

Abstract
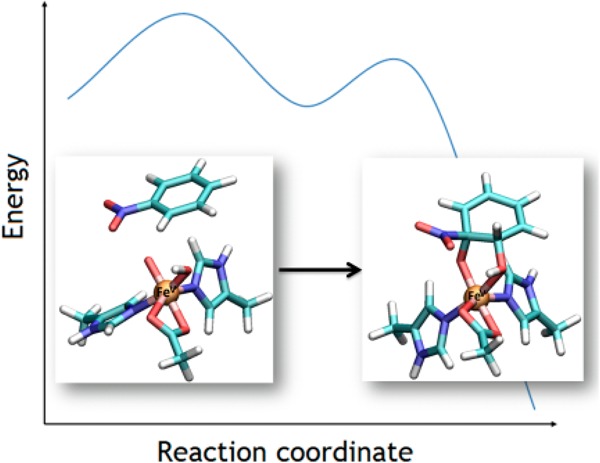
The mechanism of cis-dihydroxylation of nitrobenzene and 2-nitrotoluene catalyzed by nitrobenzene 1,2-dioxygenase (NBDO), a member of the naphthalene family of Rieske non-heme iron dioxygenases, was studied by means of the density functional theory method using four models of the enzyme active site. Different possible reaction pathways for the substrate dioxygenation initiated either by the FeIII–OOH or HO–FeV=O attack on the aromatic ring were considered and the computed activation barriers compared with the Gibbs free energy of activation for the oxygen–oxygen cleavage leading to the formation of the iron(V)–oxo species from its ferric hydroperoxo precursor. The mechanism of the substrate cis-dihydroxylation leading to the formation of a cis-dihydrodiol was then investigated, and the most feasible mechanism was found to be starting with the attack of the high-valent iron–oxo species on the substrate ring yielding a radical intermediate, which further evolves toward the final product.
Introduction
Oxidative degradation of recalcitrant nitroaromatic pollutants is initiated by the formation of a cis-dihydroxylated metabolite that spontaneously rearranges to catechol with accompanying release of nitrite (Figure 1).1−7 An understanding of the cis-dihydroxylation mechanism is thus valuable in the development of bioremediation measures, particularly protein engineering of dioxygenases to modify selectivity and enhance efficiency.8−10 The theoretical kinetic isotope effect derived using knowledge of the transition state structure can also serve as a reference value for compound-specific isotope analysis, which enables identification of the predominant transformation pathway of pollutants at a particular contaminated site.11,12 The reaction is catalyzed by nitrobenzene 1,2-dioxygenase (NBDO), as well as 2-nitrotoluene dioxygenase (2NTDO), which differ in catalytic site residue at positions 204, 251, and 293.1 These two enzymes are members of the naphthalene family of Rieske non-heme iron dioxygenases (RDOs), which consist of a Rieske [2Fe–2S] ferredoxin, an NADH-dependent flavoprotein reductase, and an α3β3 oxygenase.
Figure 1.

General reaction scheme for cis-dihydroxylation of nitroaromatic compounds catalyzed by NBDO.
RDOs also catalyze monohydroxylation, desaturation, sulfoxidation, O- and N-dealkylation, and amine oxidation.13−18 NBDO, in particular, can oxidize both the aromatic ring and the alkyl side chain of nitroaromatic compounds to yield a mixture of catechol and nitrobenzyl alcohol.1 Monohydroxylation is an undesirable competing oxidation pathway, as it does not eliminate the nitro group that prevents further degradation. The active site of RDOs is a high-spin mononuclear FeII bound to two histidines and a bidentate aspartate residue, forming the recurring 2-His-1-carboxylate facial triad motif of non-heme iron-containing oxygenases. A Rieske [2Fe–2S] cluster located within 12 Å of FeII in an adjacent α subunit transfers two electrons from NAD(P)H to the active site during the catalytic cycle. O2 binding and activation is triggered by one-electron reduction of the Rieske cluster and formation of the enzyme–substrate complex.1,8,19−22
Extensive studies on naphthalene 1,2-dioxygenase (NDO), including crystallographic data,23 single turnover24 and peroxide shunt experiments,25 and theoretical calculations,26,27 indicate a side-on bound ferric (hydro)peroxo complex as a key reaction intermediate. 18O-labeling studies on NBDO, NDO, 2NTDO, and toluene dioxygenase (TDO) further suggest that both hydroxyl groups of the product mainly originate from a single 18O2/H218O2 molecule.2,25,28−30 A theoretical study of naphthalene cis-dihydroxylation by NDO proposed that this FeIII–OOH complex directly oxidizes the substrate via an epoxide intermediate. Subsequent ring-opening leads to a carbocation, which is then attacked by the hydroxo ligand to yield cis-diol. The active site remains in the ferric state throughout the process, indicating that Fe is not involved in the redox reaction.27 On the other hand, an indole–O2 adduct fitted to an electron density map of NDO crystals suggests that the substrate is attacked by a peroxo ligand bound end-on to Fe.31
An FeIV=O oxidant is implicated in the activity of enzymes such as 2-oxo acid dioxygenases and aromatic amino acid hydroxylases, which also possess the 2-His-1-carboxylate facial triad motif.19,32,33 This was confirmed by spectroscopic studies on taurine-α-ketoglutarate dioxygenase (TauD),34−36 tyrosine hydroxylase (TyrH),37 and phenylalanine hydroxylase (PheH).38 O–O bond cleavage to form this high-valent intermediate is facilitated by additional electron donation from a cofactor or cosubstrate in contrast to RDOs, where only one electron is provided by the Rieske center.19,32,33 Oxygen–oxygen bond heterolysis in the latter yields HO–FeV=O, for which the calculated energy barrier is quite high (26.5 kcal mol–1).27 RDOs have a high-spin ligand environment,20 and theoretical studies on similar FeIII–OOR complexes show that the strong O–O bond can be attributed to σ-donation from O–O π* antibonding orbitals.39 Moreover, the non-heme ligands of RDOs do not stabilize high oxidation states (through spin delocalization and ligand oxidation) as porphyrin and thiolate ligands do for heme enzymes,39,40 which could explain the endothermicity of heterolytic O–O bond cleavage.27 Nevertheless, minor 18O incorporation (3%) observed in naphthalene dioxygenation in the presence of H218O indicates the involvement of the HO–FeV=O intermediate, which would allow solvent exchange to occur.25 Monooxygenation of indan by TDO was also found to yield 18O-labeled products.30
Heme enzymes and methane monooxygenase, which also formally have an FeV=O oxidant,32,33 catalyze olefin epoxidation41−43 instead of cis-dihydroxylation, which is a reaction unique to RDOs.8,44 An insight into the cis-dihydroxylation mechanism of high-valent iron complexes can be gained instead from functional models, which are designed to mimic the ligand environment and reactivity of RDOs.45−59 These biomimetic complexes are being developed as green alternatives to toxic or expensive oxidation catalysts.60−62 Notable examples are [FeII(6-Me3-TPA)(CH3CN)2](ClO4)2 (TPA being tris(2-pyridylmethyl)amine), which has a tetradentate ligand and two cis labile sites and is the first reported biomimetic catalyst for olefin oxidation;45 [FeII(L1)(Cl)], where the HL1 [3-(dipyridin-2-ylmethyl)-1,5,7-trimethyl-2,4-dioxo-3-azabicyclo[3.3.1]nonane-7-carboxylic acid] ligand is modeled after the 2-His-1-carboxylate motif;46 and [FeII(TPA)(NCMe)2]2+, which catalyzes the dihydroxylation of an aromatic double bond (naphthalene).47 Direct evidence of the HO–FeV=O oxidant in olefin cis-dihydroxylation has been reported for the synthetic system [FeV(O)(OH)(Me,HPytacn)](CF3SO3)+ (tacn = triazacyclononane) using variable-temperature mass spectrometry.48 Oxygen–oxygen bond cleavage is mediated by water as inferred from the significant oxygen exchange with labeled water in the cis-diol product. An important difference between these biomimetic catalysts and RDOs is the low-spin ligand environment in the former, which leads to a weaker oxygen–oxygen bond39 and, consequently, a lower barrier for heterolysis.48,49,63 Theoretical studies on Fe–Pytacn48 and Fe–TPA64 suggest an unsymmetrical concerted mechanism for cis-dihydroxylation initiated by the attack of the hydroxo ligand on the olefin to form a radical, followed by barrierless diol formation.
