

Abstract
The baculovirus expression system is a powerful tool for expression of recombinant proteins. Here we use it to produce correctly folded and glycosylated versions of the influenza A virus surface glycoproteins - the hemagglutinin (HA) and the neuraminidase (NA). As an example, we chose the HA and NA proteins expressed by the novel H7N9 virus that recently emerged in China. However the protocol can be easily adapted for HA and NA proteins expressed by any other influenza A and B virus strains. Recombinant HA (rHA) and NA (rNA) proteins are important reagents for immunological assays such as ELISPOT and ELISA, and are also in wide use for vaccine standardization, antibody discovery, isolation and characterization. Furthermore, recombinant NA molecules can be used to screen for small molecule inhibitors and are useful for characterization of the enzymatic function of the NA, as well as its sensitivity to antivirals. Recombinant HA proteins are also being tested as experimental vaccines in animal models, and a vaccine based on recombinant HA was recently licensed by the FDA for use in humans. The method we describe here to produce these molecules is straight forward and can facilitate research in influenza laboratories, since it allows for production of large amounts of proteins fast and at a low cost. Although here we focus on influenza virus surface glycoproteins, this method can also be used to produce other viral and cellular surface proteins.
Keywords: Infection, Issue 81, Influenza A virus, Orthomyxoviridae Infections, Influenza, Human, Influenza in Birds, Influenza Vaccines, hemagglutinin, neuraminidase, H7N9, baculovirus, insect cells, recombinant protein expression
Introduction
As a first response to the recent emergence of the novel H7N9 influenza virus strain in China, we acquired plasmids carrying the genomic sequences for the hemagglutinin (HA) and neuraminidase (NA) genes of the first two isolates. Using these plasmids we were able to quickly construct baculoviral expression vectors for production of recombinant HA and NA in insect cells. This expression system is well established in our laboratory and has proven pivotal to many of our ongoing research projects1-8. Insect cells are able to correctly fold and post-translationally modify complex proteins. These modifications include N-linked glycosylation, which is important for many viral glycoproteins. As such, it is not surprising that the baculovirus system is quite established for expressing influenza virus surface antigens. In fact, many of the HA and NA crystal structures have been solved using insect cell expressed proteins9-11. Another advantage of the system lies in its reliable high protein yields of up to 30 mg of HA/L cell culture (based on our experience with more than 50 different HA proteins) - a consequence of virus-infection driven expression from the strong polyhedrin promoter.
Removal of the transmembrane and endodomain of the HA and NA proteins allows for expression of secretable, soluble versions of the proteins and thus greatly facilitates the purification process. In addition, these ectodomains are hexahistidine tagged and can therefore be purified using affinity chromatography using Ni2+-resin. The transmembrane domains of HA and NA proteins contribute to homotrimer and homotetramer formation, respectively. In order to preserve these oligomerizations, we add a C-terminal trimerization domain to the HA-, and a N-terminal tetramerization domain to the NA constructs (Figure 1). We have shown conclusively that such a trimerization domain stabilizes functional, conserved conformational epitopes on the stalk-domain of HA1. The correct tetramerization of the NA might also contribute to correct folding and NA function10. Sequences and baculo-transfer vectors harboring trimerization or tetramerization domains can be requested from the authors or from other laboratories in the field ,9,10,12.
Another important issue for expression of secreted proteins in insect cells is the choice of the right cell line. While Spodoptera frugiperda derived Sf9 cells support baculovirus replication very well and are generally used for virus rescue and propagation, they have limited capacity to secrete large amounts of protein. Trichoplusia ni derived BTI-TN-5B1-4 cells (commonly known as High Five) have a higher secretion capacity and are the cell line of choice for expression in this protocol13,14. Furthermore, it is helpful to reduce or even remove fetal bovine serum (FBS) from expression cultures. We therefore use media with reduced (3%) FBS content for growing the virus working stocks, and we perform the protein expression in serum free media.
