

Abstract
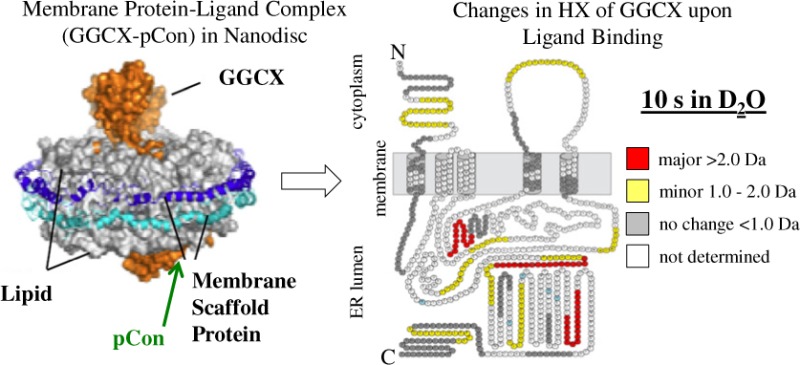
Gamma (γ)-glutamyl carboxylase (GGCX) is an integral membrane protein responsible for the post-translational catalytic conversion of select glutamic acid (Glu) residues to γ-carboxy glutamic acid (Gla) in vitamin K-dependent (VKD) proteins. Understanding the mechanism of carboxylation and the role of GGCX in the vitamin K cycle is of biological interest in the development of therapeutics for blood coagulation disorders. Historically, biophysical investigations and structural characterizations of GGCX have been limited due to complexities involving the availability of an appropriate model membrane system. In previous work, a hydrogen exchange mass spectrometry (HX MS) platform was developed to study the structural configuration of GGCX in a near-native nanodisc phospholipid environment. Here we have applied the nanodisc–HX MS approach to characterize specific domains of GGCX that exhibit structural rearrangements upon binding the high-affinity consensus propeptide (pCon; AVFLSREQANQVLQRRRR). pCon binding was shown to be specific for monomeric GGCX-nanodiscs and promoted enhanced structural stability to the nanodisc-integrated complex while maintaining catalytic activity in the presence of carboxylation co-substrates. Noteworthy modifications in HX of GGCX were prominently observed in GGCX peptides 491–507 and 395–401 upon pCon association, consistent with regions previously identified as sites for propeptide and glutamate binding. Several additional protein regions exhibited minor gains in solvent protection upon propeptide incorporation, providing evidence for a structural reorientation of the GGCX complex in association with VKD carboxylation. The results herein demonstrate that nanodisc–HX MS can be utilized to study molecular interactions of membrane-bound enzymes in the absence of a complete three-dimensional structure and to map dynamic rearrangements induced upon ligand binding.
Vitamin K-dependent γ-glutamyl carboxylase (GGCX) is a 758-amino acid integral membrane glycoprotein1 with five transmembrane domains and a disulfide bond between residues 99 and 450.2,3 Topological studies reveal that each transmembrane domain passes through the endoplasmic reticulum (ER) with the N-terminus of the protein in the cytoplasm and C-terminus in the lumen.2 In the presence of co-substrates—vitamin K hydroquinone (KH2), oxygen, and carbon dioxide—GGCX modifies select glutamic acid (Glu) residues to γ-carboxy glutamic acid (Gla) in vitamin K-dependent (VKD) proteins. Concomitant with VKD carboxylation and the formation of vitamin K 2,3-epoxide (KO) is the catalytic regeneration of KH2 involving vitamin K epoxide reductase (VKOR). As a result of the interdependence of proteins in the vitamin K cycle, depletion of KH2 and consequently reduced carboxylation are shown to result when VKOR is inhibited by Warfarin, a commonly used anticoagulant.4
GGCX recognizes VKD protein substrates through an 18-amino acid region, termed the propeptide. An alignment of multiple propeptide sequences is illustrated in Supplemental Figure S1, Supporting Information where the height of stacked amino acids indicates the frequency of that amino acid at a particular position.5,6 If one of the highly conserved amino acid residues (phenylalanine at −16, alanine at −10, or leucine at −6) is mutated, substrate carboxylation is diminished or eliminated, under most circumstances.7,8 Despite the high degree of propeptide sequence homology, substrate binding affinity to GGCX varies over 100-fold with the consensus and factor X propeptides showing the highest affinity (lowest Kd) and prothrombin the lowest affinity.9 The explanation for this variability is not yet known; however, the binding of a VKD protein to GGCX is concluded to be a cooperative event between the propeptide and the Gla domain.10
Current understanding suggests the propeptide region tightly binds GGCX between residues 495–51311,12 initiating a structural reorientation by which the Gla domain of the VKD substrate (approximately 45 amino acids)13−15 is positioned at the catalytic active site of GGCX. In the presence of KH2, select glutamic acid residues in the Gla domain are carboxylated to γ-carboxy glutamic acid. Carboxylation of the VKD protein continues in a processive manner whereby all impending carboxylations for a given Gla domain take place in a single binding event.16,17 Once GGCX converts the selected Glu residues in the VKD protein to Gla residues, the propeptide is proteolytically excised and the protein is secreted from the cell.18 In the presence of calcium, the mature VKD protein undergoes a conformational reorientation allowing association with phospholipids on the membrane surface near damaged vascular tissue. An elegant theory explaining the mechanism of carboxylation using a base amplification chemical model was proposed by Dowd and colleagues.19 That work spawned decades of mutational studies3,20−27 and quantum modeling28−30 in search of potential GGCX active site residues shown to affect enzymatic activity.
Important clues concerning the location of the glutamate binding site in GGCX have come from a naturally occurring mutant form of GGCX with a point mutation at position 394 (L394R).31,32 Patients with this mutation exhibit decreased coagulation activity of VKD clotting factors resulting in bleeding diathesis. In vitro substitutions of L394 and neighboring residues Y395 and W399 resulted in defective glutamate binding and significant inhibition of carboxylation.32 That work provided evidence that glutamate recognition is the primary function of the highly conserved domain between residues 393–404 in GGCX. In addition to playing a major role in the binding of the glutamate substrate, L394R and W399A were also shown to be involved in the propeptide binding.32
Extensive research has been conducted on the biochemistry of GGCX, as described above, yet unraveling the structural associations of protein complexes in the vitamin K cycle has been severely limited due to complexities in sample preparation including protein aggregation, oligomerization, or precipitation in solution. To surmount the challenges of investigating the structural characterization of GGCX, hydrogen exchange mass spectrometry (HX MS) of membrane protein-embedded nanodiscs has recently been reported.33 Formation of nanodiscs is an empirical process that, when optimized, promotes stabilization of a target membrane protein into a nanoscale phospholipid bilayer encompassed by an amphipathic membrane scaffold protein (MSP). Previous studies investigating membrane proteins by HX MS have been reported using lipid vesicles or detergent micelles.34−41 The use of nanodiscs, however, enables selective labeling methods such as hydrogen exchange to occur within an environment that mimics native membrane conditions. In this paper, we report nanodisc–HX of GGCX as compared with nanodisc–HX of GGCX–propeptide complexes. Comparing deuterium uptake signatures for GGCX with and without the presence of propeptide provides a dynamic visual mapping of localized structural fluctuations upon binding to the substrate. Utilizing HX MS to investigate membrane protein–ligand interactions in controlled single-molecule nanodisc environments uniquely enables investigation of conformational changes in membrane proteins induced by substrate, drug, or protein interactions.