The cis-dihydroxylation of nitroaromatic compounds, represented by nitrobenzene and 2-nitrotoluene, is examined in the present study by means of hybrid density functional theory (DFT). Nitrobenzene dioxygenation via the previously proposed epoxide pathway is investigated and compared with that of naphthalene and benzene. Alternative mechanisms arising from direct reaction with FeIII–OOH are also explored to compare the activation barriers of substrate oxidation (with or without concerted oxygen–oxygen bond cleavage) with that of the oxygen–oxygen bond cleavage to form HO–FeV=O. Finally, the mechanism of cis-dihydroxylation by HO–FeV=O is determined on the basis of experimentally observed 18O incorporation in the cis-diol product.
Computational Methods
The possible reaction mechanisms for cis-dihydroxylation of nitrobenzene and 2-nitrotoluene were analyzed using four models of the nitrobenzene dioxygenase active site, presented in Figure 2, which were constructed based on the available crystal structure of NBDO with nitrobenzene bound in the active site (PDB ID: 2BMQ).1 The smallest model included the metal ion and side chains of its first-shell ligands, namely, His211, His206, and Asp360 (model 1, Figure 2a). The active site model was then expanded to include (a) Asn258, which forms a hydrogen bond with the substrate (model 2, Figure 2b); (b) H2O molecule, which forms a hydrogen bond with the hydroperoxo ligand (model 3, Figure 2c); and (c) Asn199 (model 3, Figure 2c). A water molecule was incorporated in the active site model based on the results of the molecular dynamics simulation of NBDO (data not shown), which indicated the presence of water at the entrance to the substrate pocket. The position of the dioxygen bound side-on to the mononuclear iron was determined through an overlay with the crystal structure of the NDO-O2-indole complex (PDB ID: 1O7N).23 In all calculations, constraints were imposed on the atomic coordinates of Cα or Cβ carbon atoms of the included residues, to account for the rigidity of the protein backbone. Calculations for the other substrate were performed using the two smallest models of the active site, by replacing nitrobenzene with 2-nitrotoluene.
Figure 2.

Four models of NBDO active site used in this study: model 1 (a) comprising only first-shell iron ligands and a substrate; model 2 (b) additionally including Asn258 residue; model 3 (c) and model 4 (d) constructed by adding a water molecule and Asn199 residue, respectively. Atoms marked with an asterisk were frozen according to the nitrobenzene dioxygenase crystal structure1 during all calculations.
Relaxed potential energy surface (PES) scans were carried out to approximately locate possible transition states. All stationary points were then optimized, and vibrational analysis was performed for all minima and transition states to confirm that they have zero and exactly one imaginary frequency, respectively. All optimizations were performed using the DFT hybrid B3LYP65,66 functional and LACVP* basis set, combining the 6-31G* basis set on C, H, N, O atoms and Hay and Wadt effective core potential (ECP)67 for the description of the Fe atom. To mimic the polarization effects of the protein environment, the SMD continuum solvent model with a dielectric constant of 5.7 was used in all reported calculations, unless stated otherwise. The final energies were recomputed using the B97D Grimme functional including dispersion68 and LACV3P+* basis set (ECP for Fe and 6-311+G* for all other atoms). The final energies were also computed using dispersion-corrected B3LYP-D2 and B3LYP-D369 functionals (see Table 1 and Tables S1, S2, and S3 in the Supporting Information). Despite visible differences, the overall conclusions drawn from the values obtained using different functionals are similar. The largest discrepancy between the barriers calculated using the B97D and B3LYP functionals is observed in the case of the O–O cleavage (Table 1). As can be seen through comparison between the dispersion-corrected and noncorrected B3LYP values, dispersion effects are negligible in this particular reaction. From this it can be also inferred that significantly different description of the energy of the system provided by the B97D functional is not related to dispersion effects, but rather to the nature of the functional itself. B97D is a general-purpose functional parametrized for various systems, including those containing transition metals, and it has been shown to outperform other commonly used functionals, including B3LYP, especially in the case of the reactions involving transition metals.68 Thus, we believe it is more suitable for the description of the reaction under study, and therefore the final energies calculated using this functional will be mainly used in the discussion of the results, with B3LYP-D2 and B3LYP-D3 values referred to when formulating relevant conclusions. The effect of reoptimization of the stationary points using the dispersion-corrected functional was examined using model 1 with nitrobenzene. The geometries of the transition states optimized with or without dispersion correction were found to be very similar and energy barriers calculated for the species optimized at B97D/LACVP* level close to the values obtained through single-point corrections calculated for the B3LYP/LACVP* geometries (see Table S4 and S5 in the Supporting Information).
Table 1. Gibbs Free Energies of Activation (ΔG⧧) and Gibbs Free Energies of Reaction (ΔGr) for the O–O Cleavage Leading from FeIII–OOH to HO–FeV=O Species Calculated Using B3LYP, B97D, B3LYP-D2, and B3LYP-D3 Functionals with the LACV3P+* Basis Set for Geometries Optimized at the B3LYP/LACVP* Level.
| B3LYP |
B97D |
B3LYP-D2 |
B3LYP-D3 |
|||||
|---|---|---|---|---|---|---|---|---|
| ΔG⧧ | ΔGr | ΔG⧧ | ΔGr | ΔG⧧ | ΔGr | ΔG⧧ | ΔGr | |
| model 1 | 24.7 | 16.3 | 15.5 | 6.1 | 24.6 | 15.5 | 26.6 | 17.4 |
| model 2 | 28.4 | 19.5 | 14.6 | 8.8 | 24.1 | 19.2 | 25.6 | 21.0 |
| model 3 | 27.8 | 19.1 | 16.7 | 6.6 | 26.0 | 16.9 | 27.8 | 18.1 |
| model 4 | 26.4 | 16.7 | 15.0 | 4.1 | 24.9 | 14.7 | 26.4 | 16.1 |
Spin populations derived from the Natural Bond Orbital analysis from triple-ζ basis set are reported. Where needed, Mulliken atomic spin densities are also provided for comparison of the presented results with the data reported in the literature. The dioxygen–iron complex was modeled in the sextet state (S = 5/2) as a positively charged protonated species, i.e. [FeIII–OOH]2+, which was found to be the most energetically favorable arrangement of the dioxygen–enzyme complex in naphthalene dioxygenase.27 Additionally, the feasibility of the attack of the HO–FeIV=O species (S = 2) on the aromatic ring of nitrobenzene was assessed using the smallest model of the active site. All calculations were performed using the Gaussian 09 electronic structure package,70 and structures were produced with VMD.71
Results and Discussion
cis-Dihydroxylation through Direct FeIII–OOH Attack, with an Epoxide Intermediate
In their computational study of the cis-dihydroxylation mechanism in naphthalene 1,2-dioxygenase (NDO), Bassan et al.27 investigated how the hydroperoxo–iron(III) species may directly attack the substrate, naphthalene, to form a cis-diol. Based on the results obtained for the three different models of the active site using gas phase optimization at the B3LYP/LACVP level, they concluded that the most feasible reaction pathway involves the formation of an epoxide intermediate from the direct attack of the hydroperoxo moiety on the substrate double bond. In their proposed mechanism, the proximal O atom of the hydroperoxo ligand initially attacks the C=C bond, with distances of 2.16 and 2.20 Å. Two electrons are transferred from the substrate to the O–O σ* antibonding orbital directly as confirmed by the lack of significant change in the Fe Mulliken spin density at the transition state. Thus, the O–O bond is broken concomitantly with C–O bond formation. An epoxide intermediate is formed, but the reaction is not complete, as indicated by the long C–O bond distances (1.56 and 1.58 Å) and the residual spin density (0.08) on the reacting O atom. The reaction further evolves toward a carbocation, which originates from the cleavage of the epoxide C–O bond. At this stationary point, the spin density of Fe remains at around 4 while that of the proximal O atom increases to 0.26. Eventually the product, cis-diol, is formed by binding of the cation to the OH ligand of the iron intermediate. The attack of OH on the other carbon to form the cis-diol has a very small barrier (∼1 kcal/mol), consistent with the early transition state (C–OH bond distance of 2.60 Å). The rate-determining step was found to be the formation of an epoxide, with the concomitant cleavage of the oxygen–oxygen bond in the ferric hydroperoxo moiety. The activation energy for this concerted step calculated for the medium-size model was 17.5 kcal mol–1 (ΔG⧧ = 19.1 kcal mol–1), which was confirmed using the larger model.