Recombinant HA and NA proteins are used for many immunological techniques. Perhaps the most common one is in ELISA assays for measuring serum conversion upon vaccination in humans or animals4,6,7,15. HAs and NAs are also used in ELISPOT assays as both stimulatory molecules and detection reagents for antibody secreting cells16. Their use as molecular baits allows to specifically sort for plasmablasts or other types of B-cells that can then be used to isolate (therapeutic) monoclonal antibodies (mAbs). Recombinant HA and NA molecules are further used in the characterization of these mAbs8. Other examples include study the pH stability or receptor specificity of HAs expressed by different virus isolates, or quantitatively assessing specific NA enzymatic activity or resistance to inhibitors. Furthermore, recombinant, purified HA can be used to standardize HA content of inactivated influenza virus vaccines.
Importantly, rHA (and to a certain degree NA) vaccine candidates are currently being tested in animal models and human clinical trials with one vaccine candidate being licensed by the FDA in 20132,17-19. The advantage of these novel vaccines is that, due to the recombinant nature of these proteins, the labor-intensive process of generating of high growth reassortant viruses could be avoided. Perhaps even more relevant is the fact that their HA yield is high and reproducible from strain to strain. Also, since these vaccines are produced in an egg-free system, they do not contain protein contaminants that are problematic for individuals with egg allergies.
Here we chose to express the HA and NA proteins from two isolates of the novel Chinese H7N9 influenza virus, A/Anhui/1/13 and A/Shanghai/1/1320,21. These are timely examples, but the described protocol can be used for expression of any influenza A and B HA or NA proteins and can be adapted to express any other secreted viral or cellular proteins. Trimeric or tetrameric transmembrane proteins can be cloned into the expression cassettes used for HA and NA, respectively. However, most soluble secreted cellular proteins, like interferons for example, do not need an additional domain for stabilization and can be expressed solely with a C-terminal hexahistidine tag.
Protocol
1. Generation of Recombinant Baculovirus
- Cloning into baculovirus transfer-plasmids
- Clone H7 HA and N9 NA ectodomain (or any other HA or NA gene) into modified pFastBac transfer plasmids (see introduction). Vectors for HA containing a C-terminal trimerization domain and a hexahistidine tag, as well as for NA containing an N-terminal tetramerization domain and hexahistidine tag, have been described1,10,11,22 (Figure 1) and can be requested from the authors. Explanatory note: HA ectodomains expressed without a trimerization domain will be folded incorrectly, leading to loss of conformational neutralizing epitopes, especially in the stalk domain. Similarly, the NA enzymatic function relies on proper tetramerization.
- Generation of bacmids Transform baculovirus transfer-plasmids containing HA and NA genes into DH10Bac bacteria according using a baculovirus expression system per manufacturer's instructions. Isolate and purify bacmids using a Midiprep kit according to manufacturer's instructions (Figure 2).
- Rescue of recombinant baculovirus and verification of protein expression Explanatory note: The following steps have to be performed aseptically in a laminar flow hood. Perform this procedure independently for HA and NA (or any other gene of interest). Insect cells are usually grown at 28 °C without CO2. Sf9 cells for this protocol are grown in full TNM-FH insect cell media supplemented with 10% FBS, 1% Pen-Strep* and 0.1% Pluronic F68 solution, if not indicated otherwise, and passaged 1:4 twice a week. High Five cells are grown in SFX serum free media and passaged 1:15 twice a week. * Addition of antibiotics should be considered optional throughout the protocol and adjusted according to the practices used in each laboratory.
- Plate Sf9 insect cells in 6-well plates at a density of 2 x 105 cells/cm2 and let sit for 20 min in a 28 °C incubator (without CO2).
- Mix 2 μg (conc. 0.5-2 μg/ul) of recombinant bacmid with 100 μl of TNM-FH medium (serum free, 1% Pen-Strep). Mix 6 μl of transfection agent with 100 μl of TNM-FH medium (serum free, Pen-Strep). Combine the two volumes, mix gently and let sit for 20 min at room temperature (transfection mixture). Include one mock-transfection as control (Figure 3).
