

Abstract
The Rv1217c–Rv1218c multidrug efflux system, which belongs to the ATP-binding cassette superfamily, recognizes and actively extrudes a variety of structurally unrelated toxic chemicals and mediates the intrinsic resistance to these antimicrobials in Mycobacterium tuberculosis. The expression of Rv1217c–Rv1218c is controlled by the TetR-like transcriptional regulator Rv1219c, which is encoded by a gene immediately upstream of rv1218c. To elucidate the structural basis of Rv1219c regulation, we have determined the crystal structure of Rv1219c, which reveals a dimeric two-domain molecule with an entirely helical architecture similar to members of the TetR family of transcriptional regulators. The N-terminal domains of the Rv1219c dimer are separated by a large center-to-center distance of 64 Å. The C-terminal domain of each protomer possesses a large cavity. Docking of small compounds to Rv1219c suggests that this large cavity forms a multidrug binding pocket, which can accommodate a variety of structurally unrelated antimicrobial agents. The internal wall of the multidrug binding site is surrounded by seven aromatic residues, indicating that drug binding may be governed by aromatic stacking interactions. In addition, fluorescence polarization reveals that Rv1219c binds drugs in the micromolar range.
Keywords: Mycobacterium tuberculosis, transcriptional regulation, Rv1219c multidrug efflux regulator, multidrug resistance
Introduction
Tuberculosis (TB) caused by Mycobacterium tuberculosis is responsible for the death of approximately 2 million people each year and remains one of the most deadly diseases.1,2 Standard treatment of TB requires 6–9 months with multiple antibiotics.3 Noncompliance to this lengthy regimen results in relapses and selects for multidrug resistant (MDR) and extensively drug resistant (XDR) strains. MDR-TB are defined as strains that are resistant to at least the front-line antibiotics isoniazid and rifampicin, and XDR-TB is defined as resistant to rifampicin, isoniazid, fluoroquinolones, and at least one of the injectible second-line drugs. Totally drug-resistant TB was recently identified, indicating that even second-line drugs can be misused and become ineffective.4–8 Combined, the emergence of drug-resistant TB complicates treatment, poses a significant risk to global public health, and challenges our expectation for TB control and elimination.
Recent research indicates that multidrug resistance of M. tuberculosis is linked to constitutive or inducible expression of multidrug efflux pumps.9 Drug efflux pumps of bacteria have been categorized into five basic families: the ATP-binding cassette (ABC), resistance-nodulation-division (RND), multidrug and toxic compound extrusion, major facilitator, and small multidrug resistance families.10 In Gram-negative bacteria, efflux systems of the RND superfamily play major roles in the intrinsic and acquired tolerance of antibiotics and noxious chemicals.11 They are key mechanisms by which these pathogens survive in the presence of a variety of structurally unrelated, toxic compounds unfavorable for their survival. However, the ABC-type efflux transporters are not commonly linked to multidrug resistance. In M. tuberculosis, the ABC transporters Rv1217c–Rv1218c12 and Rv019413 are among the very few efflux proteins involved in resistance to structurally diverse antimicrobials.
Roughly 2.5% of the M. tuberculosis H37Rv genome encodes transport proteins of the ABC superfamily, but only a few of these transporters have been characterized. Recent work demonstrated that the ABC-type Rv1217c–Rv1218c efflux system functions as a multidrug efflux pump, extruding a wide range of structurally unrelated drugs, including novobiocins, pyrazolones, biarylpiperazines, bisanilinopyrimidines, pyrroles, and pyridones.12 Our work is focused on elucidating how M. tuberculosis drug efflux systems are regulated. We previously determined the crystal structure of the Rv3066 efflux regulator,14 which controls the expression of the Mmr efflux pump.15 We report here the crystal structure of the Rv1219c regulator, which represses the transcriptional regulation of the Rv1217c–Rv1218c multidrug efflux transport system. The rv1219c gene is the first gene of the Rv1217c–Rv1218c operon and encodes a 212 amino acid protein that shares sequence homology to members of the TetR family of transcriptional repressors. Our data suggest that Rv1219c is a multidrug binding protein that interacts with a variety of toxic aromatic compounds, such as bisquinolinium cyclophanes, phosphoramidites, porphyrins, and pyridazines.
Results
Overall structure of Rv1219c
The crystal structure of the M. tuberculosis Rv1219c transcriptional regulator was determined to a resolution of 2.99 Å using single isomorphous replacement with anomalous scattering (Supporting Information Table S1 and Fig. S1), revealing that only one Rv1219c molecule is present in the asymmetric unit. However, a dimeric arrangement of the regulator was found by applying a twofold crystallographic symmetry operator (Fig. 1). As a TetR-family regulator, Rv1219c consists of two functional motifs: the N-terminal DNA-binding and C-terminal ligand-binding domains. Each subunit of Rv1219c is composed of 10 helices (α1–α10 and α1′–α10′, respectively). The helices of Rv1219c are designated numerically from the N-terminus as α1 (7–23), α2 (29–36), α3 (40–47), α4 (50–74), α5 (78–86), α6 (92–104), α7 (108–130), α8 (139–160), α9 (166–188), and α10 (194–206). In this arrangement, the smaller N-terminal domain includes helices α1 through α3 and the N-terminal end of α4 (residues 50–60), with α2 and α3 forming a typical helix–turn–helix motif. However, the larger C-terminal domain comprises the C-terminal end of helices α4 (residues 61–74) through α10, and helices α8, α9, and α10 are involved in the dimerization of the regulator.
Figure 1.
