. [DOI] [PubMed] [Google Scholar]
- Brannstrom T, Kellerth JO. Changes in synaptology of adult cat spinal alpha-motoneurons after axotomy. Exp Brain Res. 1998;118:1–13. doi: 10.1007/s002210050249. [DOI] [PubMed] [Google Scholar]
- Brannstrom T, Kellerth JO. Recovery of synapses in axotomized adult cat spinal motoneurons after reinnervation into muscle. Exp Brain Res. 1999;125:19–27. doi: 10.1007/s002210050653. [DOI] [PubMed] [Google Scholar]
- Bui TV, Ter-Mikaelian M, Bedrossian D, Rose PK. Computational estimation of the distribution of L-type Ca(2+) channels in motoneurons based on variable threshold of activation of persistent inward currents. J Neurophysiol. 2006;95:225–41. doi: 10.1152/jn.00646.2005. [DOI] [PubMed] [Google Scholar]
- Bullinger KL, Nardelli P, Pinter MJ, Alvarez FJ, Cope TC. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. II. Loss of functional connectivity with motoneurons. J Neurophysiol. 2011;106:2471–85. doi: 10.1152/jn.01097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–7. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Carlin KP, Jones KE, Jiang Z, Jordan LM, Brownstone RM. Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. Eur J Neurosci. 2000;12:1635–46. doi: 10.1046/j.1460-9568.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- Carp JS, Tennissen AM, Mongeluzi DL, Dudek CJ, Chen XY, Wolpaw JR. An in vitro protocol for recording from spinal motoneurons of adult rats. J Neurophysiol. 2008;100:474–81. doi: 10.1152/jn.90422.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda O, Trimmer JS. Analysis and functional implications of phosphorylation of neuronal voltage-gated potassium channels. Neurosci Lett. 2010;486:60–7. doi: 10.1016/j.neulet.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda O, Trimmer JS. Activity-dependent phosphorylation of neuronal Kv2.1 potassium channels by CDK5. J Biol Chem. 2011;286:28738–48. doi: 10.1074/jbc.M111.251942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope TC, Clark BD. Motor-unit recruitment in self-reinnervated muscle. J Neurophysiol. 1993;70:1787–96. doi: 10.1152/jn.1993.70.5.1787. [DOI] [PubMed] [Google Scholar]
- Cope TC, Bonasera SJ, Nichols TR. Reinnervated muscles fail to produce stretch reflexes. J Neurophysiol. 1994;71:817–20. doi: 10.1152/jn.1994.71.2.817. [DOI] [PubMed] [Google Scholar]
- Cudmore RH, Turrigiano GG. Long-term potentiation of intrinsic excitability in LV visual cortical neurons. J Neurophysiol. 2004;92:341–8. doi: 10.1152/jn.01059.2003. [DOI] [PubMed] [Google Scholar]
- Deardorff AS, Romer SH, Deng Z, Bullinger KL, Nardelli P, Cope TC, Fyffe RE. Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar -motoneurons. J Physiol. 2013;591:875–897. doi: 10.1113/jphysiol.2012.240879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Fyffe RE. Expression of P2X7 receptor immunoreactivity in distinct subsets of synaptic terminals in the ventral horn of rat lumbar spinal cord. Brain Res. 2004;1020:53–61. doi: 10.1016/j.brainres.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Duflocq A, Chareyre F, Giovannini M, Couraud F, Davenne M. Characterization of the axon initial segment (AIS) of motor neurons and identification of a para-AIS and a juxtapara-AIS, organized by protein 4.1B. BMC Biol. 2011;9:66. doi: 10.1186/1741-7007-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Libet B, Young RR. The behaviour of chromatolysed motoneurones studied by intracellular recording. J Physiol. 1958;143:11–40. doi: 10.1113/jphysiol.1958.sp006041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbasiouny SM, Bennett DJ, Mushahwar VK. Simulation of dendritic CaV1.3 channels in cat lumbar motoneurons: spatial distribution. J Neurophysiol. 2005;94:3961–74. doi: 10.1152/jn.00391.2005. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Sypert GW, Munson JB. Properties of self-reinnervated motor units of medial gastrocnemius of cat. II. Axotomized motoneurons and time course of recovery. J Neurophysiol. 1986a;55:947–65. doi: 10.1152/jn.1986.55.5.947. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Sypert GW, Munson JB. Properties of self-reinnervated motor units of medial gastrocnemius of cat. I. Long-term reinnervation. J Neurophysiol. 1986b;55:931–46. doi: 10.1152/jn.1986.55.5.931. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Strittmatter SM. Regenerating nerves follow the road more traveled. Nat Neurosci. 2002;5:821–2. doi: 10.1038/nn0902-821. [DOI] [PubMed] [Google Scholar]