- Remove supernatant from plated Sf9 cells (cells will now stick to the plate) and replace with 2 ml of serum free TNM-FH medium containing Pen-Strep (1%). Add the complete volume of the transfection mixture (from step 1.2.2) and rock the 6-well plate carefully for 5 sec. Incubate in a 28 °C incubator without CO2 for 6 hr (Figure 3).
- Replace media with fresh full TNM-FH media (containing 10% FBS, 1% Pen-Strep and 0.1% Pluronic F68). Incubate in a 28 °C incubator without CO2 for 6 days. Check cells occasionally under a microscope. Infected cells usually appear bigger, rounder, have enlarged nuclei, and start to detach (Figure 4).
- Harvest of cells and confirmation of protein expression
- Harvest cells 6 days post transfection by centrifugation at 2,000 x g for 5 min at room temperature. The supernatant contains the recombinant baculovirus - this will be used to generate working stocks right away, or can be stored at 4 °C for years.
- To check protein expression collect pellets and resuspend in 500 μl of PBS. Mix 50 μl of the suspension with 50 μl of SDS-PAGE loading buffer (containing a reducing agent). Run SDS-PAGE and perform a Western blot analysis with an anti-hexahistidine antibody to confirm gene expression. Include cells from the mock transfection as negative controls (Figure 3).
- Preparation of working stocks
- Plate 5 x 105 Sf9 cells/cm2 in a T175 cm2 flask and let sit for 20 min allow them to attach to the dish. Replace complete TNM-FH media with TNM-FH media containing 3% FBS (and 1% Pen-Strep, 0.1% Pluronic F68).
- Infect cells with 100 μl of the rescue supernatant. Incubate in a 28 °C incubator without CO2 for 6 days and check occasionally if cells get infected (as described in step 1.2.5.).
- Spin at 2,000 x g at room temperature for 5 min and collect supernatant. This is now your P2 stock. This stock can be stored indefinitely at 4 °C. Repeat the infection procedure using 100 μl of the P2 stock to infect cells. The resulting stock is called P3 or working stock, which can be stored at 4 °C or used directly for protein expression. The P3 stock usually contains 107-109 plaque forming units/ml.
2. Protein Expression and Purification
Explanatory note: Sf9 cells support the growth of baculovirus very well. However, their capacity to secrete large amounts of recombinant protein is limited. Therefore we use another insect cell line, High Five cells, for expression of secreted recombinant proteins. These cells do not support baculovirus replication to very high titers13 but have high secretory capacity.
Growing up High Five cells Grow High Five cells in SFX insect cell culture medium in T175 cm2 flasks. Passage every other day 1:3. For a 200 ml expression culture you will need to grow 5 confluent flasks.
- Harvesting and infecting High Five cells
- Detach High Five cells from 5 T175 cm2 flasks by tapping the flasks with your hand. Collect the cell suspension and transfer into 50 ml centrifugation tubes.
- Spin at 1,200 x g for 7 min at room temperature. Remove supernatant by decanting and unite the pellets by resuspending them in 15 ml of P3 stock.
- Let sit for 15 min in the flow hood (Figure 5).
- Transfer 200 ml of Hyclone SFX culture medium into a clean, sterile 1,000 ml shaker flask (without buffles, sealed with aluminium foil before autoclaving, should not have been used for bacterial cultures before).
- Transfer the P3/cell suspension into the shaker flask. Seal with sterile aluminium foil and transfer flask into a shaker. Shake at 70 rpm at 28 °C for 72-96 hr.
- Harvesting recombinant protein from High Five cell expression supernatants
- 72-96 hr post infection transfer culture supernatants into 500 ml centrifugation buckets. Spin at 5,500 x g for 20 min at 4 °C. In the meantime rinse the shaker flasks 3x with double distilled water (ddH2O).
- Transfer 3 ml Ni-resin slurry into a 50 ml tube with 45 ml of PBS (one needed per 200 ml culture). Shake well and spin at 3,000 x g at room temperature for 10 min. Decant the PBS and keep the pellet. Mix the insect cell supernatant with the Ni-resin pellet and transfer into the already rinsed shaker flasks. Incubate at 4 °C for 2-4 hr, while shaking (Figure 6).