Experimental Procedures
GGCX Purification
Wild-type human GGCX with a carboxyl-terminal 12-amino acid epitope tag (EDQVDPRLIDGK) was isolated from Sf9 cells, as previously described.9 Solubilized GGCX microsomes were purified using a calcium-dependent HPC4 antibody resin (Dr. Charles T. Esmon, Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK)42 and eluted in 25 mM MOPS pH 7.5, 0.1 M NaCl, 15% glycerol, 0.5% CHAPS, 0.2% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 10 mM EDTA, stabilized by a protease inhibitor cocktail (aprotinin, leupeptin, pepstatin, and phenylmethylsulfonyl fluoride). DOPC phospholipid was purchased from Avanti Polar Lipids (Alabaster, AL), and protease inhibitors were from Roche Applied Science (Indianapolis, IN). Purified protein was stored at −80°C. Protein concentration and purity were determined by enzymatic activity assay measurements and SDS-PAGE.
Membrane Scaffold Protein (MSP1D1) Expression and Purification
MSP1D1 expression and purification were performed as previously described.43 Briefly, the MSP1D1 plasmid with a polyhistidine tag (Addgene plasmid 20061) was expressed in Escherichia coli BL21 Codon Plus (DE3) cells. The protein was isolated by Ni-NTA Agarose (QIAGEN, Valencia, CA) affinity purification, and purity was confirmed by polyacrylamide gel electrophoresis (PAGE). Fractions containing MSP1D1 were pooled and dialyzed against standard buffer (20 mM Tris-HCl pH 7.4, 0.1 M NaCl, 0.5 mM EDTA, and 0.01% NaN3). Purified protein was lyophilized and stored at −20°C. Protein concentration was determined by absorbance at 280 nm using calculated extinction coefficients (ε280nm = 21000 M–1 cm–1 for MSP1D1).
Self-Assembly of Nanodiscs
Self-assembly of GGCX-nanodiscs was initiated as previously reported.33 Phospholipid stock solutions were prepared in chloroform, and the concentration was determined by phosphate analysis.44,45 A DOPC/deoxycholate (2:1) solubilized mixture was added to the purified microsomal GGCX target protein. The equilibrated mixture was added to an excess of purified MSP1D1 and 50 mM Tris-HCl pH 7.4, 0.15 M NaCl, and 0.02% NaN3 (TBS) buffer. Samples were incubated at 4°C. Self-assembly was initiated upon detergent removal during a 2 h gentle rotation with damp SM-2 BioBeads (BioRad, Hercules, CA). For GGCX-nanodisc preparations in the presence of consensus propeptide [pCon] (AVFLSREQANQVLQRRRR) or fluorescein-labeled consensus propeptide [FpCon] (Fl-AVFLSREQANQVLQRRRR), the reaction mixture was first incubated with a 10-fold excess of propeptide for 45 min at 4°C prior to the addition of solubilized phospholipid.
Nanodisc preparations were purified by size-exclusion chromatography (SEC) monitoring the absorbance of the sample eluent at 280 nm. Separation was completed on a Tosoh TSKgel BioAssist G3SWXL 7.8 × 300 mm (5 μm particles, 250 Å) column using 50 mM Tris-HCl pH 7.0, 0.15 M NaCl, 0.02% NaN3 (TBS) mobile phase at 0.5 mL/min. Fractions containing purified GGCX-nanodiscs were isolated and concentrated by Millipore (Bedford, MA) Microcon YM-30 centrifugal filters. Purified nanodiscs were stored at 4°C for up to one month. Protein concentration and purity were determined by enzymatic activity assay measurements and SDS-PAGE.
SDS-PAGE
Purified nanodiscs were analyzed by SDS-PAGE on NuPAGE Novex 4–12% Bis–Tris gradient gels (Invitrogen Life Sciences, Grand Island, NY). SDS-PAGE buffers were prepared according to a protocol derived from Invitrogen Life Sciences including a 4× SDS-PAGE loading buffer and a 20× MES running buffer. PAGE gels were stained using SYPRO Ruby stain (Invitrogen Life Sciences) and imaged using a 300 nm blue light UV transilluminator (Alpha Innotech AlphaImager 2200) with interference wavelength filtering. For protein molecular weight referencing, the Mark 12 unstained protein ladder (Invitrogen Life Sciences) was loaded on each gel.
In Vitro Carboxylation
GGCX in vitro activity was determined by measuring the carboxylation of a synthetic pentapeptide substrate FLEEL (Phe-Leu-Glu-Glu-Leu) in the presence of excess factor IX propeptide [pFIX] (TVFLDHENANKILNRPKR) or incorporated consensus propeptide [pCon] (AVFLSREQANQVLQRRRR) and co-substrates KH2, oxygen, and radiolabeled CO2 in the form of NaH14CO3 (specific activity 55 mCi/mmol; MP Biomedicals, Solon, OH). Vitamin K1(25) phylloquinone, detergent-stabilized emulsion (Hospira Incorporated, Lake Forest, IL), or vitamin K3 menadione, water-soluble sodium bisulfite analogue, (Sigma Aldrich, St. Louis, MO) was utilized in reaction mixtures containing microsomal GGCX or GGCX-embedded nanodiscs, respectively. Vitamin K was reduced to KH2 by overnight incubation with buffer containing 25 mM Tris-HCl pH 8.5, 0.5 M NaCl, and 0.2 M dithiothreitol at 37°C.
The reagent mixture (25 mM MOPS pH 7.5, 0.50 M NaCl, CHAPS, DOPC, 30 mM FLEEL, and 2.4 μM pFIX) was allowed to incubate with microsomal GGCX or GGCX-nanodiscs for 30 min on ice. The concentration of CHAPS and DOPC in the assay reaction mixture was modified to optimize conditions for sample carboxylation such that 0.16% CHAPS/DOPC was incorporated into the assay reconstitution mixture for microsomal GGCX, and no additional CHAPS/DOPC was included for assays evaluating GGCX-nanodiscs. An excess of 222 μM (5.5 mM stock) KH2 phylloquinone or 111 μM (2.75 mM stock) KH2 menadione with 5 μL of an 18.4 mM NaH14CO3 stock solution was added to the reaction mixture for a final volume of 125 μL. The carboxylation assay was allowed to proceed for 120 min at 20°C. The carboxylated product was precipitated with 5% trichloroacetic acid and boiled to remove excess 14CO2. The radioactivity was determined by scintillation counting (Beckman LS5000TD).
Hydrogen Exchange Mass Spectrometry
Deuterium exchange of GGCX-embedded nanodiscs was carried out as described previously.33 A total of 50 pmol of GGCX-nanodiscs (100 pmol of MSP1D1; 6 nmol of DOPC) was diluted 10-fold in 99% deuterium oxide buffer (50 mM Tris-HCl, 0.15 M NaCl, 0.02% NaN3, D2O, pD 7.0) at room temperature. At times ranging from 10 s to 4 h after the introduction of D2O, the sample was quenched to pH 2.5 by the addition of formic acid and placed on ice. To retain the deuterium that exchanged into the protein, the sample was maintained under quench conditions (pH 2.5; 0°C) for all sample handling steps prior to analysis by mass spectrometry.46 Upon quenching, nanodiscs were immediately disassembled with the addition of 25:1 sodium cholate/DOPC. Digestion was completed for 5 min on ice with porcine pepsin-immobilized Poros 20AL resin (Applied Biosystems, Carlsbad, CA).47 In the last minute of digestion, 3 mg of ZrO2-coated silica resin was added to the digestion mixture to remove DOPC phospholipids. The sample was passed across a chilled filter (0.45 μm cellulose acetate) by centrifugation at 4°C for 1 min and loaded immediately onto a refrigerated UPLC-MS system for chromatography and mass spectral analysis. Empty nanodiscs and GGCX-pCon-nanodiscs were analyzed in a similar manner. For purified propeptide, 25 μL of a 2 μM solution was used in the continuous labeling HX reaction. The digestion protocol followed that of the nanodisc procedure as outlined above.