Nitrobenzene dioxygenase shares 80% sequence identity with naphthalene 1,2-dioxygenase and is also capable of cis-dihydroxylation of naphthalene.1 Hence, we decided to investigate whether the chemical pattern proposed for the reaction occurring in the NDO active site is feasible for the formation of cis-dihydrodiol from nitrobenzene catalyzed by NBDO. The transition state leading to the formation of an epoxide was located for the two smallest models of the NBDO active site used in this study, namely, the one including only first shell iron ligands (model 1) and a larger system, which additionally contains Asn258 residue (model 2). Model 1 used here corresponds to the active site model used in ref (27), but unlike the latter study geometry optimizations for both models were performed at the B3LYP/LACVP* level with continuum solvation (dielectric constant of 5.7) to account for electrostatic effects from the protein environment. Using model 1, we also obtained the transition state involving concerted O–O bond cleavage and C–O bond formation. In our case, C–O bond distances are 1.86 and 2.45 Å while the Fe Mulliken spin density is 4.07. In contrast to Bassan et al., epoxide formation is complete (as indicated by C–O bond distances of 1.39 and 1.45 Å, and zero spin density on the O atom). The activation barrier for the formation of an epoxide was computed to be 20.6 kcal mol–1 (15.3 kcal mol–1 relative to separate reactants). Similarly to what was observed for naphthalene, the reaction is exothermic, with the epoxide intermediate lying 14.9 kcal mol–1 lower in energy than the reactant complex (20.1 kcal mol–1 lower relative to separate reactants), but contrary to the results reported for NDO, we do not observe the formation of an epoxide intermediate bound to mononuclear iron, which may further evolve into a carbocation. Instead, in the intermediate state originating from the direct attack of FeIII–OOH on the nitrobenzene C1–C2 double bond (see Figure 4 for atom numbering), the O–Fe bond is broken and the epoxide species dissociates from the iron cluster. Dissociation of the epoxide results in a change of the iron coordination sphere, as illustrated in Figure 3a. The geometry of the optimized intermediate is confirmed using model 2. Even in the presence of the Asn258 residue, which forms a hydrogen bond with the nitro group of nitrobenzene, the distance between the arene oxide and the iron atom elongates upon optimization, leaving the hydroxo group as the only ligand coordinating the face of the Fe cluster. The epoxide intermediate obtained with model 2 is shown in Figure 3b.
Figure 4.

Three routes of the attack of FeIII–OOH (a) or HO–FeV=O (b) intermediate on the substrate considered as possible initial steps of its cis-dihydroxylation.
Figure 3.
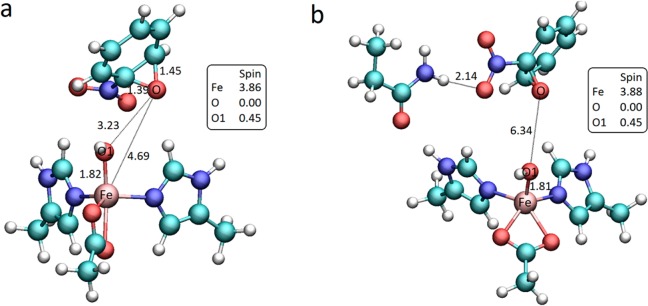
Optimized geometries, relevant spin populations, and bond lengths (Å) of (a) epoxide intermediate obtained for the smallest model of NBDO active site; (b) epoxide intermediate for model 2, additionally containing Asn258 residue.
In order to examine whether our findings are independent of the choice of the methodology, i.e., gas phase vs continuum solvent optimization, and to compare the feasibility of the mechanism involving an epoxide intermediate for nitroaromatic and nonsubstituted aromatic substrates, we additionally reoptimized the stationary points for both models in the gas phase using the B3LYP functional and LACVP basis set as in ref (27). We have reproduced the transition state preceding epoxide formation obtained by Bassan et al. for naphthalene, and additionally investigated the same step using benzene as a substrate, both in the gas phase and continuum solvent model. In the two cases where the substrate does not contain a nitro group, the reaction proceeds via the chemical pattern reported for NDO. In the case of the gas phase reaction of nitrobenzene, the epoxide dissociation from the Fe center is still observed and the activation energy calculated at the B3LYP/LACV3P**//B3LYP/LACVP level is 33.2 and 38.7 kcal mol–1 for model 1 and model 2, respectively These activation barriers are much higher than the value reported by Bassan et al. (17.5 kcal mol–1), which may be partially attributed to neglecting solvent effects in energy calculations and spurious interactions found in the gas phase optimized reactant complexes, especially in model 2. The B3LYP activation barriers calculated for the species optimized in continuum solvent model are 31.2 and 33.6 kcal mol–1 for model 1 and model 2, respectively, and further decrease to ∼21–26 kcal mol–1 when dispersion correction is included in B3LYP calculations (Tables S1, S2, and S3 in the Supporting Information). At the same time, gas phase activation energies calculated for nonsubstituted aromatic substrates, benzene and naphthalene, are much lower than the barrier obtained for nitrobenzene and in close agreement with the values previously calculated for naphthalene27 (for details see the Supporting Information). As already mentioned, in the epoxide mechanism, two electrons from the substrate are transferred to the O–O σ* antibonding orbital leading to concerted O–O bond cleavage and C–O bond formation. The increased barrier for our substrate can be attributed to the nitro substituent, which tends to make electrons less available for transfer to the active site.
The dissociation of the epoxide intermediate from the active site suggests that the previously proposed mechanism for naphthalene dihydroxylation by NDO is not feasible for nitroaromatic compounds. However, in principle one could consider a possible return of the epoxide intermediate to the metal cluster, or its hydrolysis, which could take place outside the active site. The latter reaction would yield nonstereospecific products, as water can attack at any side of the ring, hence it is an unlikely scenario for the cis-diol formation from the dissociated epoxide intermediate. The fact that 18O labeling studies show that both O atoms originate from the same source also discounts the possibility of cis-diol formation from the hydrolysis of the dissociated epoxide.2,25,28−30 On the other hand, the return of the epoxide intermediate to the active site could possibly allow the reaction to proceed to the next step, i.e., formation of a cation intermediate through the opening of the epoxide ring (Scheme 1).
Scheme 1.
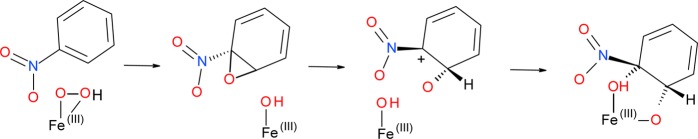
Indeed, using model 1 we were able to locate a transition state for the epoxide ring-opening (see Figure S2 in the Supporting Information), which lies 18.6 kcal mol–1 above the preceding (dissociated) epoxide intermediate and is lower in energy than the transition state for the epoxide formation by 16.8 kcal mol–1. The reaction pathway followed from this transition state by integrating the intrinsic reaction coordinate72 leads to an intermediate with a single C–O bond, optimization of which, however, does not yield an expected cation intermediate. Instead, we observe a barrierless hydride shift resulting in the formation of a ketone (Scheme 2, Figure S2 in the Supporting Information).
Scheme 2.
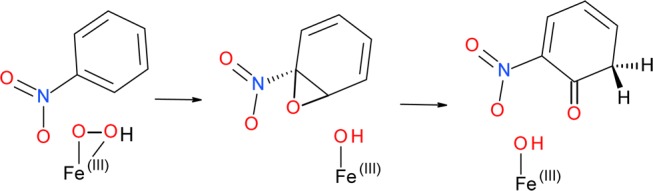
Such transformation was also observed by Bassan et al., who also mentioned that epoxide ring-opening can lead to a 1,2-hydride shift to form a ketone, for which the barrier is almost zero. They attributed formation of the cis-diol through a cationic species, despite the slightly larger barrier (∼1 kcal/mol), to protein effects. Possibly due to the presence of the electron-withdrawing nitro group, the cation intermediate proposed by Bassan et al. is even less stable in the case of nitrobenzene, and we were not able to obtain such in our calculations. Hence, the final product could not be obtained from a nitroaromatic substrate through an epoxide pathway. It should be noted, however, that we acknowledge the possibility of the current findings being related to the limitations of the adopted model that only partially accounts for the protein environment, which in this particular case might be crucial for the stabilization of the intermediate products formed along the reaction pathway. Considering the discussed differences found between the results presented here, and those reported for naphthalene,27 and a relatively low barrier of the epoxide formation compared to alternative ways of the attack on the ring (see the next section), the discussed pathway should and will be examined as a possible cis-dihydroxylation mechanism in the hybrid QM/MM calculations, which will follow the current study.