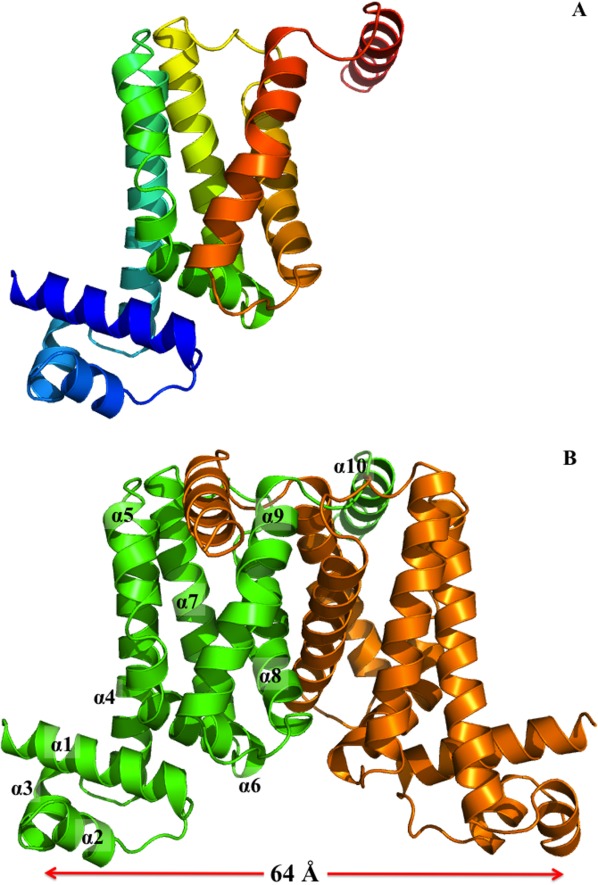
Structure of the M. tuberculosis Rv1219c regulator. (A) Ribbon diagram of a protomer of Rv1219c. The molecule is colored using a rainbow gradient from the N-terminus (blue) to the C-terminus (red). An interactive view is available in the electronic version of the article. (B) Ribbon diagram of the Rv1219c dimer. Each subunit of Rv1219c is labeled with a different color (green and orange). The figure was prepared using PyMOL (http://www.pymol.sourceforge.net). An interactive view is available in the electronic version of the article.
N-terminal domain
The smaller N-terminal domain of Rv1219c shares considerably high sequence and structural similarities with the other TetR family members.16 This is evident through protein sequence alignment that residues 7–60 of Rv1219c possess 25%, 24%, and 31% amino acid identity to TetR,17 QacR,18 and Rv3066,14 respectively. In addition, superimposition of the Cα atoms of this N-terminal region, between residues 7 and 60, with those of AcrR19 and Rv306614 results in overall rms deviations of 3.1 and 3.2 Å.
Perhaps, the most striking difference between the structures of Rv1219c and other TetR members is its large center-to-center distance, which is approximately 64 Å, between the two N-termini of the dimer (Fig. 1). This center-to-center distance is by far the longest among all known structures of the TetR-family regulators. The corresponding distances between the two recognition helices of the DNA-binding domains are 35, 39, and 42 Å in the apo forms of TetR,17 QacR,18 and AcrR.19 Given that the separation between two successive major grooves of a B-form DNA is 34 Å, the large center-to-center distance of Rv1219c may allow this regulator to span three consecutive major grooves of the promoter DNA.
C-terminal domain
The C-terminal domain of Rv1219c consists of six α helices (α4–α10), with helices α4, α5, α7, α8, and α9 forming an antiparallel five-helix bundle (Fig. 1). Like QacR, the dimerization surface mainly comprises helices α8 and α9, although helices α6 and α7 are also involved in the formation of the dimer. These helices make contacts with their counterparts to stabilize the dimerization. It should be noted that helix α10 of the C-terminal end of Rv1219c forms a long arm feature. This feature is unique in Rv1219c and was not found in other members of the TetR family. The elongated helical arm extends its length to the next subunit of the regulator and is anchored into the deep groove created by helices α5′ and α9′. Presumably, this long arm and deep groove within the dimer are engaged to generate an interlocking system, securing the dimerization state of Rv1219c. The interlocking system may also allow the two N-terminal DNA-binding domains of the dimer to shift away from each other while still maintaining the dimeric form of the regulator. Although the C-terminal region displays no primary sequence conservation among members of the TetR family, the overall structure of the C-terminal domain of Rv1219c exhibits topological similarity to those of AcrR,19 Rv3066,14 EthR,20,21 QacR,18 and CmeR.22
The crystal structure of Rv1219c also revealed that the C-terminal α-helical bundle of each subunit of the regulator forms a large internal cavity, with an internal volume of ∼802 Å3. Superimposition of the C-terminal domain of Rv1219c with that of QacR indicates that this large internal cavity overlaps with the multidrug binding pocket of QacR.18 Thus, this cavity, assembled by helices α4–α9, presumably creates a substrate-binding site of the regulator. The interior of the cavity is surrounded by several aromatic residues, including W81, Y91, W113, Y123, F154, Y174, and Y186 (Fig. 2). These residues may interact with the bound substrate via aromatic stacking and hydrophobic interactions.
Figure 2.
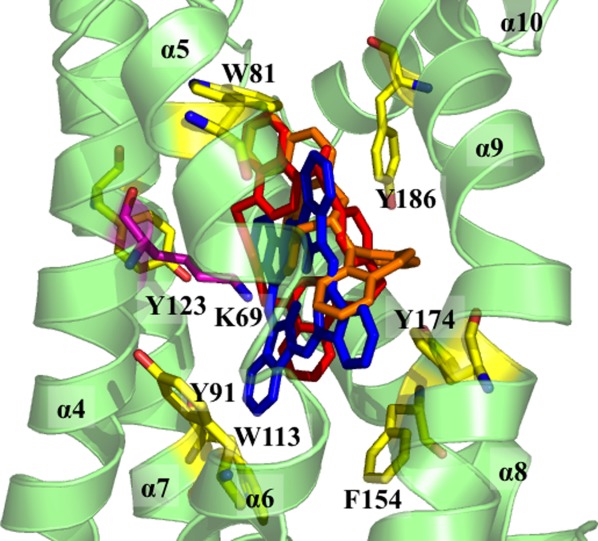
The C-terminal multidrug binding site. The seven aromatic residues (W81, Y91, W113, Y123, F154, Y174, and Y186) that surround the interior of the multidrug binding cavity of Rv1219c are in yellow sticks. The cationic residue K69, which is thought to be important for interacting with the bound drug, is in magenta stick. The top three Rv1219c substrates are also included (red, UCL 1684; orange, (S)-(+)-N-(3,5-Dioxa-4-phosphacyclohepta[2,1-a;3,4-a′]dinaphthalen-4-yl)-dibenzo[b,f]azepine; blue, phthalocyanine).