Chromatography was completed on a Waters nanoAcquity UPLC system (Milford, Massachusetts) designed for HX MS.48 The peptic peptides were trapped on a Waters VanGuard column packed with 1.7 μm C18 particles (2.1 mm × 5.0 mm) and desalted with 0.1% formic acid in water for 5 min at 100 μL/min. The trap column was placed in line with a lipid trap column (Waters VanGuard; 1.7 μm C18 particles, 2.1 mm × 5.0 mm) directly connected to the analytical column (Waters BEH 1.7 μm C18 particles, 1.0 mm × 100 mm). The peptides were eluted with an 8–40% gradient of acetonitrile in 0.05% formic acid (pH 2.5) over 12 min at a flow rate of 40 μL/min.
Mass spectral analyses were carried out on a Waters QToF Premier equipped with a standard ESI source and lock mass calibration using [Glu1]-fibrinopeptide B (200 fmol/μL). The mass spectrometer was operated with a capillary voltage of 3.5 kV, a cone voltage of 40 V, a source temperature of 80°C, a desolvation temperature of 175°C, and desolvation gas at 600 L/h. Each 0.5 s scan spanned m/z 200–2000 with an interscan delay time of 0.05 s. Spectra were collected in positive ion mode and processed with MassLynx software. Undeuterated sample controls for both GGCX-nanodiscs and empty nanodiscs were analyzed in triplicate using a Waters MSE acquisition method. Identification of peptic peptides from MSE data was performed using ProteinLynx Global Server 2.4 processing software operated with a custom protein sequence library and nonspecific enzyme digestion specificity. Relative deuterium uptake curves were generated using HX-Express software49 for peptides identified in two out of three replicate injections. Deuterium incorporation was not corrected for back-exchange, representing relative, rather than absolute changes in deuterium levels.48 The error between biological replicates with this experimental system was characterized previously33,50 and similarly here did not exceed ±0.15 Da at each time point. For this work, differences in the relative deuterium level between propeptide-bound and unbound GGCX greater than ±1.0 Da were reported as having a distinguishable change in protein conformation. Relative deuterium uptake curves for all identified GGCX peptides are reported in Supplemental Figure S2, Supporting Information for sample conditions with and without the incorporation of consensus propeptide.
Results
Formation of GGCX-pCon-Nanodiscs
GGCX-nanodiscs33 and GGCX-pCon-nanodisc complexes were prepared using a 1200:20:1 ratio of DOPC/MSP1D1/GGCX protein. The consensus propeptide (Kd = 0.083 ± 0.005 nM at 4°C)51 was chosen as a target substrate based on its high binding affinity for GGCX. Homogenous self-assembly of GGCX-pCon-nanodiscs was evaluated by size-exclusion chromatography (Figure 1) where propeptide-bound GGCX-nanodiscs (peak II) were isolated from an unassociated aggregate (peak I) and empty nanodiscs (peak III). Purified protein standards for GGCX (∼94 kDa, lane A) and MSP1D1 (24 kDa, lane B) were provided to identify protein components in concentrated size-exclusion fractions (Figure 1, gel inset). A fluorescently labeled consensus propeptide analogue (FpCon; 2.6 kDa) was used to demonstrate incorporation of pCon into GGCX-nanodiscs. A comparison of chromatographic features between the visible spectrum at 492 nm (orange) and an overlaid absorbance spectrum at 280 nm (blue) revealed that the high affinity fluorescently labeled substrate selectively incorporates into GGCX-nanodiscs (peak II) with only a small fraction shown to aggregate in an unassociated complex at the void volume of the chromatographic separation (peak I).
Figure 1.
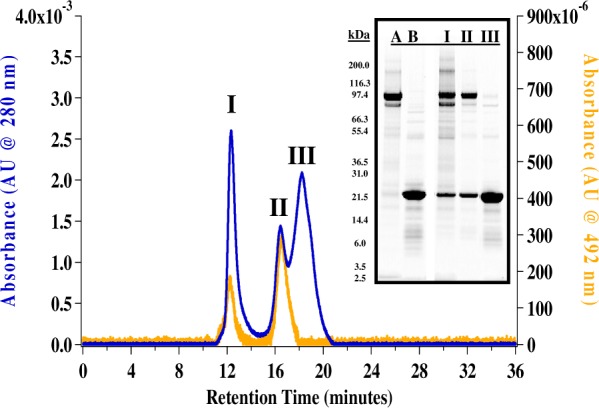
Size-exclusion chromatograms (SEC) overlaying self-assembled GGCX-FpCon-nanodiscs at 280 nm (blue) and 492 nm (orange). Chromatographic peaks labeled (I), (II), (III) correspond to labeled SDS-PAGE gel lanes (inset). GGCX (∼94 kDa) and MSP1D1 (24 kDa) purified protein standards are in gel lanes (A) and (B), respectively.
Fluorescence anisotropy was used to determine the stoichiometric ratio of pCon to GGCX in embedded nanodiscs.52 In the absence of a buffer solution containing detergent or excess phospholipid, the titration of purified GGCX-nanodiscs against a constant concentration of FpCon followed an analogous stoichiometric anisotropy profile to that of microsomal GGCX, suggesting one propeptide molecule is incorporated into one active GGCX-nanodisc (data not shown). The ability to incorporate and isolate monoisotopic GGCX-pCon-nanodiscs provides a sophisticated near-native working environment for elucidating dynamic conformational changes of GGCX and co-substrate interactions involved in the vitamin K cycle. For the remainder of the work presented, GGCX-nanodiscs were prepared with the non-fluorescein-labeled consensus propeptide (pCon).
Modifications to In Vitro GGCX Activity Assay
GGCX-nanodisc catalytic functionality was confirmed by monitoring carboxylation of the pentapeptide substrate FLEEL after protein incubation at 25°C for up to 24 h (Figure 2). For microsomal GGCX, the assay was carried out with optimized ratios of lipid, detergent, and detergent-solubilized KH2 phylloquinone to maximize catalytic efficiency. The formation of GGCX-nanodiscs, however, provides a water-soluble environment no longer stable in the presence of excess detergent.53 As a result, the water-soluble KH2 menadione analogue was utilized in the absence of excess lipid and detergent to improve catalytic efficiencies by increasing the solubility and reactivity with the active domain of the nanodisc-embedded GGCX. While GGCX microsomes lose catalytic functionality almost immediately upon exposure to ambient conditions, GGCX-nanodiscs remain in a protected environment preventing loss of activity for several hours at 25°C (Figure 2). In both microsomal and GGCX-nanodisc samples, incorporation of the high-affinity pCon substrate maximized levels of carboxylation activity at 25°C.
Figure 2.
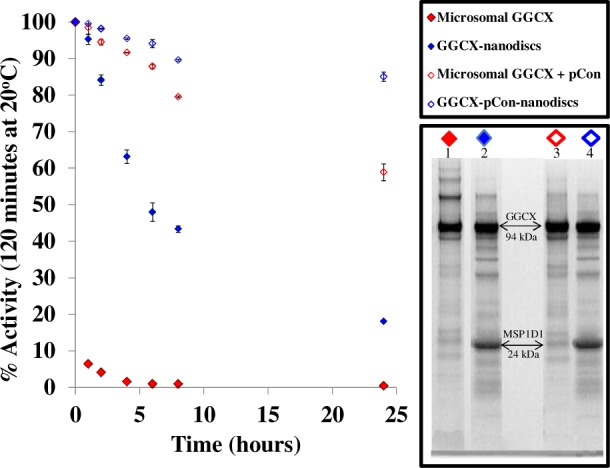
GGCX stability at 25°C monitored by catalytic activity. Activity was measured by the incorporation of radiolabeled 14C as GGCX carboxylates a small pentapeptide substrate FLEEL in the presence of propeptide, KH2, oxygen, and NaH14CO3. In all sample systems, the 100% active control is recorded at time zero. Stability is compared between microsomal GGCX (red) and GGCX-embedded nanodiscs (blue) with (◊) and without (⧫) pCon. The fractionated proteins from the 24-h time point at 25°C are represented by SDS-PAGE for each sample.