- Fox PD, Loftus RJ, Tamkun MM. Regulation of Kv2.1 K(+) conductance by cell surface channel density. J Neurosci. 2013;33:1259–70. doi: 10.1523/JNEUROSCI.3008-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG, Ramer MS. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 2011;8:110. doi: 10.1186/1742-2094-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Forero D, Portillo F, Sunico CR, Moreno-Lopez B. Nerve injury reduces responses of hypoglossal motoneurones to baseline and chemoreceptor-modulated inspiratory drive in the adult rat. J Physiol. 2004;557:991–1011. doi: 10.1113/jphysiol.2003.059972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Pinter MJ. Effects of axotomy on the distribution of passive electrical properties of cat motoneurones. J Physiol. 1984;356:433–42. doi: 10.1113/jphysiol.1984.sp015474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haftel VK, Bichler EK, Wang QB, Prather JF, Pinter MJ, Cope TC. Central suppression of regenerated proprioceptive afferents. J Neurosci. 2005;25:4733–42. doi: 10.1523/JNEUROSCI.4895-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He BP, Tay SS, Leong SK. Macrophage and microglial cell response after common peroneal nerve cut and crush in C57BL/6J mice. Neurodegeneration. 1996;5:73–80. doi: 10.1006/neur.1996.0010. [DOI] [PubMed] [Google Scholar]
- Hughes DI, Polgar E, Shehab SA, Todd AJ. Peripheral axotomy induces depletion of the vesicular glutamate transporter VGLUT1 in central terminals of myelinated afferent fibres in the rat spinal cord. Brain Res. 2004;1017:69–76. doi: 10.1016/j.brainres.2004.05.054. [DOI] [PubMed] [Google Scholar]
- Jancalek R, Dubovy P, Svizenska I, Klusakova I. Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. J Neuroinflammation. 2010;7:11. doi: 10.1186/1742-2094-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancalek R, Svizenska I, Klusakova I, Dubovy P. Bilateral changes of IL-10 protein in lumbar and cervical dorsal root ganglia following proximal and distal chronic constriction injury of peripheral nerve. Neurosci Lett. 2011;501:86–91. doi: 10.1016/j.neulet.2011.06.052. [DOI] [PubMed] [Google Scholar]
- Kihira Y, Hermanstyne TO, Misonou H. Formation of heteromeric Kv2 channels in mammalian brain neurons. J Biol Chem. 2010;285:15048–55. doi: 10.1074/jbc.M109.074260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–7. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- Kuno M, Llinas R. Alterations of synaptic action in chromatolysed motoneurones of the cat. J Physiol. 1970a;210:823–38. doi: 10.1113/jphysiol.1970.sp009244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Llinas R. Enhancement of synaptic transmission by dendritic potentials in chromatolysed motoneurones of the cat. J Physiol. 1970b;210:807–21. doi: 10.1113/jphysiol.1970.sp009243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Miyata Y, Munoz-Martinez EJ. Properties of fast and slow alpha motoneurones following motor reinnervation. J Physiol. 1974a;242:273–88. doi: 10.1113/jphysiol.1974.sp010706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M, Miyata Y, Munoz-Martinez EJ. Differential reaction of fast and slow alpha-motoneurones to axotomy. J Physiol. 1974b;240:725–39. doi: 10.1113/jphysiol.1974.sp010631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno M. Responses of spinal motor neurons to section and restoration of peripheral motor connections. Cold Spring Harb Symp Quant Biol. 1976;40:457–63. doi: 10.1101/sqb.1976.040.01.043. [DOI] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–62. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Leah JD, Herdegen T, Bravo R. Selective expression of Jun proteins following axotomy and axonal transport block in peripheral nerves in the rat: evidence for a role in the regeneration process. Brain Res. 1991;566:198–207. doi: 10.1016/0006-8993(91)91699-2. [DOI] [PubMed] [Google Scholar]