Electrophoretic mobility shift assays
The organization of the genetic locus containing the rv1219c gene is depicted in Figure 3(A). Based on the genetic organization and preliminary ChIPSeq data from the TB Systems Biology Consortium, it was likely that Rv1219c regulated itself and downstream genes via binding of multiple sites. To demonstrate direct transcriptional regulation, we performed electrophoretic mobility shift assays (EMSA) using probes that comprised the intergenic region upstream of rv1219c and intragenic regions immediately upstream of the rv1218c gene that corresponded to additional ChIP-seq peaks [Fig. 3(A)]. We observed a concentration-dependent shift of the Rv1219c probe and the two Rv1218c probes [Fig. 3(B)]. Therefore, Rv1219c directly regulates expression of Rv1219c and the Rv1217c–Rv1218c transporter. The affinity of Rv1219c for the probe encompassing the rv1219c promoter was greatest, shifting completely upon addition of 1 μM Rv1219c. The Rv1218-1 and Rv1218-2 probes did not completely shift even upon addition of 10 μM Rv1219c. Rv1219c bound the Rv1218-2 probe better than the Rv1218-1 probe, suggesting that high-, medium-, and low-affinity binding sites exist. We identified indirect repeats in each of the probes that are likely binding sequences for Rv1219c [Fig. 3(C)]. To demonstrate the minimal binding motif for Rv1219c, 74 bp oligonucleotide duplexes corresponding to the indirect repeat encompassed by probe Rv1219c were synthesized [Fig. 3(D)]. This probe shifted upon addition of purified Rv1219c protein. A mutated 74-mer oligo was also synthesized with mutations within the indirect repeat. The mutated probe shifted with reduced efficiency compared to the wild-type probe [Fig. 3(D)]. Combined these data provide strong evidence that Rv1219c recognizes the indirect repeat in the Rv1219c promoter [green highlight, Fig. 3(B)]. Our EMSA data suggest that Rv1219c regulates its own expression and the rv1219c operon primarily through the high-affinity binding site highlighted in green in Figure 2(C). The additional medium and low-affinity binding sites highlighted in yellow and blue in Figure 3(C), respectively, allow Rv1219c to further modulate the expression of rv1218c.
Figure 3.
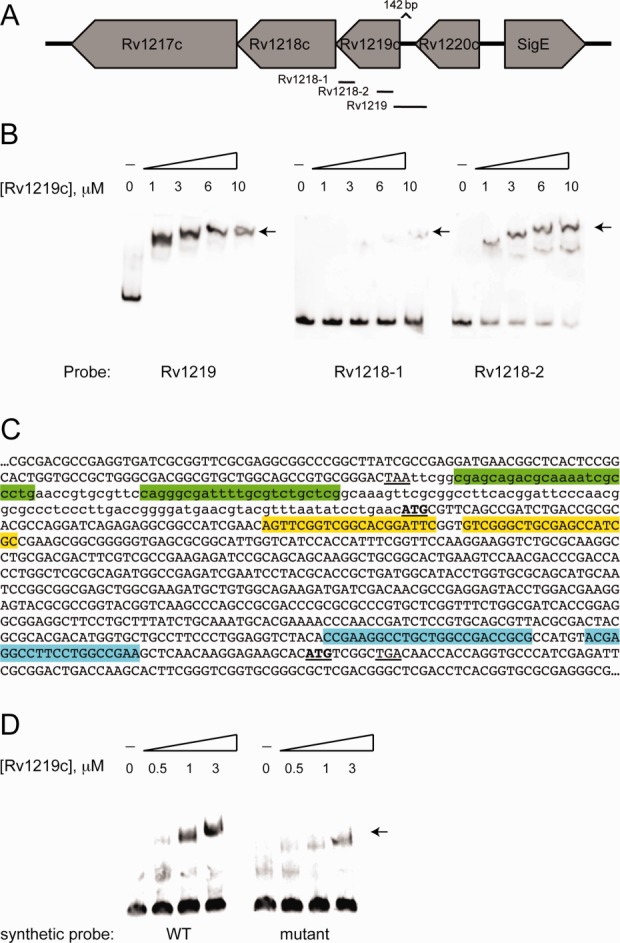
Rv1219c binds to promoter regions of rv1219c and upstream of the rv1218c gene. (A) A schematic depicting the DNA probes used in electrophoretic mobility shift assays (EMSAs) to examine the promoter region of rv1219c and rv1218c. (B) EMSAs were performed using 12 nM Dig-labeled probe and the indicated micromolar concentrations of protein. An arrow denotes the shifted probes. (C) The reverse complement sequence of rv1219c-rv1218c region. The stop codon of rv1220c is underlined, the start codons of rv1219c and rv1218c are bold and underlined. The 142 bp intergenic region between rv1220c and rv1219c is in lowercase, coding sequences are in upper case. Indirect repeats were identified in each of the probes using the MEME algorithm. The high-affinity binding site in the rv1219c promoter probe is highlighted in green. The low-affinity Rv1219c binding motifs in the rv1218-1 and rv1218-2 probes are highlighted in blue and yellow, respectively. (D) EMSAs were performed using 12 nM Dig-labeled probe and the indicated micromolar concentrations of protein. An arrow denotes the shifted probes. Synthetic oligonucleotide duplexes comprising the high affinity binding site plus eight nucleotides on either side (74 bp total) were used as probes.
