

Abstract
This work is aimed at engineering photosensitizer embedded nanoparticles (NPs) that produce optimal amount of reactive oxygen species (ROS) for photodynamic therapy (PDT). A revised synthetic approach, coupled with improved analytical tools, resulted in more efficient PDT. Specifically, methylene blue (MB) conjugated polyacrylamide nanoparticles (PAA NPs), with a polyethylene glycol dimethacrylate (PEGDMA, Mn 550) cross-linker, were synthesized so as to improve the efficacy of cancer PDT. The long cross-linker chain, PEGDMA, increases the distance between the conjugated MB molecules so as to avoid self-quenching of the excited states or species, and also enhances the oxygen permeability of the NP matrix, when compared to the previously used shorter cross-linker. The overall ROS production from the MB–PEGDMA PAA NPs was evaluated using the traditional way of monitoring the oxidation rate kinetics of anthracence-9,10-dipropionic acid (ADPA). We also applied singlet oxygen sensor green (SOSG) so as to selectively derive the singlet oxygen (1O2) production rate. This analysis enabled us to investigate the ROS composition mix based on varied MB loading. To effectively obtain the correlation between the ROS productivity and the cell killing efficacy, a microfluidic chip device was employed to provide homogeneous light illumination from an LED for rapid PDT efficacy tests, enabling simultaneous multiple measurements while using only small amounts of NPs sample. This provided multiplexed, comprehensive PDT efficacy assays, leading to the determination of a near optimal loading of MB in a PAA matrix for high PDT efficacy by measuring the light-dose-dependent cell killing effects of the various MB–PEGDMA PAA NPs using C6 glioma cancer cells.
Keywords: photodynamic therapy, polyacrylamide nanoparticle, methylene blue, microfluidic chip, reactive oxygen species (ROS)
1. INTRODUCTION
The reactive oxygen species (ROS) are several highly oxidative molecules and radicals such as singlet oxygen (1O2), hydroxyl radical, superoxide anion radical, hydrogen peroxide, etc.1 Because of their high reactivity, ROS not only function as physiological regulators of intracellular signal pathways but are also cytotoxic, whereby they induce cell death, either by apoptosis or necrosis through oxidation.1–4 ROS can be generated by visible light with the help of appropriate photosensitizers.1,5,6 This mechanism underlies photodynamic therapy (PDT), which is a clinically approved non-invasive localized therapeutic modality for cancer, cardiovascular, ophthalmic, dermatological and dental diseases.6–12 The interest in optimizing nano-photosensitizers is associated with their advantages as PDT agents, due to their potential for increased efficiency, targetability and biocompatibility, as well as their theranostic (therapy + diagnostics) operation.1,13–15
The utility of PDT in medical applications is restricted due to the following factors: 1) wavelength-dependent tissue penetration depth of the light, 2) inefficient delivery of photosensitizer (PS) to targeted area, 3) loss of PDT efficacy due to aggregation, degradation, or reduction of photosensitizers, and 4) dark toxicity of the photosensitizer.1,13,16 Despite these limitations, PDT is a promising and attractive cancer therapeutic modality because of the following advantages: minimal long-term toxicity effects, precise localized treatment, ability of repeated treatment at the same site, and suitability for patients too old or sick for conventional surgery.17 Notably, PDT efficacy can be significantly improved when nanoparticles (NPs) are applied as PS carriers, as the use of NPs can help overcome the limitations 2), 3) and 4) listed above.1,8,18–21 Methylene blue (MB) is a promising PDT dye due to its high quantum yield (ΦΔ ≈ 0.5) and long absorption wavelength (λmax = 664 nm), enabling a better light penetration depth in live tissue, as well as due to its high solubility in aqueous media.13,17 However, MB's use for in vivo PDT has been limited because of its conversion into an inactive form (leuko MB) by enzyme reduction reactions that occur in the blood stream. At higher concentration, MB may form aggregates that reduce the fluorescence quantum yield; furthermore, the MB molecule lacks specificity towards target tissues.17,22
In our previous studies, MB loaded polyacrylamide nanoparticles (PAA NPs) have been developed by two different loading methods, encapsulation and chemical conjugation.13,23 The PAA NPs were found to be biocompatible and biodegradable nano-carriers; moreover, the NP matrix was easily modified with targeting moieties and successfully protected the MB from reduction/conversion by bioenzymes.22,24–27 For making the MB NPs, the conjugation approach was found to be the most efficient method, demonstrated by higher loading, negligible leaching of MB from the nanocarrier, and by better PDT efficacy, probably due to a more homogeneous distribution of the high amounts of conjugated MB inside the NP matrix.13 These MB conjugated PAA NPs showed a PDT efficacy (per 1 mg of NPs) of almost 9 times that of MB encapsulated PAA NPs. This PDT efficacy was estimated by the traditional kinetics-based method: i.e., by determining the rate constant, k, of the first order decay kinetics of anthracence-9,10-dipropionic acid (ADPA), under oxidative quenching by the produced ROS.13,23,28
Notably, upon increasing the photosensitizer loading, the MB conjugated PAA NPs showed an upper limit for the k value. The k value initially increased with MB loading, but then peaked and decreased.13,24 This limitation on the PDT efficacy of the MB conjugated NPs — probably due to aggregation of MB moieties and self-quenching of produced excitations and/or ROS inside the nanocarriers — may be overcome by modifying the NP matrix structure through the use of longer cross-linkers. This is expected to allow (1) longer distances (gaps) between the conjugated MB moieties, (2) a higher oxygen permeability, and (3) a lower collision probability between the produced ROS.
Herein, we present the preparation of a modified MB–PAA NP, which uses polyethylene glycol dimethacrylate (PEGDMA, Mn = 550), a cross-linker that is longer than the 3-(Acryloyloxy)-2-hydroxypropyl methacrylate, (AHM, MW = 214), which was used for previously developed MB conjugated PAA NPs.13 The new PEGDMA PAA NP also uses a smaller mole fraction of the cross-linker per nanoparticle, so as to further enlarge the average pore size of the NP matrix. The PEGDMA PAA NPs were conjugated with different amounts of MB, and then characterized. Their ROS productivity was investigated using two ROS sensing dyes, ADPA and singlet oxygen sensor green (SOSG), so as to determine the optimal MB loading for PDT.