Gel electrophoresis protein banding patterns of microsomal GGCX at the 24-h time point (Figure 2, lane 1) indicates that enzymatic inactivation coincides with the formation of high molecular weight protein aggregates in the absence of protein stabilization (nanodisc or propeptide). Incorporation of GGCX into nanodiscs, however, overcomes many of the inherent complications notorious for microsome assemblies, enabling monomeric solubilization and conservation of catalytic activity with and without pCon (lanes 2 and 4, respectively). Nanodiscs thus provide a platform in which to study the structural and functional recognition of membrane protein complexes in a catalytically active and structurally protected phospholipid environment. To maintain optimal GGCX activity and limit the formation of oligomers, all HX labeling experiments were performed within 4 h of nanodisc assembly.
Hydrogen Exchange Mass Spectrometry Analysis of GGCX-Nanodiscs
The continuous labeling nanodisc–HX MS workflow, as previously reported,33 was optimized to include cholate disassembly of the nanodiscs, ZrO2 removal of sample phospholipid, and temperature-controlled UPLC separation. In the current work, the nanodisc–HX MS platform was used to characterize protein–substrate interactions by measuring local changes in HX (i.e., deuterium uptake) for the GGCX-pCon-nanodisc complex relative to GGCX-embedded nanodiscs in the absence of pCon. Time-resolved deuterium incorporation into GGCX was monitored for duplicate biological preparations of GGCX-nanodisc complexes. Forty-nine peptic peptides were unambiguously identified in GGCX with quality MS deuterium profiles for all time points in both sample forms (see Supplemental Figure S2, Supporting Information), constituting a linear sequence coverage of 42% (see Figure 3). Overlapping peptides were identified in multiple regions, providing additional support for uptake trends in small sequence intervals. Signature peptic profiles of the membrane scaffold protein (MSP1D1)50 were monitored to provide a system control for evaluating experimental performance and distinguishing peptides unique to the embedded protein, in this case GGCX (data not shown).
Figure 3.

Sequence coverage plotted on the predicted topology map of GGCX. With the exception of six colored peptides of interest, sequence coverage is indicated by amino acids colored in dark gray. Amino acids in white were not observed in these experiments. A single disulfide bond is indicated between amino acid residues cysteine 99 and cysteine 450. Glycosylation sites are highlighted in light blue. Deuterium uptake curves are provided for six representative peptic peptides where the highlighted color of the peptide sequence is matched to the color of the box surrounding the curve. The average deuterium incorporation from duplicate biological replicates is shown for GGCX-nanodiscs without (blue) and with (orange) pCon.
Among the GGCX peptides identified, six noteworthy regions (Figure 3, colored amino acid residues) were evaluated to assess changes in solvent accessibility and/or hydrogen bonding resulting from GGCX–pCon association. Comparing the magnitude and rate of deuterium exchange into the same peptide under two different conditions (in this case with and without pCon) highlights local changes in protein conformation and dynamics as a result of that binding. Whereas some regions of GGCX (e.g., residues 8–21) are shown to have a nearly indistinguishable difference (<1.0 Da) in deuterium uptake after interaction with propeptide, other regions have subtle, yet important changes in deuteration at early (e.g., residues 643–660) or late (e.g., residues 701–713) labeling times throughout the analysis.
Peptic peptides from the predicted transmembrane helices within the phospholipid bilayer were identified. The GGCX peptide LFLPYSHF (residues 375–382) is located in the exit region of the fifth transmembrane domain. Consistent with structural predictions, the representative transmembrane domain peptide displays little exchange, indicative of a solvent-protected environment and a stable α-helical structure that spans the lipid membrane. Small changes in deuteration between free and pCon-associated-GGCX were cumulatively below the experimental cutoff (<1.0 Da) and thus classified as a nonimportant change in the context of this work (see Experimental Procedures for explanation of experimental cutoff).
In the presence of propeptide, GGCX residues 395–401 display a modest, yet reproducible decrease in deuterium uptake, indicating a more solvent-protected conformation in this region across all time points. The substrate-induced reconfiguration of GGCX as measured by HX MS indicates either a direct or indirect propeptide contact. These findings correlate well with previous reports31,32 implicating the involvement of GGCX residues within the 393–404 sequence region in both glutamate and propeptide substrate binding.
The most noteworthy change in GGCX HX upon pCon association was found for peptides spanning GGCX residues 491–507. This region had relatively high levels of deuteration in the unbound state, but upon addition of propeptide there was a 66% decrease in deuterium uptake at the 10 s time point. Despite evidence of a more solvent-protected environment in the pCon-associated GGCX complex, a steady increase in deuterium content toward the unbound protein form was shown at longer labeling times, suggesting flexibility in the propeptide-bound conformation.
The topology diagrams in Figure 4 were compiled to provide a visualization of conformation changes between free and pCon-bound GGCX-nanodiscs over a continuous labeling experiment (10 s to 4 h). GGCX peptides are denoted by color in each diagram to represent a difference in the magnitude of deuterium exchange between each condition. Differences are classified as major (>2.0 Da), minor (1.0–2.0 Da), and no distinguishable change (<1.0 Da) where larger values represent greater protection from exchange in the propeptide-bound state. At the 10 s and 1 min time points, binding of the propeptide to GGCX results in many minor conformational changes. By the 5 min time interval, residues 180–186, 328–342, 395–401, 423–431, 484–490, 491–507, 507–516, 532–543, and 717–730 each continue to be characterized by a minor (1.0–2.0 Da) difference in deuterium incorporation. As the labeling experiment progressed, the uptake in deuterium for the majority of identified peptides leveled off to a steady-state value similar to that of the unbound form of the protein. This trend can most notably be seen as the topology coloration shifts from an abundance of minor deuterium differences (yellow) at the early labeling time-points to a population occupied mostly by changes in deuteration below our threshold for differentiation (gray) at the 30 min and 4 h time points. The exception to this trend is for GGCX peptide 491–507, which appears highly protected throughout the entirety of the deuterium labeling experiment. Thus, in addition to the known propeptide binding domain, these experiments support the notion that many regions within GGCX are influenced by the binding of the propeptide.
Figure 4.
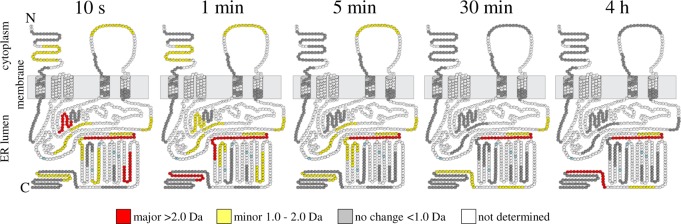
Deuterium difference diagrams illustrating GGCX topology images at consecutive HX labeling intervals from 10 s to 4 h. The difference in deuterium incorporation for each GGCX peptide was calculated at individual labeling time points by subtracting the deuterium level for the pCon-bound GGCX from the deuterium level of unbound GGCX. The change in deuterium incorporation is plotted on the predicted topology map of GGCX where a major (>2.0 Da), minor (1.0–2.0 Da), and no distinguishable change (<1.0 Da) corresponds to the colors red, yellow, and gray, respectively. Residues in white were not observed with confidence in duplicate biological and sample preparations for both experimental conditions (with or without propeptide). Neighboring and overlapping peptides were used for deuterium uptake pattern validation (see also Supplemental Figure S2, Supporting Information). Glycosylation sites are highlighted in light blue.