- Leao KE, Leao RN, Deardorff AS, Garrett A, Fyffe R, Walmsley B. Sound stimulation modulates high-threshold K(+) currents in mouse auditory brainstem neurons. Eur J Neurosci. 2010;32:1658–67. doi: 10.1111/j.1460-9568.2010.07437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Richardson PM. Responses of macrophages in rat dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1993;22:334–41. doi: 10.1007/BF01195557. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–90. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–8. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184–93. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Menegola M, Mohapatra DP, Guy LK, Park KS, Trimmer JS. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J Neurosci. 2006;26:13505–14. doi: 10.1523/JNEUROSCI.3970-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Thompson SM, Cai X. Dynamic regulation of the Kv2.1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J Neurosci. 2008;28:8529–38. doi: 10.1523/JNEUROSCI.1417-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H. Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist. 2010;16:51–64. doi: 10.1177/1073858409341085. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Weerasuriya A. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta Neuropathol. 2011;121:291–312. doi: 10.1007/s00401-010-0783-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan AS, Benton DC, Bahia PK, Hosseini R, Shah YA, Haylett DG, Moss GW. The SK3 subunit of small conductance Ca2+-activated K+ channels interacts with both SK1 and SK2 subunits in a heterologous expression system. J Biol Chem. 2004;279:1003–9. doi: 10.1074/jbc.M308070200. [DOI] [PubMed] [Google Scholar]
- Muennich EA, Fyffe RE. Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J Physiol. 2004;554:673–85. doi: 10.1113/jphysiol.2003.056192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Carpenter-Hyland EP, Hearing MC, Becker HC, Woodward JJ, Chandler LJ. Glutamate transporters regulate extrasynaptic NMDA receptor modulation of Kv2.1 potassium channels. J Neurosci. 2008;28:8801–9. doi: 10.1523/JNEUROSCI.2405-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakoshi H, Shi G, Scannevin RH, Trimmer JS. Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol. 1997;52:821–8. doi: 10.1124/mol.52.5.821. [DOI] [PubMed] [Google Scholar]
- Murphy TH. Activity-dependent synapse development: changing the rules. Nat Neurosci. 2003;6:9–11. doi: 10.1038/nn0103-9. [DOI] [PubMed] [Google Scholar]
- Nakanishi ST, Cope TC, Rich MM, Carrasco DI, Pinter MJ. Regulation of motoneuron excitability via motor endplate acetylcholine receptor activation. J Neurosci. 2005;25:2226–32. doi: 10.1523/JNEUROSCI.5065-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataraj K, Le Roux N, Nahmani M, Lefort S, Turrigiano G. Visual deprivation suppresses L5 pyramidal neuron excitability by preventing the induction of intrinsic plasticity. Neuron. 2010;68:750–62. doi: 10.1016/j.neuron.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New GA, Hendrickson BR, Jones KJ. Induction of heat shock protein 70 mRNA in adult hamster facial nuclear groups following axotomy of the facial nerve. Metab Brain Dis. 1989;4:273–9. doi: 10.1007/BF00999773. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Sanes JR, Lichtman JW. Pre-existing pathways promote precise projection patterns. Nat Neurosci. 2002;5:861–7. doi: 10.1038/nn905. [DOI] [PubMed] [Google Scholar]
- Nielsch U, Bisby MA, Keen P. Effect of cutting or crushing the rat sciatic nerve on synthesis of substance P by isolated L5 dorsal root ganglia. Neuropeptides. 1987;10:137–45. doi: 10.1016/0143-4179(87)90015-1. [DOI] [PubMed] [Google Scholar]