In addition, a new microfluidic chip was introduced for quick and reliable in vitro PDT tests. The microfluidic chip is a promising analytic device for screening PDT efficiency due to its low cost, high-throughput and ease of controlling the micro-environment.29,30 It should be noted that the therapeutic effect of PDT depends not only on the dose of the drugs but also on the doses of light and oxygen, making a reliable estimation of the therapeutic efficacy difficult and lengthy. Regarding in vitro PDT tests, usually they are performed in chambered cover glasses, for one specific test condition per experiment, and it is difficult to increase the number of tests done simultaneously, using several NP samples, due to the limited illumination area of the laser light source.13,22,24 It would be expensive to design an intense and monochromatic light source with a large enough illumination area; on the other hand, if multiple small light sources were used, it would be challenging to have the same flux of the light source on each PDT test area. Thus, employing a new approach, batches of NPs with three different MB loadings were tested simultaneously on the microfluidic chip, so as to determine their cell killing efficacy by PDT. Also, this device allowed to perform several PDT efficacy tests under exactly identical test conditions (the same NP concentration, oxygen concentration, and homogeneous light illumination) while using only small amounts of test sample. Based on this, results for light illumination at half maximal inhibitory time (IT50) were derived by measuring cell survival at varying light illumination time periods, for given MB–PEGDMA PAA NP doses, using C6 glioma cancer cells.
2. EXPERIMENTAL SECTION
Materials
Acrylamide (AA), poly(ethylene glycol) dimethacrylate, Mn 550, (PEGDMA), ammonium persulfate (APS), N,N,N',N'- tetramethylethylenediamine (TEMED), sodium dioctylsulfosuccinate (AOT), Brij 30, dimethyl sulfoxide (DMSO), phosphate buffered saline (BioReagent, pH 7.4, for molecular biology), β-nicotinamide adenine dinucleotide, reduced dipotassium salt (NADH), diaphorase from Clostridium kluyveri (NADH dehydrogenase), Trichloro(1H,1H,2H,2H-perfluorooctyl)silane and hexane were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-(3-aminopropyl)methacrylamide hydrochloride (APMA) was purchased from Polysciences (Warrington, PA, USA). Dicarboxymethylene blue NHS ester (DCMB-SE) was purchased from European Molecular Precision Biotech (Berlin, Germany). Singlet oxygen sensor green (SOSG), 9,10-dipropionic acid disodium salt (ADPA), Dulbecco's Modified Eagle Medium (DMEM),colorless DMEM (no phenol red), 100X Antibiotic-Antimycotic and LIVE/DEAD® Viability/Cytotoxicity Kits were purchased from Life Technologies (Carlsbad, CA, USA). The water was purified with a Milli-Q system from Millipore Coporation (Billerica, MA, USA). SU8 2050 was purchased from MicroChem Co. (Newton, MA, USA). Poly(dimethylsiloxane) (PDMS) kit (Sylgard 184) was purchased from Dow Corning Co. (Midland, MI, USA). Glass slides were purchased from Fisher Scientific (Hanover Park, IL, USA). All chemicals were used without further purification.
Preparation of MB–PEGDMA PAA NPs
The MB–PEGDMA PAA NPs were prepared similarly to a previously reported MB–PAA NPs method.13 A 100 μL of DCMB-SE solution (50 μg/μL in DMSO) was added to a monomer solution including 368 mg of AA and 28 mg of APMA in 0.93 mL of PBS (pH 7.4). After 2 hr incubation for MB conjugation with APMA, 53.4 μL of PEGDMA cross-linker was added into the monomer solution. The resultant monomer solution was then added into a 100 mL round bottomed flask containing 30 mL of deoxygenated hexane, 1.07 g of AOT and 2.2 mL of Brij 30 surfactants. To initiate radical polymerization, 100 μL of TEMED and 100 μL of APS (15% w/v) were added to the reaction mixture under inert conditions and stirred for 2 hrs at room temperature. The polymerized NPs were concentrated by rotary evaporation, to remove hexane, and washed with enough ethanol and pure water, using Amicon Stirred Ultrafiltratoin Cells (200mL, equipped with a Biomax 300,000 MWCO membrane). The washed MB–PEGDMA PAA NPs were freeze-dried and stored at −20 °C.
Characterization
The absorption spectra were taken on a UV-1601 UV-vis spectrometer (Shimadzu, Kyoto, Japan) and a FluoroMax-3 Spectrofluorometer (Jobin Yvon Horiba, Kyoto, Japan) was used for fluorescence spectra. For both spectra, 1 mg/mL of MB–PEGDMA PAA NPs solution in PBS (pH 7.4) was used. For the absorbance spectra, background absorbance from the scattering of PEGDMA PAA NPs was subtracted, using Origin program. Using the absorbance spectrum of MB–PEGDMA PAA NPs, the actual amount of MB content per NP was calculated, based on the calibration curve of MB-SE in PBS (pH 7.4). The differential intensity based averaged size distribution and the surface charge of the MB–PEGDMA PAA NPs were determined by dynamic light scattering (DLS) using a Delsa Nano C particle analyzer instrument (Beckman Coulter, Brea, CA, USA). Transmission electron microscopy (TEM) images of MB–PEGDMA PAA NPs were obtained on a Philips CM-100 transmission electron microscope, using uranyl-acetate staining on the grid. All of the measurements were triplicated.
Detection of ROS (k and S value tests)
The ROS produced from the MB–PEGDMA PAA NPs was detected by using ADPA. A 2 mL of MB–PEGDMA PAA NPs solution (1 mg/mL in PBS (pH 7.4)), containing 80 μL of ADPA (100 μM in pure water), was irradiated at 660 nm, over different time periods (0, 60, 120, 180, 240, 300, 480 and 660 sec), under constant stirring and temperature (25°C). The fluorescence spectra of ADPA, excited at 370 nm, were taken right after each irradiation time period. The ADPA decay constant, the “k value,” was calculated by the equation reported previously.23,28 For the detection of 1O2 emanating from the NPs, a specific chemical probe, SOSG (Singlet Oxygen Sensor Green) was used.31–34 A 10 μL aliquot of SOSG (0.5 mM in MeOH) was added to a 2 mL MB–PEGDMA PAA NPs solution (1 mg/mL in PBS (pH 7.4)), under constant stirring and temperature (25 °C). Note that the free SOSG can be activated in a pH 7.4 solvent; thus, a “blank spectrum” of SOSG fluorescence (without singlet oxygen) was taken by irradiating at 504 nm. After taking a blank spectrum, the photosensitizer sample was irradiated for 5 min at 660 nm. The enhanced SOSG fluorescence spectrum (λex = 504 nm) was obtained immediately after stopping the irradiation of photosensitizer. The arbitrarily defined “S value” constant, for singlet oxygen production, was obtained by calculating the ratio of SOSG fluorescence comparing fluorescence before and after irradiation (enhanced/unenhanced).