- Mount 10 ml polypropylene column on a clamp on a support stand. Remove both caps and place a beaker below the column to collect flow through (waste). Take the shaker flask out of the shaker and start to transfer the supernatant/slurry mixture onto the column; it will retain the Ni-resin and only the supernatant will flow through. Pass the entire volume over the column (Figure 7).
- Wash the resin 4x with 15 ml of washing buffer (50 mM Na2HCO3, 300 mM NaCl, 20 mM imidazole, pH 8). Let all the washing buffer drain from the column and close it with the cap (lower side).
- Add 2 ml of elution buffer (50 mM Na2HCO3, 300 mM NaCl, 300 mM imidazole, pH 8) and let sit for 5 min. Open column and collect the eluate. Repeat this 3 more times (with a total of 8 ml of elution buffer). Eluate that contains high concentrations of protein typically shows foam when shaken (Figure 7). Keep eluate on wet ice whenever possible.
- Buffer exchange and concentration
- Prespin 15 ml ultrafiltration centrifugation units (30 kDa cut-off for HA/NA but may vary depending on the purified protein size) with PBS (pH 7.4-7.6) at 3,000 x g at 4 °C for 20 min. Discard flow through and load eluate onto the spin column. Fill up with 5 ml of sterile, ice cold PBS and spin for 60 min at 3,000 x g at 4 °C.
- Discharge flow through again, refill with 15 ml of ice cold PBS and spin for 60 min at 3,000 x g at 4 °C. Repeat this procedure one more time.
- Take the spin column out of the centrifuge and collect the buffer exchanged concentrate (typically 200 μl). This is highly concentrated recombinant protein. Rinse the spin column with 400 μl of sterile ice cold PBS and add this to the concentrate. Keep the concentrate on ice at all times.
3. Characterization and Quality Control
Measuring protein concentration Take an aliquot of the concentrate and measure it by using your protein quantification method of choice. Set protein concentration to 1 mg/ml by adding PBS and freeze small aliquots to -80 °C. Freeze thaw cycles harm the protein structure and can lead to aggregation.
SDS-PAGE and Coomassie staining Run 500 ng of your protein on a SDS-PAGE and perform a Coomassie staining. For fast and convenient staining use Coomassie reagent in combination with a microwave protocol. HA proteins typically appear at 64 kDa whereas NA proteins should run at a size of around 56 kDa (Figures 8A and B). Using the described protocol you should not see any degradation products. If degradation occurs it is usually a sign of yeast/fungal contamination during expression. In this case repeat the procedure and make sure to keep everything sterile.
Western blot/ELISA In order to verify protein identity we recommend do to a Western blot or ELISA assay with a specific antibody. Ideally, ELISA assays are performed with an antibody that binds to conformational epitopes (e.g. anti-HA stalk antibodies1). Using such an antibody the correct folding of the protein can be confirmed (Figure 8C).
Measuring NA activity Correct folding of the viral NA can also be determined by measuring NA activity. We recommend to perform this test using the commercially available NA*STAR assay (just follow the user guidelines).
Representative Results
HA molecules from A/Shanghai/1/13 and A/Anhui/1/13 expressed well with yields of about 20 mg/L culture. NA molecules of both strains showed moderate expression levels ranging from 0.2 mg/L culture for A/Shanghai/1/13 to 0.7 mg/L for A/Anhui/1/13. All four protein preparations had very little to no impurities (Figures 8A and B) with HAs being of higher purity than NAs which can be explained by the expression level. A/Shanghai/1/13 HA was further analyzed using ELISA with the broadly neutralizing stalk-reactive human mAb CR911423. This mAb binds to a sensitive conformational epitope in the stalk domain and is used here as a probe for correct folding of this domain. The antibody shows good binding which gives confidence that the HA is indeed folded correctly (Figure 8C). A second ELISA with anti-H7 mouse polyclonal serum was performed to confirm the identity of the obtained protein as of H7 origin (Figure 8D).