Rv1219c–DNA interaction
The binding affinity of the 58-bp DNA sequence [in Fig. 3(C), green] within the rv1219c promoter region for the Rv1219c regulator was determined using isothermal titration calorimetry (ITC), which obtained the binding affinity constant, KA, of 1.2 ± 0.1 × 10−6 M−1. The enthalpic (ΔH) and entropic (ΔS) contributions for this binding are 4.8 ± 0.1 kcal mol−1 and 44.1 cal mol deg−1 (Fig. 4). Interestingly, the molar ratio for this binding reaction based on ITC is one Rv1219c dimer per ds-DNA.
Figure 4.
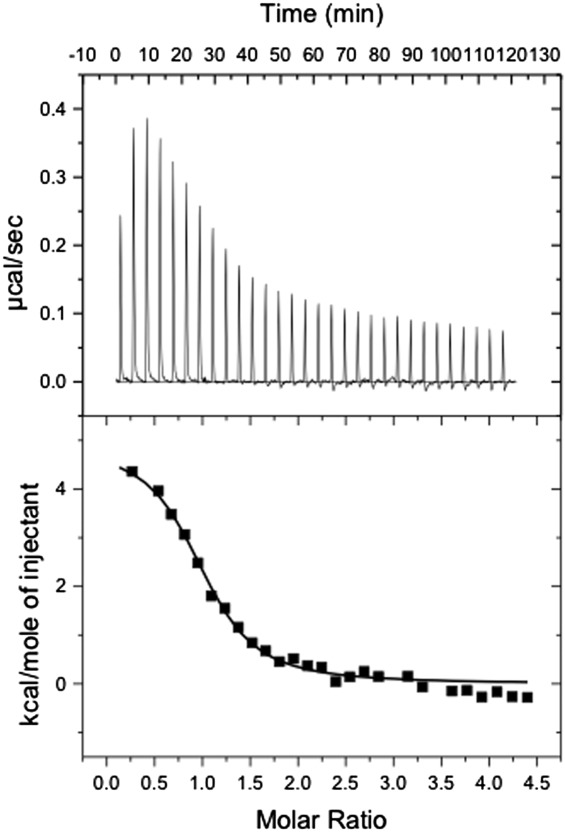
Representative isothermal titration calorimetry for the binding of the 58-bp DNA to Rv1219c regulator. (A) Each peak corresponds to the injection of 10 μL of 200 μM 58-bp DNA in buffer containing 10 mM Na-phosphate (pH 7.5) and 100 mM NaCl into the reaction cell containing 10 μM Rv1219c in the same buffer. (B) Cumulative heat of reaction is displayed as a function of the injection number. The solid line is the least-square fit to the experimental data, giving a KA of 1.2 ± 0.1 × 10−6 M−1. The molar-to-molar ratio of dimeric Rv1219c:58-bp ds-DNA is 1:1.
Rv1219c–drug interactions
The Rv3066 multidrug efflux regulator14 binds several dyes, fluorescence polarization was used to examine if Rv1219c can also bind these dyes, including rhodamine 6G, ethidium, and safranin O. We found that Rv1219c interacts with rhodamine 6G, ethidium, and safranin O with KD values of 4.6 ± 0.5, 32.4 ± 7.4, and 42.4 ± 7.6 μM, respectively (Fig. 5 and Supporting Information Fig. S2). These data also suggest that the Rv1219c protein binds these molecules with a simple binding stoichiometry of 1:1 protomer-to-drug molar ratio. Further study is needed to confirm this protein-to-drug binding ratio.
Figure 5.
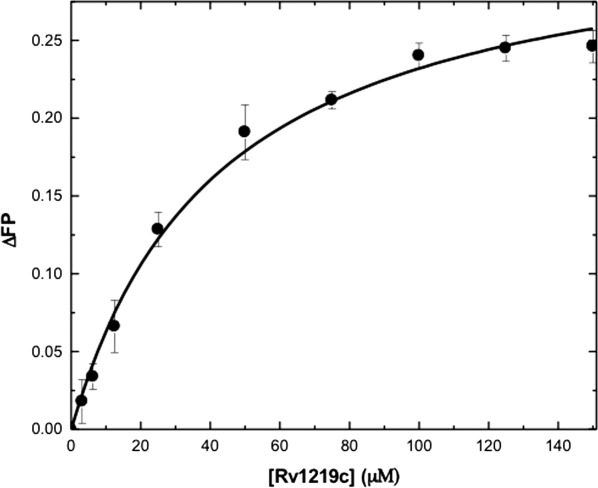
Representative fluorescence polarization of Rv1219c. The binding isotherm of Rv1219c with safranin O, showing a KD of 42.4 ± 7.6 μM. Fluorescence polarization is defined by the equation, FP = (V − H)/(V + H), where FP equals polarization, V equals the vertical component of the emitted light, and H equals the horizontal component of the emitted light of a fluorophore when excited by vertical plane polarized light. FP is a dimensionless entity and is not dependent on the intensity of the emitted light or on the concentration of the fluorophore. mP is related to FP, where 1 mP equals one thousandth of a FP.
Virtual ligand library screening
To elucidate the nature of protein–ligand interactions in the Rv1219c regulator virtual ligand screening was performed using the large internal cavity formed by the C-terminal domain of Rv1219c as a substrate binding cavity. We used AutoDock Vina23 to screen small molecules in the DrugBank24 and ZINC25 libraries. Vina23 uses the iterated local search global optimizer algorithm, which results in predicted binding free energies for these compounds ranging from −13.8 to +20 kcal mol−1. Of the 70,000 screened compounds, the best predicted substrate for Rv1219c was the bisquinolinum cyclophane compound UCL 1684 ditrifluoroacetate, which is a potassium channel blocker, with a predicted binding free energy of −13.3 kcal mol−1. Supporting Information Table S2 lists the top 11 substrates, which have the lowest predicted binding free energies, for the Rv1219c regulator.