Hydrogen Exchange Mass Spectrometry Analysis of the Consensus Propeptide
The deuterium uptake of the propeptide itself was also shown to change upon binding to GGCX of embedded nanodiscs. Figure 5A overlays a peptide isotopic distribution profile for the pCon substrate (green) and GGCX-pCon-nanodiscs (orange) as a function of deuteration time. The centroid mass value at each time point for both experimental conditions provides a graphical comparison between the unbound and bound forms of the peptide (Figure 5B). Variants (<0.5% peptide content) of the pCon sequence (additions, deletions, and single amino acid replacements as measured by amino acid analysis) were monitored. No evidence for binding or structural protection of these variants was identified in HX MS experiments, ruling out the potential for false contributions to GGCX deuterium uptake patterns.
Figure 5.
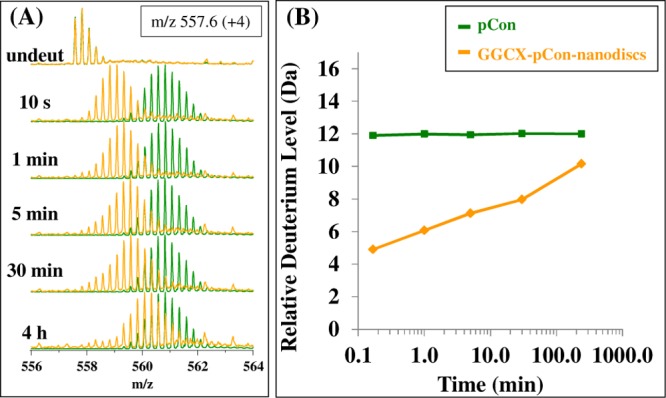
Mass spectra (A) during the HX time course for the pCon substrate (green) and GGCX-pCon-nanodiscs (orange). At each time point interval from 10 s to 4 h, the isotopic centroided mass of the peptide distribution was determined to create the deuterium uptake plot (B). The y-axis maximum represents the total possible exchangeable backbone amide hydrogens in the consensus propeptide sequence.
In the free form, pCon demonstrated high levels of deuterium incorporation indicating little protection from the solvent. This finding was further confirmed by circular dichroism (data not shown), which indicated that the consensus propeptide exhibits little or no secondary structural features. Upon association with GGCX-embedded nanodiscs, deuterium incorporation of the pCon peptide changes from that of an unstructured form to one that is protected from exchange, indicating the formation of new backbone amide hydrogen bonds or shielding from solvent of amide hydrogens in the propeptide upon binding to GGCX. While characterized by decreased deuteration in the protein-bound form, deuterium uptake increased at longer labeling times, indicating that the GGCX–pCon association remains flexible and fluid. Future investigation of deuterium uptake profiles for propeptide incorporation may provide a conformational comparison of structural changes that occur in GGCX in the presence of different propeptide substrates and under conditions in which other co-substrates are added to the GGCX-nanodisc platform.
Discussion
This study of GGCX-pCon-nanodiscs demonstrates the application of HX MS for the investigation of membrane protein–ligand interactions in a nanodisc phospholipid environment. Despite the urgent need for detailed structural information of membrane proteins, biophysical characterization is severely limited by sample incompatibilities with analytical methods, the scarcity of nonperturbing model membrane systems, and the difficulties of crystal structure determination of protein–lipid samples. Nanodisc technology provides a controlled assembly mechanism in which membrane proteins are incorporated as single molecules into a near-native phospholipid environment, optimized for a given transmembrane protein of interest. By utilizing nanodisc bilayers to supply a native-like membrane environment, HX MS can provide an analytical platform to investigate dynamic structural changes of membrane proteins in response to natural fluctuations or environmental stimuli in solution. The methodology was described in a previous report33 and continues to be recognized as an emerging technology in the advancement of membrane proteins54 with HX MS methods.55
Modeling GGCX–VKD protein interactions is of particular interest toward understanding of the complex biochemical mechanism of the vitamin K cycle. In the present study, we attempted to elucidate localized changes in GGCX protein dynamics of potential catalytic domains over a continuous deuterium-labeling experiment as a means to study structural changes that occur in GGCX upon the binding of propeptide. Our results are consistent with previous observations regarding the location of propeptide interaction with GGCX, while providing additional information on cooperative structural influences in other regions within GGCX.
To convey confidence in our model system, we specifically sought details to evaluate the binding of a propeptide to GGCX. Results indicated that the GGCX–pCon complex was successfully incorporated into nanodiscs (Figure 1). These assembled complexes were structurally stable and catalytically active over extended time periods thereby providing an advantage over traditional microsomal solubilization methods (Figure 2). Confirming propeptide specificity for GGCX-nanodiscs and extended stability of the protein complex affirmed that the conformational information derived from deuterium labeling was highly reflective of the active complex in vivo. Nanodisc analysis by HX MS thus provides conformational characterization of transmembrane protein complexes that is otherwise extremely difficult to obtain by other methods.
Changes in deuterium exchange are represented as time-dependent uptake plots constructed for individual peptides of GGCX (Figure 3; Supplemental Figure S2, Supporting Information) and the pCon substrate (Figure 5). Deuterium uptake curves were subsequently used to compile a topology map of GGCX while maintaining strict guidance to reproducibility in matched identifications (±0.15 Da) for duplicate biological sample preparations with and without pCon. Despite limited GGCX sequence coverage (42%), differential topology diagrams (Figure 4) provide a tremendous amount of information on protein conformation between free and pCon-bound GGCX-nanodiscs over the continuous labeling experiment. Ongoing improvements to the HX MS nanodisc technology, including improved sample preparation, chromatographic separation, MS sensitivity, ion mobility, and dynamic range will further increase peptide coverage and provide more information regarding protein dynamics for large transmembrane systems like GGCX.
In the absence of a three-dimensional protein structure, differential topology maps of individual GGCX peptides obtained periodically over a continuous labeling time course yields a dynamic viewpoint of substrate binding on GGCX conformation. In all experiments, the addition of pCon resulted in decreased protein flexibility (increased solvent protection or hydrogen bonding) in many regions of GGCX as indicated by a decrease in the level of deuterium uptake. For all GGCX peptides identified, 61% were characterized by changes in deuteration levels >1.0 Da for a single time point in the continuous labeling reaction. The most pronounced enhancement in deuterium exchange was primarily located around the known glutamate binding site (393–404)31,32 and the propeptide binding region (495–513).11,12 Within these domains, two GGCX peptic peptides, residues 395–401 (maximum Δ+1.5 Da) and 491–507 (maximum Δ+5.6 Da), revealed extensive protection from deuterium incorporation when the pCon substrate was present. For the 491–507 peptide, and neighboring peptides 484–490 and 507–516, pronounced changes in deuteration over longer time intervals are consistent with direct binding and sustained alteration of local solvent accessibility or hydrogen bonding networks,56 indicating little variability in the GGCX–pCon-bound conformation. Interestingly, the appearance of important and major protection at a distance from the known propeptide binding site, namely, in the glutamate binding site (residues 393–404), provides direct biophysical evidence for the relationship between the anchoring of the VKD propeptide and the catalytic carboxylation of glutamic acid residues. This observation is likely due to either a close proximity of these two parts of the protein or an allosteric mechanism that connects them.16,52,57 Evaluation of the rate and magnitude of change in GGCX deuteration upon propeptide binding provides evidence of conformational changes that modify solvent exposure or hydrogen bonding interactions as the protein adopts a catalytically stable structural state.