- O’Connell KM, Rolig AS, Whitesell JD, Tamkun MM. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci. 2006;26:9609–18. doi: 10.1523/JNEUROSCI.1825-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell KM, Loftus R, Tamkun MM. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci USA. 2010;107:12351–6. doi: 10.1073/pnas.1003028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, van Rossum MC, Wyllie DJ. Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J Physiol. 2010;588:157–70. doi: 10.1113/jphysiol.2009.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachter BR, Eberstein A. Nerve sprouting and endplate growth induced in normal muscle by contralateral partial denervation of rat plantaris. Brain Res. 1991;560:311–4. doi: 10.1016/0006-8993(91)91249-z. [DOI] [PubMed] [Google Scholar]
- Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–9. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Hirschberg K, Polishchuk RS, Gerlich D, Phair RD, Lippincott-Schwartz J. Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell. 2008;133:1055–67. doi: 10.1016/j.cell.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prather JF, Nardelli P, Nakanishi ST, Ross KT, Nichols TR, Pinter MJ, Cope TC. Recovery of proprioceptive feedback from nerve crush. J Physiol. 2011;589:4935–47. doi: 10.1113/jphysiol.2011.210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman PT, He K, Hartnett KA, Jefferson BS, Hu L, Rosenberg PA, Levitan ES, Aizenman E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci USA. 2007;104:3568–73. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanes GJ. The motor cell columns of the lumbo-sacral spinal cord of the cat. J Comp Neurol. 1951;94:313–63. doi: 10.1002/cne.900940209. [DOI] [PubMed] [Google Scholar]
- Schulz DJ, Baines RA, Hempel CM, Li L, Liss B, Misonou H. Cellular excitability and the regulation of functional neuronal identity: from gene expression to neuromodulation. J Neurosci. 2006;26:10362–7. doi: 10.1523/JNEUROSCI.3194-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–85. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- Strassmaier T, Bond CT, Sailer CA, Knaus HG, Maylie J, Adelman JP. A novel isoform of SK2 assembles with other SK subunits in mouse brain. J Biol Chem. 2005;280:21231–6. doi: 10.1074/jbc.M413125200. [DOI] [PubMed] [Google Scholar]
- Sumner BE. A quantitative analysis of boutons with different types of synapse in normal and injured hypoglossal nuclei. Exp Neurol. 1975;49:406–17. doi: 10.1016/0014-4886(75)90097-7. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Foehring R. A mechanism for homeostatic plasticity. Nat Neurosci. 2004;7:691–2. doi: 10.1038/nn0704-691. [DOI] [PubMed] [Google Scholar]
- Tiraihi T, Rezaie MJ. Synaptic lesions and synaptophysin distribution change in spinal motoneurons at early stages following sciatic nerve transection in neonatal rats. Brain Res Dev Brain Res. 2004;148:97–103. doi: 10.1016/j.devbrainres.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–6. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Waite PM, Cragg BG. The peripheral and central changes resulting from cutting or crushing the afferent nerve supply to the whiskers. Proc R Soc Lond B Biol Sci. 1982;214:191–211. doi: 10.1098/rspb.1982.0004. [DOI] [PubMed] [Google Scholar]
- Walmsley B, Berntson A, Leao RN, Fyffe RE. Activity-dependent regulation of synaptic strength and neuronal excitability in central auditory pathways. J Physiol. 2006;572:313–21. doi: 10.1113/jphysiol.2006.104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerasuriya A, Hockman CH. Perineurial permeability to sodium during Wallerian degeneration in rat sciatic nerve. Brain Res. 1992;581:327–33. doi: 10.1016/0006-8993(92)90727-q. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Rempel J, Brownstone RM. Postnatal development of cholinergic synapses on mouse spinal motoneurons. J Comp Neurol. 2004;474:13–23. doi: 10.1002/cne.20089. [DOI] [PubMed] [Google Scholar]
- Yao H, Zhou K, Yan D, Li M, Wang Y. The Kv2.1 channels mediate neuronal apoptosis induced by excitotoxicity. J Neurochem. 2009;108:909–19. doi: 10.1111/j.1471-4159.2008.05834.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McKay SE, Bewley B, Kaczmarek LK. Repetitive firing triggers clustering of Kv2.1 potassium channels in Aplysia neurons. J Biol Chem. 2008;283:10632–41. doi: 10.1074/jbc.M800253200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.