Finally, an important consideration in the discussion of the epoxide pathway is that our results are in fact consistent with an observation that RDOs generally do not catalyze epoxidation, even as an intermediate step.9 This was shown through the oxidation of styrene, which was used as a probe for epoxide formation since it is oxidized to styrene 1,2-oxide by cytochrome P-450 and other monooxygenases. Unlike the latter enzymes, NDO catalyzed the formation of (R)-1-phenyl-1,2-ethanediol. Moreover, styrene 1,2-oxide was not a substrate for NDO, indicating that the diol is formed directly and not through epoxide hydrolysis.73
Probing Alternative Mechanisms Initiated by the FeIII–OOH Attack on the Aromatic Ring
Since the mechanism involving an epoxide intermediate was found not to be plausible for the cis-dihydroxylation of nitroaromatic substrates, it was necessary to probe other chemical routes for such transformation, initiated by the direct attack of the FeIII–OOH intermediate on the ring. Three possible ways of the initial attack on the ring were considered here: the attack of the hydroxyl group on the C2 carbon, and the attack of the second oxygen atom of the hydroperoxo group on either C1 or C2 carbon, as shown in Figure 4. For each of the considered pathways, we located the corresponding transition state for the concerted attack (with concomitant O–O cleavage) and the transition state leading to the formation of a single C–O bond without the cleavage of the oxygen–oxygen bond in the ferric hydroperoxo moiety (nonconcerted attack). Concerted O–C1 attack leads to the formation of an epoxide intermediate, which was discussed in the previous section. Table 2 summarizes the activation barriers obtained for the considered scenarios.
Table 2. Gibbs Free Energies of Activation (ΔG⧧) for the Initial Attack of the FeIII–OOH Moiety on Nitrobenzene, Calculated for Different Models of the Active Sitea Relative to Reactant Complex.
| ΔG⧧ [kcal mol–1] |
||||||
|---|---|---|---|---|---|---|
| nonconcerted |
concerted |
|||||
| O1–C2 attack | O–C1 attack | O–C2 attack | O1–C2 attack | O–C1 attack | O–C2 attack | |
| model 1 | 43.9 | 38.7 | 25.7 | 20.6 | 19.5 | |
| model 2 | 43.4 | 37.5 | 22.0 | |||
| model 3 | 31.4 | 39.5 | 22.8 | |||
| model 4 | 29.1 | 37.5 | 29.9 | |||
See text for description.
In each of the probed mechanisms, the attack of the oxygen atom on the aromatic ring without the oxygen–oxygen bond cleavage was found to have a higher energetic barrier than the corresponding concerted attack. The lowest Gibbs free energy of activation was found for the concerted O–C2 attack, which leads to the formation of a single O–C2 bond or an epoxide intermediate with C2 and C3 carbon bonded to O atom, from neither of which we were able to obtain a final product. We also probed whether the nonconcerted O–C2 attack can initiate a reaction pathway leading to the formation of a diol, as in the case of model 3 we were able to locate a transition state with a relatively low barrier, compared to other nonconcerted attacks. In this particular case, an intermediate with a single C–O bond formed in the first step of the reaction can evolve toward an epoxide, which is formed upon an O–O bond cleavage. Similarly to what was observed for the pathway discussed earlier, the epoxide species dissociates from the iron cluster, despite the presence of Asn258 residue, as it is the case in model 3. Hence, within the scope of the current study, this pathway does not seem viable for cis-dihydroxylation of nitroaromatic substrates. It is worth mentioning that it resembles to a large extent an alternative mechanism for the formation of the cis-diol from naphthalene presented by Bassan et al., which involves an iron(II)–hydroperoxyl species.27 In the reaction presented therein, a similar, nonconcerted attack of the hydroperoxo ligand on the substrate leads to the formation of a radical intermediate with a single C–O bond, this step being associated with the barrier of 19–20 kcal mol–1 and endothermicity of around 18 kcal mol–1. The subsequent step of the reaction yields a cis-diol, but the associated transition state is higher in energy than the preceding one, based on which the mechanism was considered as rather unlikely. In our case, a radical is also delocalized on the substrate, evidenced by Mulliken spin density on nitrobenzene of 0.35 and 0.76 in the transition state and intermediate product, respectively. Similarly to what was observed for naphthalene, this radical intermediate has a very high energy compared to reactant complex, exhibiting a large negative driving force of this step of the reaction. The estimated energy of the transition state associated with the cleavage of the O–O bond is similar to the transition state for the formation of a radical intermediate.
A concerted C–O bond formation and oxygen–oxygen bond cleavage mechanism for the three possible modes of FeIII–OOH attack was also examined using model 1 with 2-nitrotoluene as a substrate. Activation energies are in agreement with the barriers obtained for the other substrate, with the values of 23.6, 21.2, and 21.1 kcal mol–1 for the O1–C2, O–C1, and O–C2 attack, respectively. Similarly to what was found for nitrobenzene, the resulting intermediates did not lead to the formation of a diol.
Barriers calculated using the dispersion-corrected B3LYP functional (Tables S2 and S3 in the Supporting Information) show a similar picture of the plausibility of direct reaction between the substrate and FeIII–OOH. In general, nonconcerted FeIII–OOH attacks on the ring have higher activation energies than corresponding reactions coupled with the O–O cleavage. An exception to this, as in the case of B97D results, is a barrier for the nonconcerted O–C2 attack in model 3. The barrier for the concerted O–C1 attack yielding an epoxide intermediate of around 25–26 kcal mol–1 is larger than the one obtained with the B97D functional, but at the same time according to B3LYP-D results it is the most favorable pathway involving the FeIII–OOH attack on the ring. As mentioned above, a similar barrier was found for a nonconcerted O–C2 attack, and a concerted O–C2 attack, which was excluded from further consideration as not leading to the final product, is also not energetically favored over the concerted O–C1 attack.
Oxygen–Oxygen Bond Cleavage Leading to the Formation of the HO–FeV=O Species
Subsequently, we considered the possibility of the formation of a high-valent intermediate, HO–FeV=O, prior to substrate oxidation. In low-spin biomimetic non-heme iron complexes, oxygen–oxygen bond cleavage is facilitated by the formation of a five-membered intermediate with water, as evident from exchange with 18O-labeled solvent.48,49,63 However, this would always lead to a mixed product, whereas in the cis-diol obtained from the enzyme-catalyzed reaction, both hydroxyl groups originate predominantly from a single oxygen source.2,25,28−30 It is thus more likely that HO–FeV=O is formed directly via heterolytic O–O bond cleavage in RDOs. This involves initial transfer of an α-electron from Fe to the O–O bond σ* orbital, as evidenced by the decreased spin density on Fe and increased spin density on O1. At the transition state, the O–O bond increases to ∼2 Å while the Fe=O double bond starts to form. Transfer of the second electron to the σ* orbital completes O–O bond cleavage and formation of HO–FeV=O.
The Gibbs free energy of activation for O–O bond heterolysis was also determined in the presence of the substrate using different models of the enzyme. The barrier is only 15.5 kcal mol–1 for the smallest model (model 1) and is decreased to 14.6 kcal mol–1 upon inclusion of Asn258 in model 2. Interestingly, in the case of model 3, which additionally contains a water molecule, the barrier slightly increases to 16.7 kcal mol–1, while in turn, model 4, which includes Asn199, which is within H bonding distance of the hydroperoxo ligand and is postulated to be connected to a water channel that facilitates proton transfer,23 yields a lower barrier of 15.0 kcal mol–1. In general, heterolytic oxygen–oxygen bond cleavage to form HO–FeV=O is more energetically favorable than substrate oxidation by FeIII–OOH. This suggests that the reaction proceeds through the formation of the high-valent oxo species and its subsequent attack on the ring, rather than the direct attack of the ferric hydroperoxo ligand on the substrate. It should be noted, however, that current results do not firmly elucidate the role of protein environment, including Asn258 or Asn199 residues, in facilitating heterolytic oxygen–oxygen bond cleavage, and further study of this problem is desired.
The activation energies for the O–O cleavage calculated using B3LYP-D functionals are much higher than those obtained with the B97D functional and range from 24.1 to 27.8 kcal mol–1 (Table 2). Depending on the model used, these values become comparable or larger by up to 3.9 kcal mol–1 than the barriers for the initial attack of FeIII–OOH on the substrate ring, which, considering the accuracy of the calculated energies, still allows us to consider the formation of HO–FeV=O species and its subsequent reaction with the substrate as a plausible scenario for its cis-dihydroxylation.