While the propeptide and Gla binding domains in GGCX have been extensively characterized, several subtle differences in GGCX structure were noted at varying deuteration time points, enabling predictions of associated peptide proximities within the three-dimensional structure. Major (>2.0 Da) changes in deuterium uptake upon propeptide binding are observed for peptides 257–272, 643–660, and 717–730 at the 10 s and 1 min labeling times points. While the C-terminal peptides 643–660 and 717–730 are accepted to be distal from the carboxylation active site, a recent report identified carboxylation of glutamic acid residues in GGCX peptides 625–647 and 729–758 in the presence of vitamin K.58 Although it is unlikely that C-terminal carboxylated Glas are required for catalysis,59 the present HX MS data suggest substrate-induced reorientation of GGCX peptides 643–660 and 717–730 conform to a more solvent-protected environment at initial labeling time points. Peptide 728–758, however, measured a difference in deuterium uptake below the ±1.0 Da threshold for peptide structural differentiation. Future investigations of this system in the presence of additional co-substrates (e.g., vitamin K) and a full-length VKD protein may provide information supporting the mechanism linking the location of the GGCX Gla domain in the ER lumen to enzymatic function in VKD protein carboxylation.
A quantum model reported by Wu et al.60 proposes the relative orientation of GGCX and VKOR transmembrane proteins as a heterodimer colocalized in the ER membrane allowing efficient oxidation and reduction of vitamin K during the vitamin K cycle. In accordance with predictions of transmembrane helix unit topologies for GGCX and VKOR, compatibility of the heterodimer proposed model was validated using previous biochemical and mutational data. In this system, the relative orientation of carboxylation-dependent GGCX residues L394,32 W501,61 G537,62 G558,62 and T59163 are located in close proximity to the C-terminus of GGCX and the N-terminus of VKOR.60 In correlation with this proposed structure, GGCX peptides 395–401, 491–507, 532–543, and 554–568, as identified in our HX MS experiments, exhibited structural reorientation in the form of diminished solvent exposure and/or hydrogen bonding upon the incorporation of propeptide substrate. While peptides with known involvement in glutamate (395–401) and propeptide (491–507) binding are represented by changes in deuteration over longer time periods, peptides 532–543 and 554–568 reveal changes in deuteration at shorter time intervals indicating temporary alterations of the local solvent accessibility upon the binding of propeptide.
On the opposing side of the heterodimer complex, GGCX residues W157,63 H160,64 and K21825 are predicted to reside in close proximity to the napthoquinone unit of vitamin K and the surface of the membrane in the ER lumen.60 This placement is shown to be consistent with models implicating H160 and K218 as critical residues in the deprotonation of vitamin K hydroquinone and the initiation of the carboxylation reaction.25,64,65 The close proximity of these residues to the hydrophobic transmembrane domain, however, results in minimal recovery of peptic peptides within these regions and limits evaluations by HX MS. Improvements to sample denaturation and digestion may enhance peptide recovery of potentially informative regions of GGCX.
Bridging the gap between experimental data and theoretical predictions, HX MS allows comparisons of relative deuterium incorporation for specific peptides across the GGCX polypeptide backbone, thereby providing both spatial and temporal information on structural influences and conformational changes induced by substrate introduction in previously difficult-to-explore regions of the GGCX structure. Similar to the T591K and W157R mutations, D31N is an inherited VKD coagulation factor autosomal recessive bleeding disorder associated with defects in either GGCX or VKOR.63 While wild-type GGCX activity was exhibited for the D31N mutation, W157R and T591K activities were 8% and 0% that of wild-type GGCX, respectively.63 Located on the opposite side of the membrane where propeptide binding occurs, peptide 25–40 undergoes minor (1.0–2.0 Da) changes in deuterium incorporation at the 10 s and 1 min labeling time points. Although D31 may not be directly involved in propeptide binding, HX MS data support a structural dependence on substrate association. A similar phenomenon of indirect cooperative structural influences on GGCX structure upon propeptide binding is observed for GGCX residues 180–186, 257–272, 328–342, 423–431, 685–692, and 701–713. For the majority of these peptides, a decrease in deuterium incorporation occurs at the early labeling time points. At longer time intervals, these peptides showed an eventual loss of protection consistent with long-range changes affecting the flexibility and dynamics of the entire protein. Visualization of protein dynamics through time-resolved topology maps not only provides a profile of GGCX protein dynamics over time, but also aids in understanding the mechanism by which GGCX domains cooperate to create a stable three-dimensional structure for VKD carboxylation.
In the absence of a fully resolved crystal structure for GGCX, the analysis of changes in deuterium uptake upon propeptide binding yields a structural storyline uniting conformational organization in GGCX and functional roles involving substrate binding for carboxylation. Improvements in HX MS technology will lead to the desire to analyze larger and more complex proteins and multiprotein systems. In this work, we expand upon this methodology to include an investigation of membrane protein–ligand interactions by HX MS. The investigation of the transmembrane protein GGCX aims to target the structural relationship between GGCX and co-substrates involved in the vitamin K cycle as a means to develop therapeutics for blood coagulation disorders. Using the nanodisc–HX MS platform, this study represents the first application, to our knowledge, of this strategy for investigating specific conformational changes in propeptide-bound GGCX. Future experiments aimed to improve sample throughput will not only provide a universal methodology for membrane protein investigation but also will reveal information in the mostly uncharted territory of membrane protein dynamics.
Acknowledgments
We gratefully acknowledge Mark M. Ross at the U.S. Food and Drug Administration and David L. Straight at the University of North Carolina at Chapel Hill, Department of Biology, for useful discussions. The MSP1D1 plasmid was graciously provided by the Sligar laboratory through Addgene plasmid 20061.
Supporting Information Available
Supplemental Figures S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# (C.R.M.) Genzyme, A Sanofi Company, 1 The Mountain Road, Framingham, MA 01701.
Author Present Address
⊥ (K.D.R.) Department of Pharmacy, University of Copenhagen, Copenhagen, Denmark.
The authors declare no competing financial interest.