As our fluorescence polarization data showed that Rv1219c binds rhodamine 6G, ethidium, and safranin O, Vina23 was also used to examine how these dyes interact with the regulator. These dyes also bind within the multidrug binding pocket of the protein. The predicted binding free energies for these dyes are −8.3, −8.1, and −7.8 kcal mol−1, respectively. These free energies suggest that Rv1219c binds the substrates listed in Supporting Information Table S2 more strongly than these dyes. Interestingly, Vina suggests that Rv1219c uses the same binding mode to interact with these drugs and dyes.
Discussion
With the rising incidence of MDR-TB, it is increasingly important to understand the mechanisms underlying resistance to multiple antibiotics in this pathogen. The crystal structure of Rv1219c provides direct information about how this regulator interacts with its inducing ligands. The surface of the Rv1219c multidrug binding cavity has several familiar aromatic and hydrophobic residues that are critically important in other multidrug binding proteins. These residues produce a hydrophobic environment for substrate binding in the C-terminal regulatory domain.
AutoDock Vina23 was used to study how Rv1219c binds a variety of drugs, and demonstrated that the large cavity of the multidrug binding site of each Rv1219c monomer can accommodate many different classes of drugs (Fig. 2). Surprisingly, the top substrate for Rv1219c was the positively charged heterocyclic bisquinolinium cyclophane UCL 1684 potassium channel blocker. The next top three substrates belong to the classes of phosphoramidite, tetrapyrrole, and organophosphorous, suggesting that these small molecules may also be the substrates of the Rv1217c–Rv1218c multidrug efflux pump. These top compounds are either cationic or neutral molecules. In each case, the bound drug was completely buried within the multidrug binding site of the Rv1219c protomer, and strong aromatic stacking interaction was observed between the bound drug and regulator. The docking study also indicates that residues I63, L73, W81, Y91, Y123, L146, F154, Y174, and Y186 are important for providing hydrophobic and aromatic stacking interactions with these drugs.
As fluorescence polarization experiments demonstrate that Rv1219c binds rhodamine 6G, ethidium, and safranin O in the micromolar range, we also used Vina23 to model the interactions between these positively charged dyes and the regulator. The binding affinities of these dyes by Rv1219c are much weaker than those of the top Rv1219c substrates. However, all these dyes and top Rv1219c substrates are bound within the same multidrug binding site with a similar binding mode, suggesting that the process of induction by these ligands is similar.
A distinguishing feature of multidrug binding proteins that bind cationic drugs is the presence of buried acidic glutamates or aspartates in the ligand binding pockets. This was clearly demonstrated by the structures of the QacR18,26 and Rv306614 regulators. A similar characteristic for the TetR-family regulators that recognize negatively charged antimicrobials has also been observed with TtgR27 and CmeR.28 In this case, positively charged histidines or lysines within the ligand-binding pockets are critical for the binding. In the case of Rv1219c, the predicted substrates are either neutral or positively charged. Thus, we expected a buried acidic residue was needed to participate in binding the substrate. Unexpectedly, the binding crevice of Rv1219c does not contain any glutamate or aspartate. The only charged residue found within the multidrug binding cavity is the cationic lysine K69 (Fig. 2). Based on the docking results with the positively charged UCL 1684 and neutral phthalocyanine ligands, this lysine residue is within 4.8 and 3.1 Å away from these bound ligands. It is likely that K69 is responsible for providing electrostatic interaction for the binding. However, neutralization of the formal charge of the bound ligand may not be a prerequisite for drug recognition. Intriguingly, there are three methionines, M85, M95, and M116, seemingly coordinating with each other within this hydrophobic binding cavity. These three methionines form a triad, similar to the periplasmic heavy metal binding site of the CusA efflux pump.29–31 It is possible that these three methionines may cooperate to create a metal binding site within the multidrug binding cavity. The crystal structures of Rv1219c bound with a variety of ligands will be crucial for further understanding of how this regulator recognizes multiple antimicrobials.
The structural similarity of the N-terminal domains of members of the TetR family suggests a similar mode of interaction with target DNAs. It is known that the separation between two consecutive major grooves of B-DNA is 34 Å. Based on the apo structure of Rv1219c, the two DNA recognition regions of the dimer are separated by 64 Å. Therefore, it is possible that the Rv1219c dimer is capable of spanning three successive major grooves of the double helix when it binds its IR. Rv1219c likely represses the transcription of rv1217c–rv1218c by binding to the high-affinity IR sequence in the promoter region of the efflux operon and the lower affinity sites immediately upstream of rv1218c. It is striking that the promoter probe contains a perfect IR that spans 58 bp [Fig. 3(C), green]. MEME analysis of the three probes defined a consensus binding motif corresponding to this IR that was also present in the Rv1218-2 probe. However, the putative binding site in the Rv1218-2 probe is degenerate (CGATCTGACCGCGCACGCCAGG), which may explain reduced binding of Rv1219c to the Rv1218-2 probe relative to Rv1219 probe in the EMSAs.
The control of TB has been compromised by the increasing proportion of infections due to drug-resistant strains, which are growing at alarming rate. Thus, there is a need to develop new approaches for the treatment of TB. Elucidating the structures and functions of efflux pumps and regulators of M. tuberculosis should enable researchers to explore novel avenues to combat the disease. It has been observed that the expression level of the Rv1217c–Rv1218c multidrug efflux pump is significantly increased in clinically isolated MDR-TB strains in comparison with that of the wild-type H37Rv M. tuberculosis.32 In this article, we have reported the crystal structure of the Rv1219c regulator, which controls the expression level of Rv1217c–Rv1218c. The available of this crystal structure may allow us to rationally design agents that block the function of this regulator and diminish the expression of the multidrug efflux pump, which in turn heightens the sensitivity of this pathogen to antimicrobials.