 Figure 1. Schematic of HA and NA expression constructs. The expression construct for the HA (A) contains an N-terminal signal peptide followed by the HA ectodomain, a thrombin cleavage site, a T4 trimerization domain and a hexahistidine tag. The NA expression construct (B) contains an N-terminal signal peptide followed by a hexahistidine tag, a vasodilator stimulating phosphoprotein (VASP) tetramerization domain and the NA ectodomain. Click here to view larger figure.
Figure 1. Schematic of HA and NA expression constructs. The expression construct for the HA (A) contains an N-terminal signal peptide followed by the HA ectodomain, a thrombin cleavage site, a T4 trimerization domain and a hexahistidine tag. The NA expression construct (B) contains an N-terminal signal peptide followed by a hexahistidine tag, a vasodilator stimulating phosphoprotein (VASP) tetramerization domain and the NA ectodomain. Click here to view larger figure.
 Figure 2. Scheme of bacmid generation (as described in step 1.1.2). A baculovirus-transfer vector containing HA or NA genes is transformed into DH10Bac bacteria. Recombination into the baculovirus genome that these bacteria carry produces a white colony phenotype on selection plates. A white colony is picked and restreaked. A single colony from the restreaked plate is used to inoculate a midiprep culture.
Figure 2. Scheme of bacmid generation (as described in step 1.1.2). A baculovirus-transfer vector containing HA or NA genes is transformed into DH10Bac bacteria. Recombination into the baculovirus genome that these bacteria carry produces a white colony phenotype on selection plates. A white colony is picked and restreaked. A single colony from the restreaked plate is used to inoculate a midiprep culture.
 Figure 3. Scheme of baculovirus rescue and working stock generation (as described in steps 1.2.1-6). Recombinant bacmid and transfection reagent are diluted in serum free TNM-FH medium. The two solutions are combined, the mix is incubated for 20 min and then transferred onto Sf9 cells. Six hours post transfection the transfection media is replaced by full TNM-FH media. Six days post transfection the supernatant is harvested and the cell pellet is analyzed for expression of recombinant proteins.
Figure 3. Scheme of baculovirus rescue and working stock generation (as described in steps 1.2.1-6). Recombinant bacmid and transfection reagent are diluted in serum free TNM-FH medium. The two solutions are combined, the mix is incubated for 20 min and then transferred onto Sf9 cells. Six hours post transfection the transfection media is replaced by full TNM-FH media. Six days post transfection the supernatant is harvested and the cell pellet is analyzed for expression of recombinant proteins.
 Figure 4. Healthy and infected Sf9 cells. Sf9 cells in (A) are healthy and uninfected. They are attached to the culture dish, look partially polymorphic and relatively small. Cells in (B) are infected with baculovirus, detached from the culture dish, are big and round and have enlarged nuclei. The nucleus also shows a granular appearance.
Figure 4. Healthy and infected Sf9 cells. Sf9 cells in (A) are healthy and uninfected. They are attached to the culture dish, look partially polymorphic and relatively small. Cells in (B) are infected with baculovirus, detached from the culture dish, are big and round and have enlarged nuclei. The nucleus also shows a granular appearance.
 Figure 5. Infection of High Five cells with recombinant baculovirus (working stock, as described in step 2.2). Five confluent T175 flasks with High Five cells are harvested by low-speed centrifugation and combined with 15 ml of P3 baculovirus stock. After 15 min of incubation the cell/virus mixture is transferred into 1,000 ml shaker flasks with 200 ml of SFX media.
Figure 5. Infection of High Five cells with recombinant baculovirus (working stock, as described in step 2.2). Five confluent T175 flasks with High Five cells are harvested by low-speed centrifugation and combined with 15 ml of P3 baculovirus stock. After 15 min of incubation the cell/virus mixture is transferred into 1,000 ml shaker flasks with 200 ml of SFX media.