Enzymatic Reduction test of MB–PEGDMA PAA NPs
The enzymatic reduction test of MB–PEGDMA PAA NPs was obtained based on our previous reported method.22,24 In this test, NADH and diaphorase are mixed with MB. The diaphorase catalyzes oxidation of NADH, and the H+ released from the NADH reduces the MB, converting it into the lueko-MB that neither works as a photosensitizer nor fluoresces. Into 2 mL of 12.1 nmol·mg−1 of MB conjugated PEGDMA PAA NPs solution (1 mg/mL in PBS (pH 7.4)), 133.6 μL of NADH (10 mg/mL in PBS (pH 7.4)) and 200 μL of diaphorase (1 mg/mL in PBS (pH 7.4)) were added. The fluorescence emission signal of MB–PEGDMA PAA NPs, excited at 660 nm, was monitored for 10 min, at 25 °C and under constant stirring. As a control, considering the photobleaching effect on MB, the fluorescence emission signal of the same MB conjugated PEGDMA PAA NPs solution without NADH and diaphorase was monitored. This experiment was repeated with free MB solution and the measurements were conducted in triplicate.
Microfluidic chip preparation
The microfluidic chips were fabricated using a customized soft lithography process similar to what was reported earlier.29,30 The replication mold was made of 170 μm thick negative photoresist on a silicone substrate. SU8 2050 (epoxy-based negative photoresist) is spin-coated at 1000 rpm, to reach the target thickness, and patterned with standard photolithography (UV dose 350 mJ/cm2). The resulting SU8 thickness is measured using a Dektak 6M surface profilometer (Veeco Instruments Inc. Plainview, NY, USA). Before applying PDMS, replication molds are coated with a self-assembly monolayer (SAM) of Trichloro(1H,1H,2H,2H-perfluorooctyl)silane by vacuuming for 1 hour in a desiccator. Then, a mixture of PDMS prepolymer at a 10:1 ratio (base: curing reagent) was poured onto the mold and peeled off after curing under 100 °C for 1 h. Inlet and outlet reservoirs were punched out using 2 mm and 4 mm diameter biopsy punches (MedPlus INc., Monsey, NY, USA). After an oxygen plasma treatment procedure (200 mTorr, 50 W, 30 sec) was conducted on the surface, the PDMS microfluidic layer was bonded to a glass slide. A post-bake for 20 min at 90°C was adopted for enhancing the permanent bonding between the PDMS and the glass, finalizing the assembly.
Test of LED light illumination uniformity on microfluidic chip
To test the uniformity of light illumination on the microfluidic chip, an LED lamp (625 (± 20) nm, Philips Lumileds, San Jose, CA, USA) was placed 9 cm above the chip and used as a light source, instead of the microscope light bulb, for mimicking the illumination conditions as would be used in the PDT test. Then, a x2 bright field image of the microfluidic chip was obtained on a Nikon eclipse TE2000-U microscope (Nikon Instruments Inc., Melville, NY, USA). The grayscale intensity of the whole PDT tested area in each quadrant on the bright field image was measured and averaged by ImageJ program.
Cell Culture and PDT test on the Chip
The C6 rat glioma cells (CCL-107, ATCC, Manassas, VA, USA) were cultivated in DMEM media with 10% heat inactivated fetal bovine serum (HI-FBS) and 1% 100× Antibiotic-Antimycotic (10,000 units/mL of penicillin, 10,000 μg/mL of streptomycin, and 25 μg/mL of Fungizone®). Prior to cell plating on the microfluidic chip, the chip was coated, using a solution of 0.01% collagen and 0.2% acetic acid in pure water, and incubated overnight at 37 °C. Thereafter, the chip was washed with DMEM media to remove any residual collagen solution, and the C6 cells were subsequently loaded into the microfluidic chip and cultured for about 24 hr in the incubator, under 5% CO2 at 37 °C. In 3 of the 4 wells on the chip, 50 μL of either 2.1 nmol·mg−1, 5.5 nmol·mg−1 or 12.1 nmol·mg−1 of MB conjugated PEGDMA PAA NPs (0.2 mg/mL in DMEM) were injected and the chip was covered by aluminum foil, to protect the sample from light. The remaining well of the microfluidic chip was not treated with NPs solution, and thus served as a control for each condition. After 10 min incubation, the chip was illuminated by LED light (625 (± 20) nm; 35.2 mW), for varying periods of time, ranging from 0 to 21 min, to deliver light doses of 0, 5.6, 11.2, 16.8, 22.4, 28, 33.6 and 39.2 J/cm2. For 0 min light illumination, the NPs were incubated an extra 21 min (the same condition as for the longest light illumination times) without any light source as a dark toxicity test of NPs on C6 glioma cells. The distance between the chip and the LED was kept at 9 cm. After additional 15 min incubation, without any light, the MB–PEGDMA PAA NPs solution was removed from the chip. Then, 50 μL of colorless DMEM solution containing 0.25 μL of calcein AM (2 mM in DMSO) and 0.25 μL of ethidium homodimer-1 (2 mM in DMSO) were added to each well. After 10 min incubation, the fluorescence images of calcein AM (excited at 460 – 500 nm), and ethidium homodimer-1 (excited at 530 – 560 nm) were taken, using the Nikon eclipse TE2000-U microscope. The numbers of live and dead cells in an ×10 image were counted manually, and cell viability was calculated by equation 1 (ep.1) below.
| (eq.1) |
3. RESULTS AND DISCUSSION
3.1 Characteristics of MB–PEGDMA PAA NPs
Based on the differential intensity (%) obtained by DLS, the hydrodynamic size of the MB–PEGDMA PAA NPs was 78.5 (± 5.8) nm (Figure 1). This is a larger size than the one previously reported for MB–AHM PAA NPs, 55.8 (± 5.0) nm.13 The increased size of the NPs is attributed to the use of a longer cross-linker, PEGDMA (Mn 550), compared to AHM (MW 214). Furthermore, a reduced amount of cross-linker, from 13.6% to 2% (molar ratio of total monomer), may have also helped to increase the hydrodynamic size of the NPs, possibly due to a higher swelling ratio of the hydrogel, caused by the forming of fewer networks inside the NP matrix.35 The size of the dehydrated MB–PEGDMA PAA NPs was checked by TEM imaging. Similar with previous reported hydrogel NPs, the dehydrated MB–PEGDMA PAA NP shrunk upon drying, its size being around 14 nm (Figure 2).13,27 The surface charge of the MB–PEGDMA PAA NPs was 23.0 (± 1.7) mV. This positive surface charge presumably originates from the primary amines, from the APMA monomer that was used for further modification, e.g. for PEGylation, or for attaching a targeting moiety.13,27,36
Figure 1.