These studies were supported by the National Institutes of Health (Grant GM101135 to J.R.E. and Grants HL48318 and HL06350 to D.W.S.), the Danish Council for Independent Research Natural Sciences (Grants 09-063876 and 11-104058 to K.D.R.), and Waters Corporation (J.W.J and J.R.E.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tie J. K.; Zheng M. Y.; Pope R. M.; Straight D. L.; Stafford D. W. (2006) Identification of the N-linked glycosylation sites of vitamin K-dependent carboxylase and effect of glycosylation on carboxylase function. Biochemistry 45, 14755–14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J. K.; Wu S. M.; Jin D. Y.; Nicchitta C. V.; Stafford D. W. (2000) A topological study of the human gamma-glutamyl carboxylase. Blood 96, 973–978. [PubMed] [Google Scholar]
- Tie J. K.; Mutucumarana V. P.; Straight D. L.; Carrick K. L.; Pope R. M.; Stafford D. W. (2003) Determination of disulfide bond assignment of human vitamin K-dependent gamma-glutamyl carboxylase by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Biol. Chem. 278, 45468–45475. [DOI] [PubMed] [Google Scholar]
- Whitlon D. S.; Sadowski J. A.; Suttie J. W. (1978) Mechanism of coumarin action: significance of vitamin K epoxide reductase inhibition. Biochemistry 17, 1371–1377. [DOI] [PubMed] [Google Scholar]
- Crooks G. E.; Hon G.; Chandonia J. M.; Brenner S. E. (2004) WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T. D.; Stephens R. M. (1990) Sequence logos - a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen M. J.; Cantor A. B.; Furie B. C.; Brown C. L.; Shoemaker C. B.; Furie B. (1987) Recognition site directing vitamin-K-dependent gamma-carboxylation resides on the propeptide of factor-IX. Cell 48, 185–191. [DOI] [PubMed] [Google Scholar]
- Pan L. C.; Price P. A. (1985) The propeptide of rat bone gamma-carboxyglutamic acid protein shares homology with other vitamin-K-dependent protein precursors. Proc. Natl. Acad. Sci. U.S.A. 82, 6109–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley T. B.; Jin D. Y.; Lin P. J.; Stafford D. W. (1999) The propeptides of the vitamin K-dependent proteins possess different affinities for the vitamin K-dependent carboxylase. J. Biol. Chem. 274, 16940–16944. [DOI] [PubMed] [Google Scholar]
- Lin P. J.; Straight D. L.; Stafford D. W. (2004) Binding of the factor IX gamma-carboxyglutamic acid domain to the vitamin K-dependent gamma-glutamyl carboxylase active site induces an allosteric effect that may ensure processive carboxylation and regulate the release of carboxylated product. J. Biol. Chem. 279, 6560–6566. [DOI] [PubMed] [Google Scholar]
- Lin P. J.; Jin D. Y.; Tie J. K.; Presnell S. R.; Straight D. L.; Stafford D. W. (2002) The putative vitamin K-dependent gamma-glutamyl carboxylase internal propeptide appears to be the propeptide binding site. J. Biol. Chem. 277, 28584–28591. [DOI] [PubMed] [Google Scholar]
- Wu S. M.; Mutucumarana V. P.; Geromanos S.; Stafford D. W. (1997) The propeptide binding site of the bovine gamma-glutamyl carboxylase. J. Biol. Chem. 272, 11718–11722. [DOI] [PubMed] [Google Scholar]
- Katayama K.; Ericsson L. H.; Enfield D. L.; Walsh K. A.; Neurath H.; Davie E. W.; Titani K. (1979) Comparison of amino-acid sequence of bovine coagulation Factor-IX (Christmas Factor) with that of other vitamin K-dependent plasma proteins. Proc. Natl. Acad. Sci. U.S.A. 76, 4990–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenflo J.; Suttie J. W. (1977) Vitamin K-dependent formation of gamma-carboxyglutamic acid. Annu. Rev. Biochem. 46, 157–172. [DOI] [PubMed] [Google Scholar]
- Suttie J. W. (1980) Mechanism of action of vitamin K: synthesis of gamma-carboxyglutamic acid. CRC Crit. Rev. Biochem. 8, 191–223. [DOI] [PubMed] [Google Scholar]
- Morris D. P.; Stevens R. D.; Wright D. J.; Stafford D. W. (1995) Processive post-translational modification. Vitamin K-dependent carboxylation of a peptide substrate. J. Biol. Chem. 270, 30491–30498. [DOI] [PubMed] [Google Scholar]
- Stenina O.; Pudota B. N.; McNally B. A.; Hommema E. L.; Berkner K. L. (2001) Tethered processivity of the vitamin K-dependent carboxylase: factor IX is efficiently modified in a mechanism which distinguishes Gla’s from Glu’s and which accounts for comprehensive carboxylation in vivo. Biochemistry 40, 10301–10309. [DOI] [PubMed] [Google Scholar]
- Bristol J. A.; Ratcliffe J. V.; Roth D. A.; Jacobs M. A.; Furie B. C.; Furie B. (1996) Biosynthesis of prothrombin: intracellular localization of the vitamin K-dependent carboxylase and the sites of gamma-carboxylation. Blood 88, 2585–2593. [PubMed] [Google Scholar]
- Dowd P.; Hershline R.; Ham S. W.; Naganathan S. (1995) Vitamin-K and energy transduction: a base strength amplification mechanism. Science 269, 1684–1691. [DOI] [PubMed] [Google Scholar]
- Canfield L. M. (1987) Vitamin K-dependent oxygenase/carboxylase; differential inactivation by sulfhydryl reagents. Biochem. Biophys. Res. Commun. 148, 184–191. [DOI] [PubMed] [Google Scholar]
- Mack D. O.; Suen E. T.; Girardot J. M.; Miller J. A.; Delaney R.; Johnson B. C. (1976) Soluble enzyme system for vitamin K-dependent carboxylation. J. Biol. Chem. 251, 3269–3276. [PubMed] [Google Scholar]
- Pudota B. N.; Miyagi M.; Hallgren K. W.; West K. A.; Crabb J. W.; Misono K. S.; Berkner K. L. (2000) Identification of the vitamin K-dependent carboxylase active site: Cys-99 and Cys-450 are required for both epoxidation and carboxylation. Proc. Natl. Acad. Sci. U.S.A. 97, 13033–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie J. K.; Jin D. Y.; Loiselle D. R.; Pope R. M.; Straight D. L.; Stafford D. W. (2004) Chemical modification of cysteine residues is a misleading indicator of their status as active site residues in the vitamin K-dependent gamma-glutamyl carboxylation reaction. J. Biol. Chem. 279, 54079–54087. [DOI] [PubMed] [Google Scholar]
- Rishavy M. A.; Pudota B. N.; Hallgren K. W.; Qian W.; Yakubenko A. V.; Song J. H.; Runge K. W.; Berkner K. L. (2004) A new model for vitamin K-dependent carboxylation: the catalytic base that deprotonates vitamin K hydroquinone is not Cys but an activated amine. Proc. Natl. Acad. Sci. U.S.A. 101, 13732–13737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishavy M. A.; Hallgren K. W.; Yakubenko A. V.; Shtofman R. L.; Runge K. W.; Berkner K. L. (2006) Brønsted analysis reveals Lys218 as the carboxylase active site base that deprotonates vitamin K hydroquinone to initiate vitamin K-dependent protein carboxylation. Biochemistry 45, 13239–13248. [DOI] [PubMed] [Google Scholar]
- Shimizu A.; Sugiura I.; Matsushita T.; Kojima T.; Hirai M.; Saito H. (1998) Identification of the five hydrophilic residues (Lys-217, Lys-218, Arg-359, His-360, and Arg-513) essential for the structure and activity of vitamin K-dependent carboxylase. Biochem. Biophys. Res. Commun. 251, 22–26. [DOI] [PubMed] [Google Scholar]
- Sugiura I.; Furie B.; Walsh C. T.; Furie B. C. (1996) Profactor IX propeptide and glutamate substrate binding sites on the vitamin K-dependent carboxylase identified by site-directed mutagenesis. J. Biol. Chem. 271, 17837–17844. [DOI] [PubMed] [Google Scholar]
- Davis C. H.; Deerfield D.; Stafford D. W.; Pedersen L. G. (2007) Quantum chemical study of the mechanism of action of vitamin K carboxylase (VKC). IV. Intermediates and transition states. J. Phys. Chem. A 111, 7257–7261. [DOI] [PubMed] [Google Scholar]
- Davis C. H.; Deerfield D.; Wymore T.; Stafford D. W.; Pedersen L. G. (2007) A quantum chemical study of the mechanism of action of vitamin K carboxylase (VKC) - III. Intermediates and transition states. J. Mol. Graphics Modell. 26, 409–414. [DOI] [PubMed] [Google Scholar]
- Deerfield D.; Davis C. H.; Wymore T.; Stafford D. W.; Pedersen L. G. (2006) Quantum chemical study of the mechanism of action of vitamin K epoxide reductase (VKOR). Int. J. Quantum Chem. 106, 2944–2952. [DOI] [PubMed] [Google Scholar]
- Brenner B.; Sanchez-Vega B.; Wu S. M.; Lanir N.; Stafford D. W.; Solera J. (1998) A missense mutation in gamma-glutamyl carboxylase gene causes combined deficiency of all vitamin K-dependent blood coagulation factors. Blood 92, 4554–4559. [PubMed] [Google Scholar]