Possible Ways of the HO–FeV=O Attack on the Substrate and the Mechanism of Its cis-Dihydroxylation
The lower barrier for the formation of HO–FeV=O, in comparison with a direct reaction between FeIII–OOH and the substrate, suggests that the high-valent intermediate is a possible oxidant. This would be consistent with minor 18O incorporation (3%) observed in naphthalene dioxygenation in the presence of H218O. A separate experiment with naphthalene cis-diol and H218O confirms that solvent exchange occurs with a reactive intermediate and not with the product.2518O-labeled products may be attributed to rapid intermolecular exchange between HO–FeV=O and H218O prior to substrate oxidation.74,75 Therefore, we probed possible dioxygenation pathways starting from a direct attack of HO–FeV=O on the substrate (Figure 4b).
The barriers computed for the initial formation of an oxygen–carbon bond presented in Table 3 prove this step to be in general more energetically favorable when the substrate is attacked by an HO–FeV=O species rather than by the ferric hydroperoxo intermediate discussed before.
Table 3. Gibbs Free Energies of Activation (ΔG⧧) for the Initial Attack of the HO–FeV=O Moiety on Nitrobenzene, Calculated for All Models of the Active Site Relative to Reactant Complexa.
| ΔG⧧ [kcal mol–1] |
|||
|---|---|---|---|
| O1–C2 attack | O–C1 attack | O–C2 attack | |
| model 1 | 7.0 (13.1) | 9.8 (15.8) | 6.3 (12.3) |
| model 2 | 2.4 (11.2) | 6.5 (15.3) | 4.0 (12.8) |
| model 3 | 8.0 (14.6) | 7.1 (13.7) | 4.4 (11.0) |
| model 4 | 7.4 (11.5) | 8.3 (12.4) | 5.9 (10.0) |
In parentheses, values calculated relative to the FeIII–OOH reactant complex are given.
The results obtained for the two smallest models indicate that the O1–C2 attack is the first step of the reaction, while for models 3 and 4, a noticeably smaller barrier is found for the O–C2 attack. The latter findings may be, however, affected by the presence of a water molecule in the active site model. It can be expected that the lowering of the activation barrier for the two largest models, compared to the values computed for models 1 and 2, might be the effect of the inclusion of a water molecule and its stabilization in the continuum solvent used in this study. Moreover, in the current study, we were not able to obtain a final product in the reaction starting with the O–C2 attack on the ring. A comparison between the barriers for all possible initial steps of the reaction also shows that the formation of a single C–O bond initiated by the attack of hydroxyl oxygen of the iron(V)–oxo complex on the C2 carbon is the most feasible transformation, from which the reaction can evolve toward the final product. Similar activation energies were computed for the initial step of 2-nitrotoluene oxidation, with ΔG⧧ of 0.4 and 2.4 kcal mol–1 for the O1–C2 attack in models 1 and 2, respectively. A barrier of 4.8 kcal mol–1 calculated for the O–C2 attack in model 1 of 2-nitrotoluene also agrees well with the ordering of the possible transition states located for nitrobenzene.
The barriers calculated using B3LYP-D functionals agree very well with the results presented in Table 3, and confirm low activation energies for the HO–FeV=O attack compared to a direct reaction of FeIII–OOH with the substrate (Tables S2 and S3 in the Supporting Information). Likewise, the B3LYP-D results demonstrate that the energetically most favorable step initiating cis-dihydroxylation is the formation of an O1–C2 bond through an HO–FeV=O attack on the aromatic ring.
The mechanism of cis-dihydroxylation, as shown in Figure 5, was studied in detail using models 1 and 2. Initial attack of the hydroxo ligand on the C2 carbon is the rate-determining step with a Gibbs free energy of activation of 7.0 kcal/mol in model 1 and 2.4 kcal/mol in model 2. The higher barrier in model 1 may be partly due to artificial hydrogen bonding between the substrate and the hydroperoxo ligand in the reactant complex, which is eliminated in the larger models upon inclusion of Asn258. The increase in the barrier to 8.0 and 7.4 kcal/mol in models 3 and 4, respectively, suggests that structural contributions from the protein environment have a significant effect on reaction energetics. Steric, as well as electrostatic, effects will be investigated further in a QM/MM study. As the C2–O1 bond forms in the transition state (dC–O = 2.12 Å in both models, Figures 6 and 7), an electron is transferred to the active site, resulting in radical formation in the substrate (spin density of 0.5). Radical formation, as confirmed by the spin density of ∼1 on the substrate in the intermediate state, is exothermic by 5.5 and 7.4 kcal/mol in models 1 and 2 (Table 4). Fe is reduced to the +4 oxidation state in the first step of the reaction, leading to a spin density of 2.9. In the final step, the oxo ligand attacks the C1 carbon with an activation barrier of 3.2 and 2.8 kcal/mol in models 1 and 2 (Table 4). The low barrier is consistent with an early transition state as evidenced by the long C1–O bond distance (2.48 Å in model 1, Figure 6, and 2.49 Å in model 2). The other electron from the substrate is transferred to the active site during this step, which is reflected in the decreased spin density on nitrobenzene (0.8) and increased spin density on Fe (3.1). cis-Diol formation is exothermic by 42.9 and 47.1 kcal/mol in models 1 and 2 (Table 4). This is accompanied by reduction of the active site back to the initial ferric state, where the spin density on Fe is about 3.9. Upon the formation of the diol, the distance between the nitro group and C1 carbon elongates to 1.6 Å, with the substituent still bonded to the aromatic ring, which agrees with the general reaction scheme of Rieske dioxygenases suggested in the literature, according to which the nitrite release occurs in the subsequent step of the catalytic cycle.
Figure 5.

Reaction profile of cis-dihydroxylation of nitrobenzene through a direct attack of the HO–FeV=O species on the aromatic ring. The figure shows Gibbs free energy diagram obtained for the smallest model of the NBDO active site.
Figure 6.

Optimized geometries, relevant spin populations, and bond lengths (Å) of the key stationary points located along the reaction pathway depicted in Figure 5: TS1, transition state for the initial attack of the iron(V)–oxo intermediate on the carbon–carbon double bond; INT1 radical intermediate formed upon the formation of a single C–O bond; TS2 transition state for the oxygen attack on the C1 carbon yielding the cis-dihydrodiol PC. Spin population for nitrobenzene (NBZ) was summed over all atoms of the ring and the nitro group.
Figure 7.
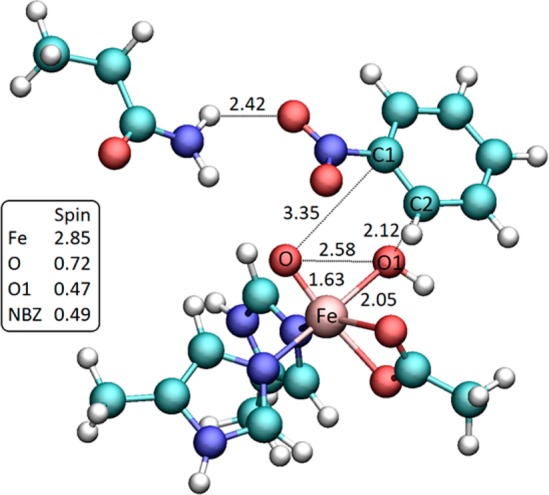
Transition state for the attack of the iron(V)–oxo species on nitrobenzene ring obtained for model 2 of NBDO active site, containing first-shell iron ligands and Asn258 residue. Key distances and relevant spin population are reported.
Table 4. Reaction Profiles for the cis-Dihydroxylation of Nitrobenzene and 2-Nitrotoluene According to the Mechanism Passing through a Radical Intermediate Formed in a Direct Attack of HO–FeV=O Species on the Substratea.
| ΔG [kcal mol–1] |
||||
|---|---|---|---|---|
| nitrobenzene |
2-nitrotoluene |
|||
| model 1 | model 2 | model 1 | model 2 | |
| reactant complex | 0.0 | 0.0 | 0.0 | 0.0 |
| TS1 | 7.0 | 2.4 | 0.4 | 2.4 |
| radical intermediate | –5.5 | –7.4 | –6.9 | –9.2 |
| TS2 | –2.4 | –4.6 | –4.3 | –5.6 |
| diol | –42.9 | –47.1 | –47.5 | –48.0 |
The Gibbs free energy values were obtained using two different models of the active site (see text for details).
Table 4 also shows the energy profiles calculated for 2-nitrotoluene undergoing the same reaction mechanism. The energy diagrams for both substrates are very similar, but in the case of 2-nitrotoluene, we do not observe lowering of the activation barrier for the first step of the reaction while moving from model 1 to model 2. The geometries of the stationary points obtained for 2-nitrotoluene in the two smallest models are analogous to the ones located for nitrobenzene, and we do not observe significant differences between the two compounds undergoing the suggested chemical pattern.