Materials and Methods
Cloning of rv1219c
The rv1219c ORF from genomic DNA of M. tuberculosis strain H37Rv was amplified by PCR using the primers 5′-CCATGGGCCGTTCAGCCGATCTGACC-3′ and 5′-GGATCCTCAGTGATGATGATGATGATGGCCGACATGTGCTTCTCC-3′. The corresponding PCR product was digested with NcoI and BamHI, extracted from the agarose gel, and inserted into pET15b as described by the manufacturer (Merck KGaA, Darmstadt, Germany) to generate a product that encodes a Rv1219c recombinant protein with a 6xHis tag at the C-terminus (Rv1219c-His6). The recombinant plasmid (pET15bΩrv1219c) was transformed into DH5α cells and the transformants were selected on LB agar plates containing 100 μg mL−1 ampicillin. The presence of the correct rv1219c sequence in the plasmid construct was verified by DNA sequencing.
Expression and purification of Rv1219c
Briefly, Rv1219c-His6 was overproduced in Escherichia coli BL21(DE3) cells carrying pET15bΩrv1219c. Cells were grown in 6 L of Luria Broth (LB) medium with 100 μg mL−1 ampicillin at 37°C. When the OD600 reached 0.5, the culture was treated with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to induce Rv1219c expression, and cells were harvested within 3 h. The collected bacterial cells were suspended in 100 mL ice-cold buffer containing 20 mM Na-HEPES (pH 7.2) and 200 mM NaCl, 10 mM MgCl2, and 0.2 mg DNase I (Sigma–Aldrich). The cells were then lysed with a French pressure cell. Cell debris was removed by centrifugation for 45 min at 4°C and 20,000 rev min−1. The crude lysate was filtered through a 0.2 μm membrane and was loaded onto a 5 mL Hi-Trap Ni2+-chelating column (GE Healthcare Biosciences, Pittsburgh, PA) pre-equilibrated with 20 mM Na-HEPES (pH 7.5) and 250 mM NaCl. To remove unbound proteins and impurities, the column was first washed with six column volumes of buffer containing 50 mM imidazole, 250 mM NaCl, and 20 mM Na-HEPES (pH 7.5). The Rv1219c protein was then eluted with four column volume of buffer containing 300 mM imidazole, 250 mM NaCl, and 20 mM Na-HEPES (pH 7.5). The purity of the protein was judged using 12.5% SDS-PAGE stained with Coomassie Brilliant Blue. The purified protein was extensively dialyzed against buffer containing 100 mM imidazole, 250 mM NaCl, and 20 mM Na-HEPES (pH 7.5), and concentrated to 12 mg mL−1.
For the SeMet-Rv1219c-His6 protein expression, a 10 mL LB broth overnight culture containing E. coli BL21(DE3)/pET15bΩrv1219c cells was transferred into 60 mL of LB broth containing 100 μg mL−1 ampicillin and grown at 37°C. When the OD600 value reached 1.2, cells were harvested by centrifugation at 6000 rev min−1 for 10 min, and then washed two times with 10 mL of M9 minimal salts solution. The cells were re-suspended in 60 mL of M9 media and then transferred into a 6 L pre-warmed M9 solution containing 100 µg mL−1 ampicillin. The cell culture was incubated at 25°C with shaking. When the OD600 reached 0.4, 100 mg L−1 of lysine, phenylalanine, and threonine, 50 mg L−1 isoleucine, leucine, and valine, and 60 mg L−1 of l-selenomethionine were added. The culture was induced with 1 mM IPTG after 15 min. Cells were then harvested within 15 h after induction. The procedures for purifying SeMet-Rv1219c were identical to those of the native protein.
Crystallization of Rv1219c
All crystals of the Rv1219c-His6 were obtained using hanging-drop vapor diffusion. The Rv1219c crystals were grown at 4°C in 24-well plates with the following procedures. A 2 μL protein solution containing 12 mg mL−1 Rv1219c protein in 20 mM Na-HEPES (pH 7.5), 250 mM NaCl, and 100 mM imidazole was mixed with a 2 μL of reservoir solution containing 5% Jeffamine M-600, 0.1 M Na-citrate (pH 5.6), and 0.6 M NaCl. The resultant mixture was equilibrated against 500 μL of the reservoir solution. Crystals grew to a full size in the drops within 2 weeks. Typically, the dimensions of the crystals were 0.2 mm × 0.2 mm × 0.2 mm. Cryoprotection was achieved by raising the glycerol concentration stepwise to 25% with a 5% increment in each step. Crystals of the tungsten derivative were prepared by incubating the crystals of Rv1219c in solution containing 5% Jeffamine M-600, 0.1 M Na-citrate (pH 5.6), 0.6 M NaCl, and 1 mM (NH4)2W6(μ-O)6(μ-Cl)6Cl6 for 24 h at 25°C.