- Mutucumarana V. P.; Acher F.; Straight D. L.; Jin D. Y.; Stafford D. W. (2003) A conserved region of human vitamin K-dependent carboxylase between residues 393 and 404 is important for its interaction with the glutamate substrate. J. Biol. Chem. 278, 46488–46493. [DOI] [PubMed] [Google Scholar]
- Hebling C. M.; Morgan C. R.; Stafford D. W.; Jorgenson J. W.; Rand K. D.; Engen J. R. (2010) Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal. Chem. 82, 5415–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi S.; Takio K. (2001) Structure of melittin bound to phospholipid micelles studied using hydrogen–deuterium exchange and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 12, 1247–1253. [DOI] [PubMed] [Google Scholar]
- Busenlehner L. S.; Salomonsson L.; Brzezinski P.; Armstrong R. N. (2006) Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 103, 15398–15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard M.; Benjamin D. R.; Tito P.; Robinson C. V.; Dobson C. M. (2000) Solvent effects on the conformation of the transmembrane peptide gramicidin A: insights from electrospray ionization mass spectrometry. Biophys. J. 78, 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmers J. A.; van Duijn E.; Haverkamp J.; Greathouse D. V.; Koeppe R. E. II; Heck A. J.; Killian J. A. (2001) Interfacial positioning and stability of transmembrane peptides in lipid bilayers studied by combining hydrogen/deuterium exchange and mass spectrometry. J. Biol. Chem. 276, 34501–34508. [DOI] [PubMed] [Google Scholar]
- Hansen R. K.; Broadhurst R. W.; Skelton P. C.; Arkin I. T. (2002) Hydrogen/deuterium exchange of hydrophobic peptides in model membranes by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 13, 1376–1387. [DOI] [PubMed] [Google Scholar]
- Pinheiro T. J.; Cheng H.; Seeholzer S. H.; Roder H. (2000) Direct evidence for the cooperative unfolding of cytochrome c in lipid membranes from H-2H exchange kinetics. J. Mol. Biol. 303, 617–626. [DOI] [PubMed] [Google Scholar]
- Man P.; Montagner C.; Vernier G.; Dublet B.; Chenal A.; Forest E.; Forge V. (2007) Defining the interacting regions between apomyoglobin and lipid membrane by hydrogen/deuterium exchange coupled to mass spectrometry. J. Mol. Biol. 368, 464–472. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Chien E. Y.; Chalmers M. J.; Pascal B. D.; Gatchalian J.; Stevens R. C.; Griffin P. R. (2010) Dynamics of the β2-adrenergic G-protein coupled receptor revealed by hydrogen–deuterium exchange. Anal. Chem. 82, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns D. J.; Kurosawa S.; Sims P. J.; Esmon N. L.; Esmon C. T. (1988) The interaction of a Ca2+ dependent monoclonal antibody with the protein-C activation peptide region. Evidence for obligatory Ca2+ binding to both antigen and antibody. J. Biol. Chem. 263, 826–832. [PubMed] [Google Scholar]
- Denisov I. G.; Grinkova Y. V.; Lazarides A. A.; Sligar S. G. (2004) Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- Chen P. S.; Toribara T. Y.; Warner H. (1956) Microdetermination of phosphorus. Anal. Chem. 28, 1756–1758. [Google Scholar]
- Düzgünes N. (2003) Preparation and quantitation of small unilamellar liposomes and large unilamellar reverse-phase evaporation liposomes. Methods Enzymol. 367, 23–27. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Q.; Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein-structure elucidation. Protein Sci. 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Pan H.; Smith D. L. (2002) Hydrogen exchange-mass spectrometry: optimization of digestion conditions. Mol Cell Proteomics 1, 132–138. [DOI] [PubMed] [Google Scholar]
- Wales T. E.; Fadgen K. E.; Gerhardt G. C.; Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal. Chem. 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis D. D.; Engen J. R.; Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 17700–11703. [DOI] [PubMed] [Google Scholar]
- Morgan C. R.; Hebling C. M.; Rand K. D.; Stafford D. W.; Jorgenson J. W.; Engen J. R. (2011) Conformational transitions in the membrane scaffold protein of phospholipid bilayer nanodiscs. Mol Cell Proteomics 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins-Gruber S. L.; Mutucumarana V. P.; Lin P. J.; Jorgenson J. W.; Stafford D. W.; Straight D. L. (2010) Effect of vitamin K-dependent protein precursor propeptide, vitamin K hydroquinone, and glutamate substrate binding on the structure and function of γ-glutamyl carboxylase. J. Biol. Chem. 285, 31502–31508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnell S. R.; Tripathy A.; Lentz B. R.; Jin D. Y.; Stafford D. W. (2001) A novel fluorescence assay to study propeptide interaction with gamma-glutamyl carboxylase. Biochemistry 40, 11723–11733. [DOI] [PubMed] [Google Scholar]
- Shih A. Y.; Freddolino P. L.; Sligar S. G.; Schulten K. (2007) Disassembly of nanodiscs with cholate. Nano Lett. 7, 1692–1696. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Robinson C. V. (2011) Advances in the mass spectrometry of membrane proteins: from individual proteins to intact complexes. Annu. Rev. Biochem. 80, 247–271. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; West G. M.; Griffin P. R. (2011) Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev. Proteomics 8, 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Kallenbach N. R. (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 16, 521–655. [DOI] [PubMed] [Google Scholar]
- Knobloch J. E.; Suttie J. W. (1987) Vitamin K-dependent carboxylase. Control of enzyme activity by the propeptide region of factor X. J. Biol. Chem. 262, 15334–15337. [PubMed] [Google Scholar]
- Hallgren K. W.; Zhang D.; Kinter M.; Willard B.; Berkner K. L. (2013) Methylation of γ-carboxylated Glu (Gla) allows detection by liquid chromatography-mass spectrometry and the identification of Gla residues in the γ-glutamyl carboxylase. J. Proteome Res. 12, 2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkner K. L.; Pudota B. N. (1998) Vitamin K-dependent carboxylation of the carboxylase. Proc. Natl. Acad. Sci. U.S.A. 95, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Liu S. B.; Davis C. H.; Stafford D. W.; Kulman J. D.; Pedersen L. G. (2011) A hetero-dimer model for concerted action of vitamin K carboxylase and vitamin K reductase in vitamin K cycle. J. Theor. Biol. 279, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soute B. A.; Jin D. Y.; Spronk H. M.; Mutucumarana V. P.; Lin P. J.; Hackeng T. M.; Stafford D. W.; Vermeer C. (2003) Characteristics of recombinant W501S mutated human γ-glutamyl carboxylase. J. Thromb. Haemostasis 2, 597–604. [DOI] [PubMed] [Google Scholar]
- Vanakker O. M.; Martin L.; Gheduzzi D.; Leroy B. P.; Loeys B. L.; Guerci V. I.; Matthis D.; Terry S. F.; Coucke P. J.; Pasquali-Ronchetti I.; De Paepe A. (2007) Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J. Invest. Dermatol. 127, 581–587. [DOI] [PubMed] [Google Scholar]
- Darghouth D.; Hallgren K. W.; Shtofman R. L.; Mrad A.; Gharbi Y.; Maherzi A.; Kastally R.; LeRicousse S.; Berkner K. L.; Rosa J. P. (2006) Compound heterozygosity of novel missense mutations in the gamma-glutamyl-carboxylase gene causes hereditary combined vitamin K-dependent coagulation factor deficiency. Blood 108, 1925–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishavy M. A.; Berkner K. L. (2008) Insight into the coupling mechanism of the vitamin K-dependent carboxylase: mutation of histidine 160 disrupts glutamic acid carbanion formation and efficient coupling of vitamin K epoxidation to glutamic acid carboxylation. Biochemistry 47, 9836–9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishavy M. A.; Hallgren K. W.; Berkner K. L. (2011) The vitamin K-dependent carboxylase generates γ-carboxylated glutamates by using CO2 to facilitate glutamate deprotonation in a concerted mechanism that drives catalysis. J. Biol. Chem. 286, 44821–44832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.