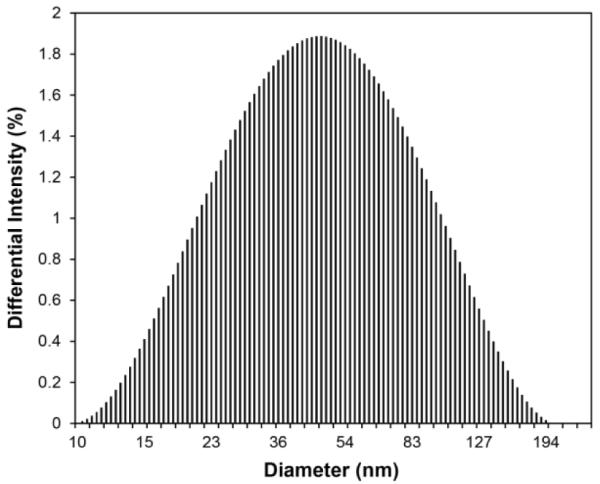
DLS of MB–PEGDMA PAA NPs
Figure 2.
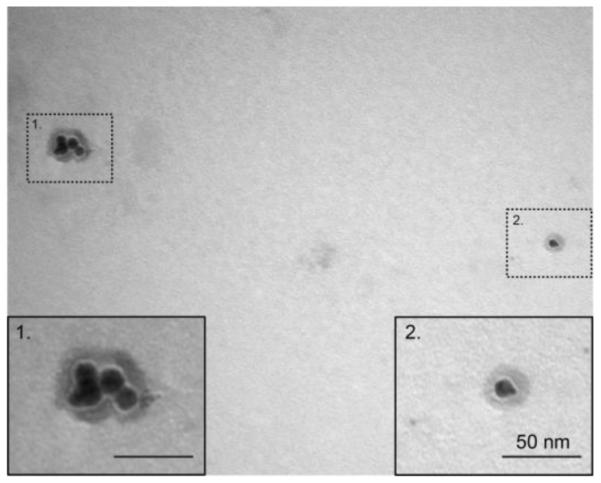
TEM of MB–PEGDMA PAA NPs
The absorption spectra of the MB–PEGDMA PAA NPs, containing different amounts of conjugated MB, were obtained (Figure 3a). The MB–PEGDMA PAA NPs have their absorbance peak at 669 nm, which is the same position as for free DCMB-SE. Increasing the conjugated MB amount, another peak appears at 619 nm, which is due to aggregation of the MBs.37–39 A significant dimer aggregate signal is observed, starting at 12.1 nmol·mg−1 of MB loaded PEGDMA PAA NPs, and at higher loadings. Under the same conditions, the excitation and emission spectra of MB–PEGDMA PAA NPs were obtained (Figure 3b). There are strong excitation and emission signal peaks at 669 nm and 690 nm, respectively. The excitation peak wavelength matches exactly with the absorbance wavelength. Both the excitation and emission signals increased as more MBs are conjugated. However, these spectral intensities rather decreased from the 12.1 nmol·mg−1 of MB loading, which is the same MB loading point where the significant MB dimer absorbance peak was observed. Because of the large amount of MB dimers inside the NP matrix, there might be self-quenching of MB fluorescence, caused by interactions between excited states of MB molecules, and indeed a very low fluorescence signal was obtained, at the highest MB loading, 29.6 nmol·mg−1, in PEGDMA PAA NPs.13,17
Figure 3.
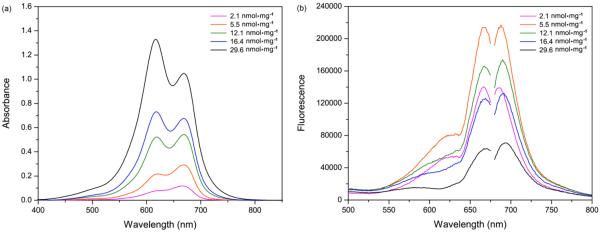
(a) Absorbance spectra of MB–PEGDMA PAA NPs,as function of matrix concentration (SD =±5.2%). (b) Fluorescence excitation (left peak) and emission (right peak) spectra of MB–PEGDMA PAA NPs, λex = 660 nm (SD = ± 4.6%, n = 3).
3.2 Advantages of MB–PEGDMA PAA NPs
According to our previous studies, the PAA nanomatrix protected the embedded MB molecules from undergoing reduction into a photo-inactive form, leuko-MB, due to an isomerization reaction catalyzed by bio-enzymes.22,24 Although the MB–PEGDMA PAA NPs may have a larger pore size, due to having longer chains and reduced amounts of cross-linker, we believe that the pore size is still small enough to prevent the enzymes from entering into the NP matrix — to prevent the embedded MB from reduction. The efficiency of protection of MB from reduction was tested for the MB–PEGDMA PAA NPs by the enzyme reduction test. While less than 20% of the fluorescence intensity of free MB remained in the presence of NADH and diaphorase, more than 90% of MB fluorescence intensity in the PEGDMA PAA NPs remained under the same conditions (Figure 4). The 8% of fluorescence decrease of MB–PEGDMA PAA NPs is presumably caused by the MBs conjugated onto the surface of the PEGDMA PAA NPs, which are not well protected by the matrix, while most of the conjugated MBs residing deeper inside the NPs are protected by the nanomatrix.
Figure 4.
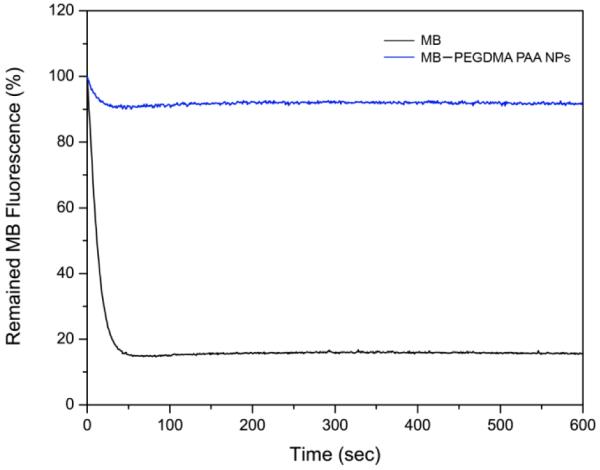
Enzyme reduction test of MB–PEGDMA PAA NPs: Free vs PAA encapsulated MB(SD = ± 7.1%, n = 3).
In addition, the MB–PEGDMA PAA NPs have an advantage based on optical considerations. In the PDT experiment, it is very important to optimize the local concentration of the photosensitizer in the blood stream, or inside organs since the absorbance maxima could change depending on the MB concentration, due to dimer formation.17 Hence, the in vivo production efficiency of ROS may be very different from the in vitro experimental value. In contrast with MB encapsulated in the nanomatrix, the PEGDMA PAA NPs attach their MB moieties by a conjugation method. MB–PEGDMA PAA NPs essentially keep the same distances between MBs even when varying the NP concentrations, since the distances among the MBs are fixed by the chemical bonds within the PAA chain. In Figure 5, the absorbance spectra of MB–PEGDMA PAA NPs do not change shape even at the higher NP concentrations. Thus one can maximize the PDT efficacy of MB at a high optimal concentration by the use of these NPs.