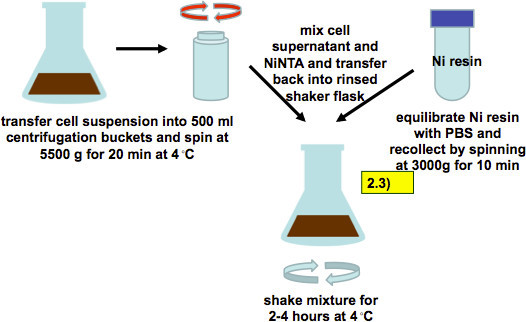 Figure 6. Harvest of cell supernatant 72-96 hr post infection and incubation with Ni-NTA resin (as described in step 2.3). Infected cell suspensions are harvested by low speed centrifugation. Culture supernatants are mixed with PBS-equilibrated Ni-resin and incubated for 2-4 hr while shaking.
Figure 6. Harvest of cell supernatant 72-96 hr post infection and incubation with Ni-NTA resin (as described in step 2.3). Infected cell suspensions are harvested by low speed centrifugation. Culture supernatants are mixed with PBS-equilibrated Ni-resin and incubated for 2-4 hr while shaking.
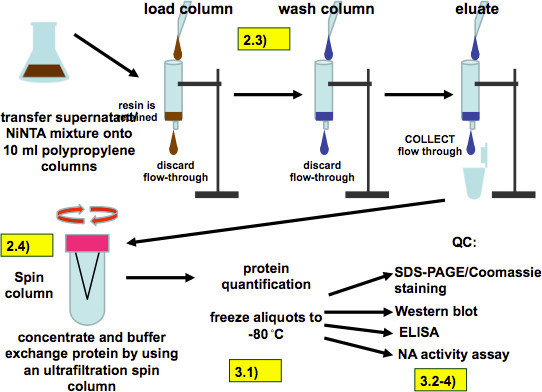 Figure 7. Purification procedure (as described in 2.3 and 2.4) and quality control (as described in steps 3.1.1-4). After incubation the all of the supernatant/resin mixture is loaded on polypropylene columns. The resin is retained by the columns and is then washed four times with washing buffer and the recombinant protein is eluted with 8 ml of elution buffer applied in 2 ml steps. The eluate is then buffer exchanged and concentrated with an ultrafiltration spin column, aliquoted and frozen. Quality control assays include SDS-PAGE, Western Blot, ELISA and measurement of the NA activity of recombinant NA.
Figure 7. Purification procedure (as described in 2.3 and 2.4) and quality control (as described in steps 3.1.1-4). After incubation the all of the supernatant/resin mixture is loaded on polypropylene columns. The resin is retained by the columns and is then washed four times with washing buffer and the recombinant protein is eluted with 8 ml of elution buffer applied in 2 ml steps. The eluate is then buffer exchanged and concentrated with an ultrafiltration spin column, aliquoted and frozen. Quality control assays include SDS-PAGE, Western Blot, ELISA and measurement of the NA activity of recombinant NA.
 Figure 8. Representative results. HA (A) and NA (B) proteins of A/Anhui/1/13 and A/Shanghai/1/13 were analyzed using SDS-PAGE and Coomassie staining. Structural integrity of the HA of A/Shanghai/1/13 was assessed using the broadly neutralizing human stalk-reactive antibody CR9114 that binds to a sensitive conformational epitope (C). The identity of the same HA was confirmed by a polyclonal anti-H7 mouse serum (D).
Figure 8. Representative results. HA (A) and NA (B) proteins of A/Anhui/1/13 and A/Shanghai/1/13 were analyzed using SDS-PAGE and Coomassie staining. Structural integrity of the HA of A/Shanghai/1/13 was assessed using the broadly neutralizing human stalk-reactive antibody CR9114 that binds to a sensitive conformational epitope (C). The identity of the same HA was confirmed by a polyclonal anti-H7 mouse serum (D).
Discussion
Although expression of influenza virus HA and NA proteins in insect cells using the baculovirus expression system is generally straight forward, there are several important points to consider. It is important, as pointed out in the introduction, that the ectodomains of HA and NA are expressed in fusion with trimerization or tetramerization domains, respectively. These domains ensure correct folding of the molecules usually assisted by the transmembrane domain, which is not present if only the ectodomain is expressed. For example, the expression of the HA ectodomain without a trimerization domain leads to oligomerization and destabilization of important neutralizing epitopes in the stalk domain1. Further, the stability of the proteins is decreased and degradation by insect cell and baculovirus proteases occurs. The choice of the trimerization or tetramerization domain is less important, a number of them ranging from leucine zipper domains22 to T4 phage foldons11 have been tested so far and all showed satisfying results. It is important to keep in mind that multibasic cleavage sites as they occur in highly pathogenic H5N124 or H7N725 isolates (but not in H7N9) have to be removed before expression since they are cleaved by furin like proteases which are present at all times in the baculovirus expression system1,12.