The cis-dihydroxylation mechanism of nitrobenzene by NBDO is similar to that of olefins catalyzed by the biomimetic complexes Fe–Pytacn48 and Fe–TPA.64 However, the second C–O bond formation is not barrierless in the present case possibly due to the stability of the nitrobenzyl radical intermediate. Heme enzymes, which also have an iron–oxo active site, catalyze epoxidation of the C=C bond instead of cis-dihydroxylation due to the absence of two cis labile sites. Other enzymes with a 2-His-1-carboxylate facial triad motif have an FeIV=O oxidant due to the presence of a cofactor or cosubstrate that donates an additional electron. As in principle this species can also be formed during the NBDO catalytic process, provided that a second electron is supplied to the active site, we have calculated the activation barriers for the possible attacks of this intermediate on the substrate as well. If HO–FeIV=O is used as an oxidant, the barriers for the O1–C2, O–C1, and O–C2 attack on the nitrobenzene ring become 24.8, 25.3, and 14.7 kcal mol–1, respectively, which indicates that HO–FeIV=O is in general less reactive than the HO–FeV=O complex. Nevertheless, the possibility of the product being formed in the most favorable pathway involving HO–FeIV=O species, i.e., the one starting from the O–C2 attack, was explored. The first step of the reaction yields a radical intermediate with a single C–O bond (Scheme 3), formed with endothermicity of 12.1 kcal mol–1.
Scheme 3.
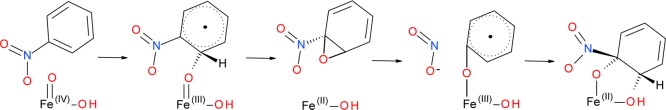
Subsequently, an epoxide intermediate is formed with a barrier of 13.7 kcal mol–1, which is comparable to the barrier associated with the first step of the reaction. The epoxide intermediate is very high in energy, lying 22.4 kcal mol–1 above the initial reactant complex, and similarly to what was observed for the epoxide mechanism described before, it dissociates from the iron center (see Figure S3 in the Supporting Information). Assuming the return to the active site was possible, the transition state for the epoxide ring-opening was located and the barrier associated with this step of the reaction was computed to be 4.3 kcal mol–1. In the resulting intermediate, upon the formation of a single C1–O bond, the nitro group of the substrate is released, as evidenced by the C1–N bond elongated to 3.1 Å. The final step of the reaction involves the attack of hydroxyl oxygen on the C2 carbon, which is associated with the activation barrier of 13.7 kcal mol–1 and yields the final product, cis-dihydrodiol, with the nitro substituent rebound to the substrate ring.
Conclusions
We have employed DFT methods to investigate the cis-dihydroxylation mechanism of nitrobenzene dioxygenase. Possible chemical patterns for dioxygenation of nitrobenzene and 2-nitrotoluene were probed using four models of NBDO active site. Based on the literature data, we assumed the iron–dioxygen ground state to be a sextet (S = 5/2) with +2 charge. Different ways of a direct attack of both hydroperoxo–iron(III) and iron(V)–oxo intermediate on the aromatic ring of the substrate were considered, and activation energies of the corresponding initial steps of the cis-dihydroxylation were calculated. The Gibbs free energy barriers where then used as a guideline in assessing the feasibility of a given mechanism. The activation barrier related to the oxygen–oxygen cleavage leading from the FeIII–OOH to the high-valent HO–FeV=O species was also evaluated. Finally, the reactivity of the HO–FeIV=O species (S = 2) toward the aromatic ring of nitrobenzene was assessed and compared with that of the iron(V)–oxo intermediate.
We have found that the Gibbs free energies of activation for the direct attack of the ferric hydroperoxo intermediate on the ring, occurring either with or without concomitant oxygen–oxygen bond rupture, are comparable to or higher than the activation barrier for the formation of the iron(V)–oxo species from its ferric precursor. This suggests the HO–FeV=O species to be the possible oxidant in the studied reaction. Moreover, the two cis-dihydroxylation pathways starting with a direct attack of the FeIII–OOH species on the ring, including the mechanism involving the epoxidation step suggested previously for NDO,27 which were recognized as energetically plausible, did not lead to the formation of the expected product, cis-diol. The reason for this is an instability of the intermediate products formed in these reactions, stemming from the presence of the nitro substituent, which might be overcome in the explicit protein environment. Thus, these mechanisms will be reconsidered as potentially viable and further investigated in our future QM/MM study. At the same time, the activation energies for the possible attacks of HO–FeV=O on the substrate aromatic ring were found to be much lower than the ones calculated for the FeIII–OOH intermediate, proving HO–FeV=O to be a stronger oxidant in the studied process. Furthermore, the most favorable reaction pathway found for this oxidant leads to the formation of a cis-dihydrodiol from both of the considered nitroaromatic substrates. The said mechanism starts with the rate-limiting attack of the hydroxyl oxygen on the C2 carbon of the substrate to form a radical intermediate, which is stabilized by the nitro substituent through spin delocalization. In the following step of the reaction, the second oxygen of the HO–FeV=O moiety attacks the C1 carbon forming a highly stabilized final product, a cis-dihydrodiol. Our proposed mechanism is also consistent with experimentally observed 18O incorporation in the cis-diol product, and will be used as a starting point in the investigation of steric and electrostatic effects accompanying the reaction taking place in the full enzyme environment.
As stated above, our conclusions were verified by performing calculations for both nitrobenzene and 2-nitrotoluene, with no significant differences found between the reactions modeled for both substrates. The calculations performed here for 2-nitrotoluene will also allow for the future comparison between the cis-dihydroxylation and monohydroxylation reactions of this substrate, both of which are catalyzed by NBDO. Lastly, the results obtained for the HO–FeIV=O species proved it to be in general less reactive than the HO–FeV=O complex. However, the pathway starting from the most favorable attack of the HO–FeIV=O on the substrate was also found to be yielding the final product.
Acknowledgments
This work is supported by these grants: PSRP-025/2010 from the Polish-Swiss Research Program and FP7-264329 from the Seventh Framework Programme, Maria Curie Action ITN “CSI:Environment”. Allocation of computer time at Academic Computer Centre CYFRONET, AGH, Krakow and Lodz University of Technology Computer Centre (under PLATON project), is acknowledged.