Figure 5.
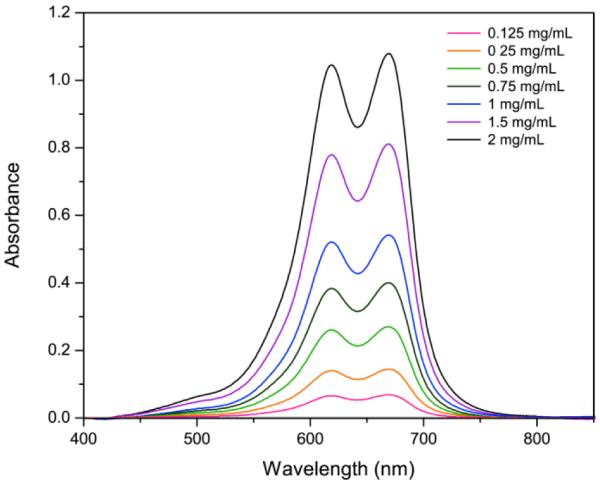
Absorbance spectra of MB–PEGDMA PAA NPs(12.1 nmol·mg−1 MB loading) with varying NP concentration. The absorbance peak of the MB dimer is at 619 nm and that of the monomer MB at 669 nm (SD = ± 4.2%, n = 3).
3.3 ROS productivity tests of MB–PEGDMA PAA NPs
Based on the k value test using ADPA, the ROS production efficiency of the MB–PEGDMA PAA NPs was estimated and compared with the previous MB–AHM PAA NPs (Figure 6). The k value increased with MB loading concentration up to 12.1 nmol·mg−1 of MB loaded PEGDMA PAA NPs, where k = 13.4 × 10−4 s−1, and this k value maximum is 1.44 times higher than the maximum value found with MB–AHM PAA NPs, namely k = 9.3 × 10−4 s−1 at 8.8 nmol·mg−1 of MB loaded sample.13 In addition, to compare the required amounts of conjugated MB between the two different NPs for producing the same amount of ROS, the MB–AHM PAA NPs were reproduced.13 The MB–AHM PAA NPs containing 3.0 nmol·mg−1 of MB have a k value of 5.9 × 10−4 s−1 while the MB–PEGDMA PAA NPs containing only 2.1 nmol·mg−1 of MB produce a similar amount of ROS, i.e., with k = 6.0 × 10−4 s−1. These improvements presumably originate from a reduced amount of longer cross-linker, which increases the distances between MBs, resulting in reduction of self-quenching while also providing a larger pore size, which allows for better oxygen permeation and lower collision probability between the produced ROS.
Figure 6.
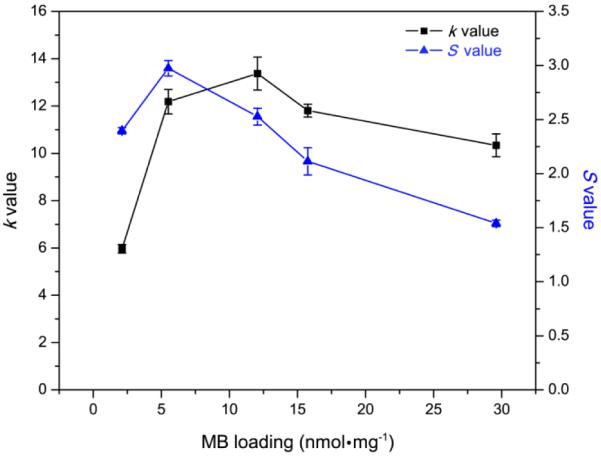
The k and S value s of MB–PEGDMA PAA NPs depend on the amount of loaded MB. Black line: k value obtained by ADPA, Blue line: S value obtained by SOSG dye.
In Figure 6 (black line), the k value of the MB–PEGDMA PAA NPs decreases after peaking at 12.1 nmol·mg−1 of MB loading, which may result from too high MB loading within the limited space of the PAA NPs. It is necessary for the produced ROS to escape from the nanomatrix in order to kill the cancer cell or, in our measurements here, to react with the external ROS detection probe. However, the over-produced ROS species could react among themselves before getting out of the PAA NPs, so the amount of ROS outside the NPs may decrease, rather. Moreover, the excited photosensitizers could be self-quenched at the highly concentrated MB loading inside the limited space of the NPs, when those MB groups are close enough due to proximity of MBs in adjacent polymer chains.
Notably, the fluorescence emission spectra are determined by the amount of MB monomer, which is involved in producing 1O2.17 However, the k value dependence on the total MB loading does not follow the trend observed in the fluorescence spectra (Figure 3b) in a simple way. The highest fluorescence intensity of the NPs was obtained at 5.5 nmol·mg−1 of MB loading, not at the MB loading of the highest k value, which was 12.1 nmol·mg−1. Furthermore, at 2.1 nmol·mg−1 of MB loading, the NPs have 2 times higher fluorescence intensity but 42% less k value than at 29.6 nmol·mg−1 of MB loading. For finer discrimination, so as to detect only the 1O2, among all the ROS produced by the MB–PEGDMA PAA NPs, a singlet oxygen probe, SOSG was applied; SOSG reacts solely with 1O2 but not with other ROS such as superoxide radical anion or hydroxyl radical.32–34 In the presence of singlet oxygen, it emits an enhanced fluorescence at 525 nm. The SOSG can be activated in a DMSO and PBS (pH 7.4) solvent, in which a blank SOSG fluorescence spectrum was first taken, i.e., without 1O2 generation from MB–PEGDMA PAA NPs. Then, with the NPs included, the enhanced SOSG fluorescence intensity, which is due to the presence of 1O2, was taken, and the enhancement ratio was calculated, so as to give the “S value”. In Figure 6 (blue line), it can be seen that the maximal amount of 1O2 was produced at a loading of 5.5 nmol·mg−1 MB in PEGDMA PAA NPs. Also, the relationship between the fluorescence of MB–PEGDMA PAA NPs and the S value was quite linear, which supports the hypothesis that 1O2 is produced overwhelmingly by the MB monomer, but not by the MB dimer (Figure 7). Due to the small size of the ADPA and SOSG molecules, these dyes can penetrate into the nanoparticles and detect the ROS inside the PAA NPs platform. Because of this, the k and S values can be overestimated due to detecting the ROS generated inside NPs. However, the overestimation equally applies to all the tested NPs and the relative k and S values should not be affected. Thus, this potential overestimation will not distort the general trend of k and S values observed with various MB loadings in PAA NPs.
Figure 7.