Data collection, structural determination, and refinement
All diffraction data were collected at 100 K at beamline 24ID-E located at the Advanced Photon Source, using an ADSC Quantum 315 CCD-based detector. Diffraction data were processed using DENZO and scaled using SCALEPACK.33 The crystals of Rv1219c belong to the space group I432 (Supporting Information Table S1). Based on the molecular weight of Rv1219c (23.18 kDa), the asymmetric unit is expected to contain one regulator protomer with a solvent content of 22.56%. The heavy-atom derivative was isomorphous with the native crystal (Supporting Information Table S1). Six tungsten cluster sites were identified using HySS as implemented in the PHENIX package.34 Single isomorphous replacement with anomalous scattering was used to obtain experimental phases using the program MLPHARE.35,36 The resulting phases were subjected to density modification by RESOLVE37 using the native structure factor amplitudes. Density modified phases were good enough to allow us to visualize the secondary structural features of the molecule. These phases were then subjected to density modification and phase extension to 2.99 Å-resolution using the program RESOLVE.37 The resulting phases were of excellent quality that enabled tracing of most of the molecule. In addition, the selenomethionyl-substituted (SeMet) crystal data were used to help in tracing the molecules by anomalous difference Fourier maps where we could ascertain the proper registry of SeMet residues. The full-length Rv1219c protein consists of eight methionine residues and all these eight selenium sites were identified in each protomer of the protein. After tracing the initial model manually using the program Coot,38 the model was refined against the native data at 2.99 Å-resolution in PHENIX,34 leaving 5% of reflections in Free-R set. Iterations of refinement using PHENIX34 and CNS39 and model building in Coot38 lead to the current model, which consists of 205 residues with excellent geometrical characteristics (Supporting Information Table S1).
Electrophoretic mobility shift assays
Rv1219c-His6 was purified from E. coli lysates using Talon resin (Novagen). Probes were amplified from the H37Rv genome using the primers listed in Supporting Information Table S3. Synthetic probes were synthesized with the following sequences: WT-TAATTCGGCGAGCAGACGCAAAATCGCCCTGAACCGTGCGTTCCAGGGCGATTTTGCGTCTGCTCGGCAAAGTT; mutant-TAATTCGGTATACGCTACGAGCATCTATATGAACCGTGCGTTCTATACGCTACAGGGCGATTATAGGCAAAGTT. All probes were labeled with Digoxigenin using the Roche DIG Gel Shift kit. For EMSA analysis, 12 nM Dig-labeled probe and the indicated micromolar concentrations of protein were incubated for 45 min at room temperature in the Roche binding buffer modified by the addition of 0.25 mg mL−1 herring sperm DNA, and 0.75 μg mL−1 poly(d[I-C]). All reactions were resolved on a 6% native polyacrylamide gel in TBE buffer, transferred to nylon membrane, and DIG-labeled DNA–protein complexes detected following the manufacturer's recommendations. Chemiluminescent signals were acquired using an ImageQuant LAS 4000 (GE).
Isothermal titration calorimetry
We used ITC to examine the binding of the DNA sequence [highlighted in green in Fig. 3(C)] to the purified Rv1219c regulator. Measurements were performed on a VP-Microcalorimeter (MicroCal, Northampton, MA) at 25°C. Before titration, the protein was thoroughly dialyzed against buffer containing 10 mM Na-phosphate pH 7.2 and 100 mM NaCl. The protein concentration was determined using the Bradford assay. The protein sample was then adjusted to a final concentration of 10 μM. DNA solution consisting of 200 μM 58-bp ds-DNA in 10 mM Na-phosphate pH 7.2 and 100 mM NaCl was prepared as the titrant. The protein and ligand samples were degassed before they were loaded into the cell and syringe. Binding experiments were performed with the protein solution (1.5 mL) in the cell and the DNA as the injectant. Ten-microliter injections of the ligand solution were used for data collection.
Injections occurred at intervals of 240 s, and the duration time of each injection was 10 s. Heat transfer (μcal s−1) was measured as a function of elapsed time (s). The mean enthalpies measured from injection of the ligand in the buffer were subtracted from raw titration data before data analysis with ORIGIN software (MicroCal). Titration curves were fitted by a nonlinear least squares method to a function for the binding of a DNA to a macromolecule. Nonlinear regression fitting to the binding isotherm provided us with the equilibrium binding constant (KA = 1/KD) and enthalpy of binding (ΔH). Based on the values of KA, the change in free energy (ΔG) and entropy (ΔS) were calculated with the equation: ΔG = −RT lnKA = ΔH − TΔS, where T is 273 K and R is 1.9872 cal K−1 mol−1. Calorimetry trials were also performed in the absence of Rv1219c in the same experimental conditions. No change in heat was observed in the injections throughout the experiment.
Fluorescence polarization assay for ligand binding affinity
Fluorescence polarization was used to determine the binding affinities of a variety of Rv1219c ligands, including rhodamine 6G, ethidium bromide, and safranin O. The experiment was done using a ligand binding solution containing 10 mM Na-phosphate (pH 7.2), 100 mM NaCl, and 1 μM ligand (rhodamine 6G, ethidium bromide, or safranin O). The protein solution consisting of Rv1219c in 10 mM Na-phosphate (pH 7.2), 100 mM NaCl, and 1 μM ligand (rhodamine 6G, ethidium bromide, or safranin O) was titrated into the ligand binding solution until the polarization (P) was unchanged. As this is a steady-state approach, fluorescence polarization measurement was taken after a 5-min incubation for each corresponding concentration of the protein and bile acid to ensure that the binding has reached equilibrium. All measurements were performed at 25°C using a PerkinElmer LS55 spectrofluorometer equipped with a Hamamatsu R928 photomultiplier. The excitation and emission wavelengths were 526 and 555 nm for rhodamine 6G, 483, and 620 nm for ethidium, and 520 and 587 nm for safranin O. Fluorescence polarization signal (in ΔP) was measured at the emission wavelength. Each titration point recorded was an average of 15 measurements. Data were analyzed using the equation, P = {(Pbound − Pfree)[protein]/(KD + [protein])} + Pfree, where P is the polarization measured at a given total protein concentration, Pfree is the initial polarization of free ligand, Pbound is the maximum polarization of specifically bound ligand, and [protein] is the protein concentration. The titration experiments were repeated for three times to obtain the average KD value. Curve fitting was accomplished using the program ORIGIN (OriginLab Corporation, Northampton, MA).
Virtual ligand screening using AutoDock Vina
AutoDock Vina was used for virtual ligand screening of a variety of compounds. The docking area was assigned visually to cover the internal cavity of the Rv1219c monomer. A grid of 35 Å × 35 Å × 35 Å with 0.375 Å spacing was calculated around the docking area for all atom types presented in the DrugBank and ZINC libraries using AutoGrid. The iterated local search global optimizer algorithm was used to predict the binding free energies for these compounds.