Supporting Information Available
Activation barriers for FeIII–OOH and HO–FeV=O attacks on nitrobenzene and 2-nitrotoluene calculated using the B3LYP and B3LYP-D functionals. Comparison between the Gibbs free activation energies and Gibbs free reaction energies for the O–O cleavage calculated for the stationary points optimized at the B3LYP/LACVP* level with single-point corrections using B97D, B3LYP-D2, or B3LYP-D3 and the corresponding values calculated for the stationary points reoptimized using a given dispersion-corrected functional. Barriers for the FeV attacks on nitrobenzene ring calculated for the structures optimized using the B97D functional. Activation energies for the formation of an epoxide intermediate for benzene and naphthalene; illustration of the transition state for the epoxide formation and the epoxide intermediate for benzene. Ketone intermediate formed from an epoxide intermediate and a preceding transition state for nitrobenzene. Stationary points along the O–C2 pathway involving HO–FeIV=O species. Comparison between Mulliken and NBO spin densities calculated for selected species. XYZ coordinates of all stationary points. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Friemann R.; Ivkovic-Jensen M. M.; Lessner D. J.; Yu C.-L.; Gibson D. T.; Parales R. E.; Eklund H.; Ramaswamy S. Structural Insight into the Dioxygenation of Nitroarene Compounds: The Crystal Structure of Nitrobenzene Dioxygenase. J. Mol. Biol. 2005, 348, 1139–1151. [DOI] [PubMed] [Google Scholar]
- Nishino S. F.; Spain J. C. Oxidative Pathway for the Biodegradation of Nitrobenzene by Comamonas sp. Strain JS765. Appl. Environ. Microbiol. 1995, 61, 2308–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju K.-S.; Parales R. E. Nitroaromatic Compounds, from Synthesis to Biodegradation. Microbiol. Mol. Biol. Rev. 2010, 74, 250–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D. R.; Bugg T. D. H. Arene Cis-Dihydrodiol Formation: From Biology to Application. Org. Biomol. Chem. 2006, 4, 181–192. [DOI] [PubMed] [Google Scholar]
- Ju K.-S.; Parales R. E. Control of Substrate Specificity by Active-Site Residues in Nitrobenzene Dioxygenase. Appl. Environ. Microbiol. 2006, 72, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parales R. E.; Huang R.; Yu C.-L.; Parales J. V.; Lee F. K. N.; Lessner D. J.; Ivkovic-Jensen M. M.; Liu W.; Friemann R.; Ramaswamy S.; Gibson D. T. Purification, Characterization, and Crystallization of the Components of the Nitrobenzene and 2-Nitrotoluene Dioxygenase Enzyme Systems. Appl. Environ. Microbiol. 2005, 71, 3806–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessner D. J.; Johnson G. R.; Parales R. E.; Spain J. C.; Gibson D. T. Molecular Characterization and Substrate Specificity of Nitrobenzene Dioxygenase from Comamonas sp. strain JS765. Appl. Environ. Microbiol. 2002, 68, 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro D. J.; Gakhar L.; Ramaswamy S. Rieske Business: Structure-Function of Rieske Non-Heme Oxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 175–190. [DOI] [PubMed] [Google Scholar]
- Wackett L. P. Mechanism and Applications of Rieske Non-Heme Iron Dioxygenases. Enzyme Microb. Technol. 2002, 31, 577–587. [Google Scholar]
- Furukawa K. Engineering Dioxygenases for Efficient Degradation of Environmental Pollutants. Curr. Opin. Biotechnol. 2000, 11, 244–249. [DOI] [PubMed] [Google Scholar]
- Wijker R. S.; Bolotin J.; Nishino S. F.; Spain J. C.; Hofstetter T. B. Using Compound-Specific Isotope Analysis to Assess Biodegradation of Nitroaromatic Explosives in the Subsurface. Environ. Sci. Technol. 2013, 47136872–6883. [DOI] [PubMed] [Google Scholar]
- Hofstetter T. B.; Spain J. C.; Nishino S. F.; Bolotin J.; Schwarzenbach R. P. Identifying Competing Aerobic Nitrobenzene Biodegradation Pathways by Compound-Specific Isotope Analysis. Environ. Sci. Technol. 2008, 42, 4764–4770. [DOI] [PubMed] [Google Scholar]
- Lee J.; Zhao H. Mechanistic Studies on the Conversion of Arylamines into Arylnitro Compounds by Aminopyrrolnitrin Oxygenase: Identification of Intermediates and Kinetic Studies. Angew. Chem., Int. Ed. 2006, 45, 622–625. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Bowers N. I.; Dalton H.; Garrett M. D.; Harrison J. S.; Sheldrake G. N. Dioxygenase-Catalysed Oxidation of Disubstituted Benzene Substrates: Benzylic Monohydroxylation versus Aryl Cis-Dihydroxylation and the Meta Effect. Org. Biomol. Chem. 2006, 4, 3343–3349. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Byrne B. E.; Haughey S. A.; Kennedy M. A.; Allen C. C. R. Dioxygenase-Catalysed Oxidation of Alkylaryl Sulfides: Sulfoxidation versus Cis-Dihydrodiol Formation. Org. Biomol. Chem. 2004, 2, 2530–2537. [DOI] [PubMed] [Google Scholar]
- Resnick S. M.; Lee K.; Gibson D. T. Diverse Reactions Catalyzed by Naphthalene Dioxygenase from Pseudomonas sp Strain NCIB 9816-4. J. Ind. Microbiol. Biotechnol. 1996, 17, 438–457. [Google Scholar]
- Gibson D. T.; Resnick S. M.; Lee K.; Brand J. M.; Torok D. S.; Wackett L. P.; Schocken M. J.; Haigler B. E. Desaturation, Dioxygenation, and Monooxygenation Reactions Catalyzed by Naphthalene Dioxygenase from Pseudomonas sp. Strain 9816. J. Bacteriol. 1995, 177, 2615–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick S. M.; Gibson D. T. Biotransformation of Anisole and Phenetole by Aerobic Hydrocarbonoxidizing Bacteria. Biodegradation 1993, 4, 195–203. [Google Scholar]
- Bruijnincx P. C. A.; van Koten G.; Gebbink R. J. M. K. Mononuclear Non-Heme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad: Recent Developments in Enzymology and Modeling Studies. Chem. Soc. Rev. 2008, 37, 2716–2744. [DOI] [PubMed] [Google Scholar]
- Ohta T.; Chakrabarty S.; Lipscomb J. D.; Solomon E. I. Near-IR MCD of the Nonheme Ferrous Active Site in Naphthalene 1,2-Dioxygenase: Correlation to Crystallography and Structural Insight into the Mechanism of Rieske Dioxygenases. J. Am. Chem. Soc. 2008, 130, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- Parales R. E. The Role of Active-Site Residues in Naphthalene Dioxygenase. J. Ind. Microbiol. Biotechnol. 2003, 30, 271–278. [DOI] [PubMed] [Google Scholar]
- Karlsson A.; Parales J. V.; Parales R. E.; Gibson D. T.; Eklund H.; Ramaswamy S. Crystal Structure of Naphthalene Dioxygenase: Side-on Binding of Dioxygen to Iron. Science 2003, 299, 1039–1042. [DOI] [PubMed] [Google Scholar]
- Wolfe M. D.; Parales J. V.; Gibson D. T.; Lipscomb J. D. Single Turnover Chemistry and Regulation of O2 Activation by the Oxygenase Component of Naphthalene 1,2-Dioxygenase. J. Biol. Chem. 2001, 276, 1945–1953. [DOI] [PubMed] [Google Scholar]
- Wolfe M. D.; Lipscomb J. D. Hydrogen Peroxide-coupled Cis-Diol Formation Catalyzed by Naphthalene 1,2-Dioxygenase. J. Biol. Chem. 2003, 278, 829–835. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Borowski T.; Siegbahn P. E. M. Quantum Chemical Studies of Dioxygen Activation by Mononuclear Non-Heme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad. Dalton Trans. 2004, 3153–3162. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Blomberg M. R. A.; Siegbahn P. E. M. A Theoretical Study of the Cis-Dihydroxylation Mechanism in Naphthalene 1,2-Dioxygenase. J. Biol. Inorg. Chem. 2004, 9, 439–452. [DOI] [PubMed] [Google Scholar]
- An D.; Gibson D. T.; Spain J. C. Oxidative Release of Nitrite from 2-Nitrotoluene by a Three-Component Enzyme System from Pseudomonas sp. Strain JS42. J. Bacteriol. 1994, 176, 7462–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. B.; Spain J. C.; Haddock J. D.; Gibson D. T. Oxidation of Nitrotoluenes by Toluene Dioxygenase: Evidence for a Monooxygenase Reaction. Appl. Environ. Microbiol. 1992, 58, 2643–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackett L. P.; Kwart L. D.; Gibson D. T. Benzylic Monooxygenation Catalyzed by Toluene Dioxygenase from Pseudomonas putida. Biochemistry 1988, 27, 1360–1367. [DOI] [PubMed] [Google Scholar]