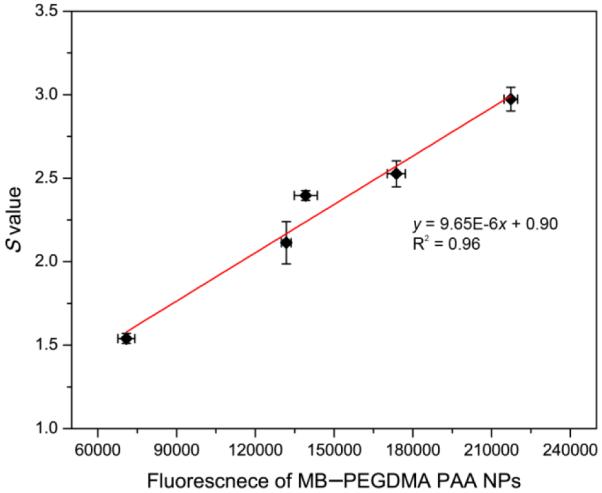
Correlation of S value with fluorescence emission intensity of MB–PEGDMA PAA NPs based on Figure 3b.
To find out the relationship of the k value and S value with the PDT effect of the MB–PEGDMA PAA NPs, the ROS producing routes of MB were investigated. Baptista's group reported that not only the MB monomer but also the MB dimers produce specific ROS, through two major photochemical pathways, Type I and II (Scheme 2).17,38–40 The MB monomer produces 1O2 by energy transfer to the oxygen molecule from the triplet excited state MB (3MB+*), Type II, while at the higher MB concentration, the dimer MB ((MB)22+) produces semi-reduced MB radicals (MB•) by redox suppression of excited ground sate dimers, and the MB• is oxidized to produce superoxide, Type I. Therefore we can explain why the higher MB loaded NPs (12.1, 15.8, 29.6 nmol·mg−1) had a higher overall ROS production, compared to the lower MB loading case; the increase in superoxide radical production also affects the ADPA quenching rate, which reflects a weighted total ROS count, even though the production of 1O2 decreases.
Scheme 2.

Schematic ROS producing routes of MB:MB monomer produces singlet oxygen (TypeII) while MB dimer produces superoxide (radical anion)(Type I). Note: Energy levels not drawn to scale.
3.4 PDT efficacy test using Microfluidic chips
To find out the relationship of ROS productivity based on the k and S values with the actual cell killing efficacy, an in vitro PDT test was performed using microfluidic chips. The chip was divided into four equal parts to increase throughput per test and it was illuminated homogeneously by the same light source. Through a gray scale test of the microfluidic chip's bright field image, the homogeneity of the LED light illumination was confirmed for the microfluidic chip under test. The averaged gray scale exhibited a very similar intensity, around 2300 (arbitrary units) for each quadrant in the microfluidic chip (Figure 8). In Figure 9, it shows cell survival data, for three different MB loadings, under a fixed NP concentration of 0.2 mg/mL, based on a live/dead cell assay, as a function of light illumination time. The MB–PEGDMA PAA NPs of the three different MB loadings (2.1, 5.5 and 12.1 nmol·mg−1) were investigated vs. a control of C6 cells without any NP treatment. Notably, based on “0 min” and “control” cases, no cell death was induced by light without NPs, nor by NPs without light. The k and S values exhibit significantly different trends in the MB loading range of 5.5 to 12.1 nmol·mg−1. As expected for this loading range, the 2.1 nmol·mg−1 MB loaded NPs which have the lowest k and S values among three samples took the longest times, 21 min, to kill all of the C6 cells. In addition, with the 5.5 nmol·mg−1 of MB loaded NPs, it took 12 min to kill the C6 cells, but only 9 min was taken for killing most of the cells when using the 12.1 nmol·mg−1 of MB loaded NPs (Figure 9a). This trend clearly corresponds with the k value, not the S value, and is expected since not just the 1O2 but also the superoxide radical anion contributes to the killing of the cells.17,41 In addition, the half maximal inhibitory time (IT50), needed to kill the C6 cells, was calculated for each batch of NPs based on Figure 9b, and it shows a linear relationship with the k value (Figure 10). This supports the hypothesis that the relative trend of the detected k values is not distorted by potential overestimation of the absolute values. Using this trend, the IT50 for different MB loaded NPs could be estimated.
Figure 8.
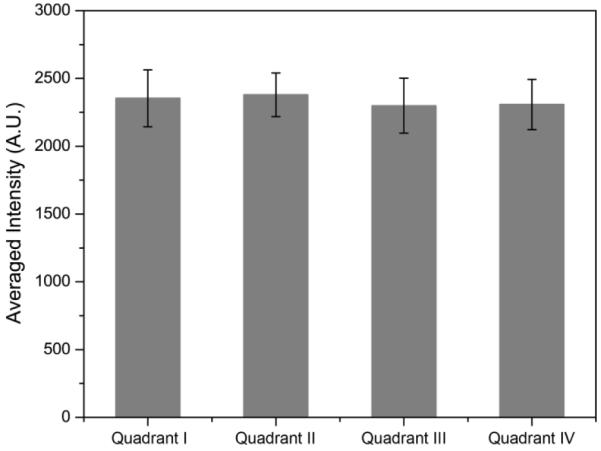
Averaged bright field image intensity on microfluidic chip illuminated by LED light source. Overall gray scale intensities in each quadrant of the chip were obtained and averaged.
Figure 9.

(A) Fluorescence images, with calcein AM and ethidium homodimer-1, for determining the viability of C6 cells, after PDT treatment using MB–PEGDMA PAA NPs, with varied MB concentration (2.1, 5.5, 12.1 nmol·mg−1) and illumination time (0 – 21 min). The green color identifies a live cell (calcein AM) and the red color a dead cell (ethidium homodimer-1). (B) Cell viability (%) of A (a–c) based on eq. 1 (n = 3).
Figure 10.
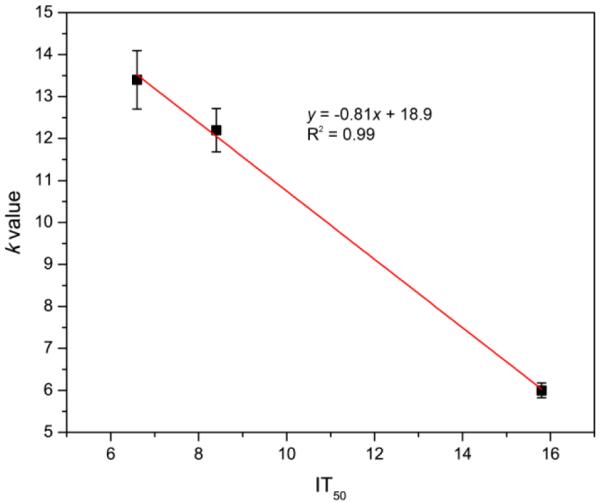
Relationship between IT50 and k value of MB–PEGDMA PAA NPs.