Another possible reason for degradational products is contamination with yeast or mold. This can happen as early as during working stock generation and can go unnoticed during storage of stocks at 4 °C. Many yeasts and molds prefer temperatures between 20-30 °C for optimal growth and are actually inhibited at 37 °C, which is common culture temperature for mammalian cells. Since most antibiotics that are used in cell culture are not efficacious against eukaryotic organisms like yeast and mold, and insect cell culture is performed at optimal growth conditions of these germs extra care has to be taken in terms of aseptically handling of cultures. In addition to overgrowing insect cells and acidifying media, molds and yeasts express many soluble proteases and can therefore completely degrade recombinant proteins.
A problem that sometimes occurs is precipitation in the elution buffer. This is rarely seen and more or less strain specific (e.g. A/Vietnam/1203/04 H5 HA tends to precipitate), but can be prevented by quick and complete buffer exchange to PBS. Storage of proteins in imidazole-containing elution buffer is not recommended. The purification procedure from harvest to the freezing of aliquots to -80 °C should be performed within one working day and storage of purified protein at temperature other than -80 °C should be avoided. Also, repeated freeze-thaw cycles have been proven to impact negatively on stability of certain epitopes1.
Yields should be expected to be in the range of 0.2-30 mg/L culture. Many HAs express in the upper range while NAs tend to express at lower levels. Also, expression levels for HAs seem to correlate inversely with the number of glycosylation sites. HAs from human seasonal H3N2 and seasonal prepandemic H1N1 isolates usually show low expression levels, whereas most HAs of avian influenza virus origin have high expression levels. To improve expression it is recommended to increase the cell density in the flask or to try to use less or more working stock per expression. 10 fold increase or decrease of the working stock can impact positively on expression levels. Another factor that can impact on expression levels is the time between infection and harvest. 96 hr are ideal for most HAs and NAs but shorter incubation times might actually increase yields of some proteins.
Insect cells are able to perform posttranslational modifications in an almost mammalian-like manner and are able to fold very complex protein structures with similar efficiencies as mammalian cells (but producing higher yields). In terms of glycosylation, they exhibit a pattern that is slightly different from mammalian cells and features mostly paucimannosidic N-glycans that lack sialic acid modifications19. However, insect cell lines that have been modified to produce mammalian like glycosylation (including terminal sialic acid) are commercially available14.
The baculovirus expression system is an extremely useful tool for expression of the recombinant influenza surface glycoproteins - HA and NA. These recombinant proteins are used for many immunological and biochemical assays, as well as for standardization of antigen content of vaccines and as antigen for vaccination experiments in animal models. Baculovirus-expressed HA and NA exhibit full biological function and correct folding if expressed in frame with trimerization and tetramerization domains respectively. This distinguishes them from bacterial expressed HAs that are non-glycosylated and lack functional epitopes in the stalk domain. Furthermore, the baculovirus expression system exhibits significantly higher protein yields than mammalian cell systems26 and handling and upscaling are easier. In summary we describe an easy and efficient protocol for expressing large amounts of recombinant influenza virus HA and NA molecules.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Patrick C. Wilson for monoclonal antibody CR9114. We also thank Jennifer Debeauchamp and Richard Webby for the original A/Anhui/1/13 plasmids. Partial support for this work was provided by the National Institute for Allergy and Infectious Diseases-funded Program Project Grant AI097092-01A1 and CEIRS (Centers of Excellence for Influenza Research and Surveillance, HHSN26620070010C). FK was supported by an Erwin Schrödinger fellowship (J 3232) from the Austrian Science Fund (FWF).