- Carredano E.; Karlsson A.; Kauppi B.; Choudhury D.; Parales R. E.; Parales J. V.; Lee K.; Gibson D. T.; Eklund H.; Ramaswamy S. Substrate Binding Site of Naphthalene 1,2-Dioxygenase: Functional Implications of Indole Binding. J. Mol. Biol. 2000, 296, 701–712. [DOI] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol. 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva E. G.; Neibergall M. B.; Chakrabarty S.; Lipscomb J. D. Finding Intermediates in the O2 Activation Pathways of Non-Heme Iron Oxygenases. Acc. Chem. Res. 2007, 40, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs-Gelasco P. J.; Price J. C.; Guyer R. B.; Brehm J. H.; Barr E. W.; Bollinger J. M. Jr.; Krebs C. EXAFS Spectroscopic Evidence for an Fe=O Unit in the Fe(IV) Intermediate Observed during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126, 8108–8109. [DOI] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Henshaw T. F.; Monterosso G. R.; Ryle M. J.; Hausinger R. P. Direct Detection of Oxygen Intermediates in the Non-Heme Fe Enzyme Taurine/α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126, 1022–1023. [DOI] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M. Jr.; Krebs C. The First Direct Characterization of a High-valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- Eser B. E.; Barr E. W.; Frantom P. A.; Saleh L.; Bollinger J. M. Jr.; Krebs C.; Fitzpatrick P. F. Direct Spectroscopic Evidence for a High-Spin Fe(IV) Intermediate in Tyrosine Hydroxylase. J. Am. Chem. Soc. 2007, 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panay A. J.; Lee M.; Krebs C.; Bollinger J. M. Jr.; Fitzpatrick P. F. Evidence for a High-Spin Fe(IV) Species in the Catalytic Cycle of a Bacterial Phenylalanine Hydroxylase. Biochemistry 2011, 50, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker A.; Solomon E. I. Dioxygen Activation by Copper, Heme and Non-Heme Iron Enzymes: Comparison of Electronic Structures and Reactivities. Curr. Opin. Chem. Biol. 2005, 9, 152–163. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Decker A.; Lehnert N. Non-Heme Iron Enzymes: Contrasts to Heme Catalysis. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier B.; de Visser S. P.; Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- Green J.; Dalton H. Further Evidence for Multiple Pathways in Soluble Methane-Monooxygenase-Catalysed Oxidations from the Measurement of Deuterium Kinetic Isotope Effects. Eur. J. Biochem. 1994, 226, 555–560. [DOI] [PubMed] [Google Scholar]
- Green J.; Dalton H. Substrate Specificity of Soluble Methane Monooxygenase. J. Biol. Chem. 1989, 264, 17698–17703. [PubMed] [Google Scholar]
- Que L. Jr. The Oxo/Peroxo Debate: a Nonheme Iron Perspective. J. Biol. Inorg. Chem. 2004, 9, 684–690. [DOI] [PubMed] [Google Scholar]
- Chen K.; Que L. Jr. Cis-Dihydroxylation of Olefins by a Non-Heme Iron Catalyst: A Functional Model for Rieske Dioxygenases. Angew. Chem., Int. Ed. 1999, 38, 2227–2229. [DOI] [PubMed] [Google Scholar]
- Oldenburg P. D.; Ke C.-Y.; Tipton A. A.; Shteinman A. A.; Que L. Jr. A Structural and Functional Model for Dioxygenases with a 2-His-1-Carboxylate Triad. Angew. Chem., Int. Ed. 2006, 45, 7975–7978. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Ke C.-Y.; Xue G.; Que L. Jr. Bio-inspired Arene Cis-Dihydroxylation by a Non-Haem Iron Catalyst Modeling the Action of Naphthalene Dioxygenase. Chem. Commun. 2009, 50–52. [DOI] [PubMed] [Google Scholar]
- Prat I.; Mathieson J. S.; Güell M.; Ribas X.; Luis J. M.; Cronin L.; Costas M. Observation of Fe(V)=O using Variable-Temperature Mass Spectrometry and its Enzyme-like C-H and C=C Oxidation Reactions. Nat. Chem. 2011, 3, 788–793. [DOI] [PubMed] [Google Scholar]
- Quiñonero D.; Morokuma K.; Musaev D. G.; Mas-Ballesté R.; Que L. Jr. Metal-Peroxo versus Metal-Oxo Oxidants in Non-Heme Iron-Catalyzed Olefin Oxidations: Computational and Experimental Studies on the Effect of Water. J. Am. Chem. Soc. 2005, 127, 6548–6549. [DOI] [PubMed] [Google Scholar]
- Feng Y.; England J.; Que L. Jr. Iron-Catalyzed Olefin Epoxidation and Cis-Dihydroxylation by Tetraalkylcyclam Complexes: The Importance of cis-Labile Sites. ACS Catal. 2011, 1, 1035–1042. [Google Scholar]
- Company A.; Gómez L.; Fontrodona X.; Ribas X.; Costas M. A Novel Platform for Modeling Oxidative Catalysis in Non-Heme Iron Oxygenases with Unprecedented Efficiency. Chem.—Eur. J. 2008, 14, 5727–5731. [DOI] [PubMed] [Google Scholar]
- Mas-Ballesté R.; Costas M.; van den Berg T.; Que L. Jr. Ligand Topology Effects on Olefin Oxidations by Bio-Inspired [FeII(N2Py2)] Catalysts. Chem.—Eur. J. 2006, 12, 7489–7500. [DOI] [PubMed] [Google Scholar]
- Oldenburg P. D.; Shteinman A. A.; Que L. Jr. Iron-Catalyzed Olefin Cis-Dihydroxylation Using a Bio-Inspired N,N,O-Ligand. J. Am. Chem. Soc. 2005, 127, 15672–15673. [DOI] [PubMed] [Google Scholar]
- Klopstra M.; Roelfes G.; Hage R.; Kellogg R. M.; Feringa B. L. Non-Heme Iron Complexes for Stereoselective Oxidation: Tuning of the Selectivity in Dihydroxylation Using Different Solvents. Eur. J. Inorg. Chem. 2004, 846–856. [Google Scholar]
- Roelfes G.; Vrajmasu V.; Chen K.; Ho R. Y. N.; Rohde J.-U.; Zondervan C.; la Crois R. M.; Schudde E. P.; Lutz M.; Spek A. L.; Hage R.; Feringa B. L.; Münck E.; Que L. Jr. End-on and Side-on Peroxo Derivatives of Non-Heme Iron Complexes with Pentadentate Ligands: Models for Putative Intermediates in Biological Iron/Dioxygen Chemistry. Inorg. Chem. 2003, 42, 2639–2653. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Costas M.; Que L. Jr. Iron-Catalyzed Olefin Cis-Dihydroxylation by H2O2: Electrophilic versus Nucleophilic Mechanisms. J. Am. Chem. Soc. 2003, 125, 9912–9913. [DOI] [PubMed] [Google Scholar]
- Chen K.; Costas M.; Kim J.; Tipton A. K.; Que L. Jr. Olefin Cis-Dihydroxylation versus Epoxidation by Non-Heme Iron Catalysts: Two Faces of an FeIII-OOH Coin. J. Am. Chem. Soc. 2002, 124, 3026–3035. [DOI] [PubMed] [Google Scholar]
- Chen K.; Costas M.; Que L. Jr. Spin State Tuning of Non-Heme Iron-Catalyzed Hydrocarbon Oxidations: Participation of FeIII-OOH and FeV=O intermediates. J. Chem. Soc., Dalton Trans. 2002, 672–679. [Google Scholar]
- Costas M.; Tipton A. K.; Chen K.; Jo D.-H.; Que L. Jr. Modeling Rieske Dioxygenases: The First Example of Iron-Catalyzed Asymmetric Cis-Dihydroxylation of Olefins. J. Am. Chem. Soc. 2001, 123, 6722–6723. [DOI] [PubMed] [Google Scholar]
- Que L. Jr.; Tolman W. B. Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- Oldenburg P. D.; Que L. Jr. Bio-Inspired Nonheme Iron Catalysts for Olefin Oxidation. Catal. Today 2006, 117, 15–21. [Google Scholar]
- Rohde J.-U.; Bukowski M. R.; Que L. Jr. Functional Models for Mononuclear Nonheme Iron Enzymes. Curr. Opin. Chem. Biol. 2003, 7, 674–682. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Blomberg M. R. A.; Siegbahn P. E. M.; Que L. Jr. A Density Functional Study of O-O Bond Cleavage for a Biomimetic Non-Heme Iron Complex Demonstrating an FeV-Intermediate. J. Am. Chem. Soc. 2002, 124, 11056–11063. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Blomberg M. R. A.; Siegbahn P. E. M.; Que L. Jr. Two Faces of a Biomimetic Non-Heme HO-FeV=O Oxidant: Olefin Epoxidation versus Cis-Dihydroxylation. Angew. Chem. 2005, 117, 2999–3001. [DOI] [PubMed] [Google Scholar]
- Hariharan P. C.; Pople J. A. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor. Chim. Acc. 1973, 28, 213–222. [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-Consistent Molecular Orbital Methods. 23. A Polarization-Type Basis Set for 2nd-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar]
- Hay P. J.; Wadt W. R. Ab Initio Effective Core Potentials for Molecular Calculations - Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar]
- Grimme S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–99. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parameterization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Fukui K. The Path of Chemical-Reactions - The IRC Approach. Acc. Chem. Res. 1981, 14, 363–68. [Google Scholar]
- Lee K.; Gibson D. T. Stereospecific Dihydroxylation of the Styrene Vinyl Group by Purified Naphthalene Dioxygenase from Pseudomonas sp. Strain NCIB 9816-4. J. Bacteriol. 1996, 178, 3353–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Company A.; Prat I.; Frisch J. R.; Mas-Ballesté D. R.; Güell M.; Juhász G.; Ribas X.; Münck D. E.; Luis J. M.; Que L. Jr.; Costas M. Modeling the Cis-Oxo-Labile Binding Site Motif of Non-Heme Iron Oxygenases: Water Exchange and Oxidation Reactivity of a Non-Heme Iron(IV)-Oxo Compound Bearing a Tripodal Tetradentate Ligand. Chem.—Eur. J. 2011, 17, 1622–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg P. D.; Feng Y.; Pryjomska-Ray I.; Ness D.; Que L. Jr. Olefin Cis-Dihydroxylation with Bio-Inspired Iron Catalysts. Evidence for an FeII/FeIV Catalytic Cycle. J. Am. Chem. Soc. 2010, 132, 17713–17723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.