4. CONCLUSIONS AND FUTURE DIRECTIONS
The PEGDMA cross-linked PAA NPs were designed for a higher PDT efficacy. Because of the longer cross-linker, PEGDMA, this novel NP matrix provides an increased distance among the conjugated MB photosensitizer groups, so as to control/minimize the aggregation of MB moieties inside the NPs reducing collision probability among the produced ROS and providing higher oxygen permeability. Such control of aggregation can serve as a tuning mechanism for the produced ROS mixture. At the same time, the PEGDMA PAA NPs matrix prevents any leaching out of the MB photosensitizer; protects it from reduction by plasma enzymes as tested in the presence of NADH with diaphorase even with increased pore size, and also preserves its spectral shape at both low and high concentrations.
The ROS productivity was measured using both k and S values, respectively obtained from the ROS sensitive ADPA probe and from the singlet oxygen sensitive SOSG probe. An excellent correlation was found between the MB monomer fluorescence peak intensity and the singlet oxygen production (S value), as well as between the k value and the total ROS production. Based on the k value, 1.44 times more ROS was produced by the new MB–PEGDMA PAA NPs, relative to the previous generation's MB–AHM PAA NPs, when comparing them at the most optimized MB loading for each NP. In addition, using the MB–PEGDMA PAA NPs saves 30% of the costly MB-SE needed to produce the same yield of ROS (k = 6.0 ×10−4 s−1) with the 3.0 nmol·mg−1 MB loaded AHM PAA NPs.
A microfluidic device optimized for in vitro PDT was developed so as to enable reliable quantitative measurements of the PDT efficacy of MB–PEGDMA PAA NPs. Because of the small size of the chip, the LED illumination could be provided homogeneously, making it possible to reliably compare several different NPs simultaneously. The highest measured k value for the MB–PEGDMA PAA NPs, k = 13.4×10−4 s−1 (at 12.1 nmol·mg−1 MB loading), correlates with the most rapid killing of the C6 cancer cells (IT50 = 6.6 min) and a linear relationship was found between this traditional k value and the IT50 for PDT cell killing. This confirms the accepted, but sometimes overlooked, notion that PDT is not just due to singlet oxygen, but a result of various ROS, and that the ROS composition/mix may be important, as well as the ability of tuning this composition/mix.
In summary, a revision in synthetic approach coupled with improved analytical methods, resulted in more efficient PDT. These optimized MB NPs are promising candidates as multifunctional photosensitive nanoplatforms for both therapy (PDT) and diagnostic purposes. Tumor targeted MB–PAA NPs can be utilized to delineate the tumor boundaries (during surgery), i.e., visually monitor the blue “painted” tumor due to the accumulation of these blue nanoparticles when targeted specifically to the tumor cells.42–44 Furthermore, the same MB NPs can also be used as sonophoric contrast agents for obtaining the 3-dimensional structure of the “blue” tumor by photoacoustic imaging, as demonstrated previously with other types of blue PAA NPs.44 Additionally, these NPs could also serve as nano-regulators by mimicking ROS production during cellular metabolism. The highly localized ROS produced by these NPs could also be used to modulate intracellular signaling pathways, enabling refined and novel methods of cellular research.2,45
ACKNOWLEDGEMENTS
This project is financially supported by NIH grants R01 EB007977 (RK) and by the NSF Engineering Research Center for Wireless Integrated Microsystems (WIMS) at the University of Michigan. The authors thank Mijin Park for help with the preparation of Scheme 1.
Scheme 1.
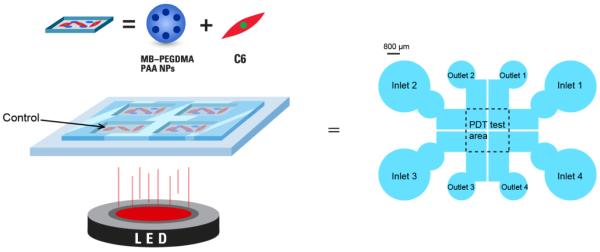
Simplified PDT test setup of MB–PEGDMA PAA NPs on microfluidic chip.
REFERENCES
- (1).Koo Lee YE, Kopelman R. Biomedical Nanoetechnology: Methods and Protocols. 2011;726:151. [Google Scholar]
- (2).Finkel T. J. Cell Biol. 2011;194:7. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Buytaert E, Dewaele M, Agostinis P. Biochim. Biophys. Acta. 2007;1776:86. doi: 10.1016/j.bbcan.2007.07.001. [DOI] [PubMed] [Google Scholar]
- (4).Theodossiou TA, Hothersall JS, De Witte PA, Pantos A, Agostinis P. Mol. Pharm. 2009;6:1775. doi: 10.1021/mp900166q. [DOI] [PubMed] [Google Scholar]
- (5).Garland MJ, Cassidy CM, Woolfson D, Donnelly RF. Future Med. Chem. 2009;1:667. doi: 10.4155/fmc.09.55. [DOI] [PubMed] [Google Scholar]
- (6).Kim S, Ohulchanskyy TY, Bharali D, Chen YH, Pandey RK, Prasad PN. J. Phys. Chem. C. 2009;113:12641. doi: 10.1021/jp900573s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. J. Natl. Cancer Inst. 1998;90:889. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chatterjee DK, Fong LS, Zhang Y. Adv. Drug. Deliv. Rev. 2008;60:1627. doi: 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- (9).Avula UM, Kim G, Lee YE, Morady F, Kopelman R, Kalifa J. Heart Rhythm. 2012;9:1504. doi: 10.1016/j.hrthm.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sibata CH, Colussi VC, Oleinick NL, Kinsella TJ. Braz. J. Med. Biol. Res. 2000;33:869. doi: 10.1590/s0100-879x2000000800002. [DOI] [PubMed] [Google Scholar]
- (11).Konopka K, Goslinski T. J. Dent. Res. 2007;86:694. doi: 10.1177/154405910708600803. [DOI] [PubMed] [Google Scholar]
- (12).Gursoy H, Ozcakir-Tomruk C, Tanalp J, Yilmaz S. Clin. Oral Investig. 2013;17:1113. doi: 10.1007/s00784-012-0845-7. [DOI] [PubMed] [Google Scholar]