References
- Krammer F, et al. A carboxy-terminal trimerization domain stabilizes conformational epitopes on the stalk domain of soluble recombinant hemagglutinin substrates. PLoS One. 2012;7:e43603. doi: 10.1371/journal.pone.0043603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F, Pica N, Hai R, Margine I, Palese P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly-protective stalk-specific antibodies. J. Virol. 2013. [DOI] [PMC free article] [PubMed]
- Leyva-Grado VH, Mubareka S, Krammer F, Cárdenas WB. Influenza virus infection in Guinea pigs raised as livestock, ecuador. Emerg. Infect. Dis. 2012;18:1135–1138. doi: 10.3201/eid1807.111930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margine I, et al. H3N2 influenza virus infection induces broadly reactive hemagglutinin stalk antibodies in humans and mice. J. Virol. 2013. [DOI] [PMC free article] [PubMed]
- Miller MS, et al. 1976 and 2009 H1N1 Influenza Virus Vaccines Boost Anti-Hemagglutinin Stalk Antibodies in Humans. J. Infect. Dis. 1976;652 doi: 10.1093/infdis/jis652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pica N, et al. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. U.S.A. 2012;109:2573–2578. doi: 10.1073/pnas.1200039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangster MY, et al. The B cell response and hemagglutinin stalk-reactive antibody production in different age cohorts following 2009 H1N1 influenza vaccination. Clin. Vaccine Immunol. 2013. [DOI] [PMC free article] [PubMed]
- Tan GS, et al. A pan-h1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J. Virol. 2012;86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 2008;82:10493–10501. doi: 10.1128/JVI.00959-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, et al. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- Wei CJ, et al. Comparative efficacy of neutralizing antibodies elicited by recombinant hemagglutinin proteins from avian H5N1 influenza virus. J. Virol. 2008;82:6200–6208. doi: 10.1128/JVI.00187-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer F, et al. Trichoplusia ni cells (High Five) are highly efficient for the production of influenza A virus-like particles: a comparison of two insect cell lines as production platforms for influenza vaccines. Mol. Biotechnol. 2010;45:226–234. doi: 10.1007/s12033-010-9268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmberger D, et al. Insect cells for antibody production: evaluation of an efficient alternative. J. Biotechnol. 2011;153:160–166. doi: 10.1016/j.jbiotec.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Santiago FW, Lambert Emo K, Fitzgerald T, Treanor JJ, Topham DJ. Antigenic and immunogenic properties of recombinant hemagglutinin proteins from H1N1 A/Brisbane/59/07 and B/Florida/04/06 when produced in various protein expression systems. Vaccine. 2012;30:4606–4616. doi: 10.1016/j.vaccine.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrammert J, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011;208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor JJ, et al. Protective efficacy of a trivalent recombinant hemagglutinin protein vaccine (FluBlok) against influenza in healthy adults: a randomized, placebo-controlled trial. Vaccine. 2011;29:7733–7739. doi: 10.1016/j.vaccine.2011.07.128. [DOI] [PubMed] [Google Scholar]
- Dalakouras T, Smith B, Platis D, Cox M, Labrou N. Development of recombinant protein-based influenza vaccine. Expression and affinity purification of H1N1 influenza virus neuraminidase. J. Chromatogr. A. 2006;1136:48–56. doi: 10.1016/j.chroma.2006.09.067. [DOI] [PubMed] [Google Scholar]
- Krammer F, Grabherr R. Alternative influenza vaccines made by insect cells. Trends Mol. Med. 2010;16:313–320. doi: 10.1016/j.molmed.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Gao R, et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013. [DOI] [PubMed]
- Goff PH, et al. Induction of cross-reactive antibodies to novel H7N9 influenza virus by recombinant Newcastle disease virus expressing a North American lineage H7 subtype hemagglutinin. J. Virol. 2013. [DOI] [PMC free article] [PubMed]
- Weldon WC, et al. Enhanced immunogenicity of stabilized trimeric soluble influenza hemagglutinin. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel J, et al. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 2009;83:1742–1753. doi: 10.1128/JVI.01920-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1356–1361. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kost T, Condreay J, Jarvis D. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 2005;23:567–575. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]