- (13).Hah HJ, Kim G, Lee YE, Orringer DA, Sagher O, Philbert MA, Kopelman R. Macromol. Biosci. 2011;11:90. doi: 10.1002/mabi.201000231. [DOI] [PubMed] [Google Scholar]
- (14).Kostron H. Methods Mol Biol. 2010;635:261. doi: 10.1007/978-1-60761-697-9_17. [DOI] [PubMed] [Google Scholar]
- (15).Zhang J, An F, Li Y, Zheng C, Yang Y, Zhang X, Zhang X. ChemComm. 2013;49:8072. doi: 10.1039/c3cc43413c. [DOI] [PubMed] [Google Scholar]
- (16).Lin J, Wang SJ, Huang P, Wang Z, Chen SH, Niu G, Li WW, He J, Cui DX, Lu GM, Chen XY, Nie ZH. ACS nano. 2013;7:5320. doi: 10.1021/nn4011686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tardivo JP, Del Giglio A, de Oliveira CS, Gabrielli DS, Junqueira HC, Tada DB, Severino D, Turchiello RDF, Baptista MS. Photodiagn. Photodyn. 2005;2:175. doi: 10.1016/S1572-1000(05)00097-9. [DOI] [PubMed] [Google Scholar]
- (18).Bechet D, Couleaud P, Frochot C, Viriot ML, Guillemin F, Barberi-Heyob M. Trends Biotechnol. 2008;26:612. doi: 10.1016/j.tibtech.2008.07.007. [DOI] [PubMed] [Google Scholar]
- (19).Simon T, Boca-Farcau S, Gabudean AM, Baldeck P, Astilean S. Journal of biophotonics. 2013;6:950. doi: 10.1002/jbio.201300058. [DOI] [PubMed] [Google Scholar]
- (20).Yan F, Zhang Y, Kim KS, Yuan HK, Vo-Dinh T. Photochem. Photobiol. 2010;86:662. doi: 10.1111/j.1751-1097.2009.00696.x. [DOI] [PubMed] [Google Scholar]
- (21).Wilson BC, Patterson MS. Phys. Med. Biol. 2008;53:R61. doi: 10.1088/0031-9155/53/9/R01. [DOI] [PubMed] [Google Scholar]
- (22).Tang W, Xu H, Park EJ, Philbert MA, Kopelman R. Biochem. Bioph. Res. Commun. 2008;369:579. doi: 10.1016/j.bbrc.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tang W, Xu H, Kopelman R, Philbert MA. Photochem. Photobiol. 2005;81:242. doi: 10.1562/2004-05-24-RA-176.1. [DOI] [PubMed] [Google Scholar]
- (24).Qin M, Hah HJ, Kim G, Nie G, Lee YE, Kopelman R. Photochem. Photobiol. Sci. 2011;10:832. doi: 10.1039/c1pp05022b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang S, Kim G, Lee YE, Hah HJ, Ethirajan M, Pandey RK, Kopelman R. ACS nano. 2012;6:6843. doi: 10.1021/nn301633m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang SY, Fan WZ, Kim G, Hah HJ, Lee YEK, Kopelman R, Ethirajan M, Gupta A, Goswami LN, Pera P, Morgan J, Pandey RK. Laser Surg Med. 2011;43:686. doi: 10.1002/lsm.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yoon H, Ray A, Koo Lee YE, Kim G, Wang X, Kopelman R. J. Mater. Chem. B. 2013;1:5611. doi: 10.1039/C3TB21060J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Moreno MJ, Monson E, Reddy RG, Rehemtulla A, Ross BD, Philbert M, Schneider RJ, Kopelman R. Sensor Actuat. B-Chem. 2003;90:82. [Google Scholar]
- (29).Lou X, Kim G, Koo Lee YE, Kopelman R, Yoon E. International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS'10) 2010:719. [Google Scholar]
- (30).Lou X, Kim G, Koo Lee YE, Kopelman R, Yoon E. International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS '11) 2011:2058. [Google Scholar]
- (31).Wang J, Zhu G, You M, Song E, Shukoor MI, Zhang K, Altman MB, Chen Y, Zhu Z, Huang CZ, Tan W. ACS nano. 2012;6:5070. doi: 10.1021/nn300694v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xiao L, Gu L, Howell SB, Sailor MJ. ACS nano. 2011;5:3651. doi: 10.1021/nn1035262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lin HY, Shen Y, Chen DF, Lin LS, Wilson BC, Li BH, Xie SS. J. Fluoresc. 2013;23:41. doi: 10.1007/s10895-012-1114-5. [DOI] [PubMed] [Google Scholar]
- (34).Flors C, Fryer MJ, Waring J, Reeder B, Bechtold U, Mullineaux PM, Nonell S, Wilson MT, Baker NR. J. Exp. Bot. 2006;57:1725. doi: 10.1093/jxb/erj181. [DOI] [PubMed] [Google Scholar]
- (35).Ilic-Stojanovic SS, Nikolic L, Nikolic V, Stankovic M, Stamenkovic J, Mladenovic-Ranisavljevic I, Petrovic S. Chem. Ind. Chem. Eng. Q. 2012;18:1. [Google Scholar]
- (36).Karamchand L, Kim G, Wang S, Hah H, Ray A, Jiddou R, Koo Lee YE, Philbert M, Kopelman R. Nanoscale. 2013 doi: 10.1039/c3nr00908d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Patil K, Pawar R, Talap P. Phys. Chem. Chem. Phys. 2000;2:4313. [Google Scholar]
- (38).Severino D, Junqueira HC, Gugliotti M, Gabrielli DS, Baptista MS. Photochem. Photobiol. 2003;77:459. doi: 10.1562/0031-8655(2003)077<0459:ioncio>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (39).Junqueira HC, Severino D, Dias LG, Gugliotti MS, Baptista MS. Phys. Chem. Chem. Phys. 2002;4:2320. [Google Scholar]
- (40).Gabrielli D, Belisle E, Severino D, Kowaltowski AJ, Baptista MS. Photochem. Photobiol. 2004;79:227. doi: 10.1562/be-03-27.1. [DOI] [PubMed] [Google Scholar]
- (41).Auten RL, Davis JM. Pediatr. Res. 2009;66:121. doi: 10.1203/PDR.0b013e3181a9eafb. [DOI] [PubMed] [Google Scholar]
- (42).Orringer DA, Koo YE, Chen T, Kim G, Hah HJ, Xu H, Wang S, Keep R, Philbert MA, Kopelman R, Sagher O. Neurosurgery. 2009;64:965. doi: 10.1227/01.NEU.0000344150.81021.AA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nie G, Hah HJ, Kim G, Lee YE, Qin M, Ratani TS, Fotiadis P, Miller A, Kochi A, Gao D, Chen T, Orringer DA, Sagher O, Philbert MA, Kopelman R. Small. 2012;8:884. doi: 10.1002/smll.201101607. [DOI] [PubMed] [Google Scholar]
- (44).Ray A, Wang XD, Lee YEK, Hah HJ, Kim G, Chen T, Orringer DA, Sagher O, Liu XJ, Kopelman R. Nano Res. 2011;4:1163. doi: 10.1007/s12274-011-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).D'Autreaux B, Toledano MB. Nat. Rev. Mol. Cell Biol. 2007;8:813. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]