Protein Data Bank accession code
Coordinates and structural factors for the structure of Rv1219c have been deposited at the RCSB Protein Data Bank with an accession code 4NN1.
Acknowledgments
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors are grateful to Louis Messerle at University of Iowa for providing us the (NH4)2W6(μ-O)6(μ-Cl)6Cl6 complex used in this study.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Maartens G, Wilkinson RJ. Tuberculosis. Lancet. 2007;370:2030–2043. doi: 10.1016/S0140-6736(07)61262-8. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2010. Fact sheet no. 104: tuberculosis. Available at http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 3.Centers for Disease Control and Prevention. 2012. Fact sheet: tuberculosis. Available at: http://www.cdc.gov/tb/topic/treatment/default.htm.
- 4.Frieden TR, Sterling T, Pablos-Mendez A, Kilburn JO, Cauthen GM, Dooley SW. The emergence of drug-resistant tuberculosis in New York City. N Engl J Med. 1993;328:521–556. doi: 10.1056/NEJM199302253280801. [DOI] [PubMed] [Google Scholar]
- 5.Pillay M, Sturm AW. Evolution of the extensively drug-resistant F15/LAM4/KZN strain of Mycobacterium tuberculosis in KwaZulu-Natal, South Africa. Clin Infect Dis. 2007;45:1409–1414. doi: 10.1086/522987. [DOI] [PubMed] [Google Scholar]
- 6.Goldman RC, Plumley KV, Laughon BE. The evolution of extensively drug resistant tuberculosis (XDR–TB): history, status and issues for global control. Infect Disord Drug Targets. 2007;7:73–91. doi: 10.2174/187152607781001844. [DOI] [PubMed] [Google Scholar]
- 7.Udwadia ZF, Amale RA, Rodrigues C. Totally drug-resistant tuberculosis in India. Clin Infect Dis. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 8.Iseman MD. Treatment of multidrug-resistant tuberculosis. N Engl J Med. 1993;329:784–791. doi: 10.1056/NEJM199309093291108. [DOI] [PubMed] [Google Scholar]
- 9.Nikaido H. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol. 2001;12:215–223. doi: 10.1006/scdb.2000.0247. [DOI] [PubMed] [Google Scholar]
- 10.Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs. 2004;64:159–204. doi: 10.2165/00003495-200464020-00004. [DOI] [PubMed] [Google Scholar]
- 11.Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development protein. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- 12.Balganesh M, Kuruppath S, Marcel N, Sharma S. Rv1218c, an ABC transporter of Mycobacterium tuberculosis with implications in drug discovery. Antimicrob Agents Chemother. 2010;54:5167–5172. doi: 10.1128/AAC.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danilchanka O, Mailaender C, Niederweis M. Identification of a novel multidrug efflux pump of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2008;52:5203–5211. doi: 10.1128/AAC.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolla JR, Do SV, Long F, Dai L, Su CC, Lei HT, Chen X, Gerkey JE, Murphy DC, Rajashankar KR, Zhang Q, Yu EW. Structural and functional analysis of the transcriptional regulator Rv3066 of Mycobacterium tuberculosis. Nucleic Acids Res. 2012;40:9340–9355. doi: 10.1093/nar/gks677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossi De E, Branzoni M, Cantoni R, Milano A, Riccardi G, Ciferri O. mmr, a Mycobacterium tuberculosis gene conferring resistance to small cationic dyes and inhibitors. J Bacteriol. 1998;180:6068–6071. doi: 10.1128/jb.180.22.6068-6071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang XD, Gallegos MT, Brennan R, Tobes R. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinrichs W, Kisker C, Duvel M, Muller A, Tovar K, Hillen W, Saenger W. Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science. 1994;264:418–420. doi: 10.1126/science.8153629. [DOI] [PubMed] [Google Scholar]
- 18.Schumacher MA, Miller MC, Brennan RG. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001;294:2158–2163. doi: 10.1126/science.1066020. [DOI] [PubMed] [Google Scholar]
- 19.Li M, Gu R, Su CC, Routh MD, Harris KC, Jewell ES, McDermott G, Yu EW. Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J Mol Biol. 2007;374:591–603. doi: 10.1016/j.jmb.2007.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dover LG, Corsino PE, Daniels IR, Cocklin SL, Tatituri V, Besra GS, Futterer K. Crystal structure of the TetR/CamR family repressor Mycobacterium tuberculosis EthR implicated in ethionamide resistance. J Mol Biol. 2004;340:1095–1105. doi: 10.1016/j.jmb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Frenois F, Engohang-Ndong J, Locht C, Baulard AR, Villeret V. Structure of EthR in a ligand bound conformation reveals therapeutic perspectives against tuberculosis. Mol Cell. 2004;16:301–307. doi: 10.1016/j.molcel.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Gu R, Su CC, Shi F, Li M, McDermott G, Zhang Q, Yu EW. Crystal Structure of the transcriptional regulator CmeR from Campylobacter jejuni. J Mol Biol. 2007;372:583–593. doi: 10.1016/j.jmb.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucl Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks BE, Piro KM, Brennan RG. Multidrug-binding transcription factor QacR binds the bivalent aromatic diamidines DB75 and DB359 in multiple positions. J Am Chem Soc. 2007;129:8389–8395. doi: 10.1021/ja072576v. [DOI] [PubMed] [Google Scholar]
- 27.Alguel Y, Meng C, Terán W, Krell T, Ramos JL, Gallegos M-T, Zhang X. Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J Mol Biol. 2007;369:829–840. doi: 10.1016/j.jmb.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lei HT, Shen Z, Surana P, Routh MD, Su CC, Zhang Q, Yu EW. Crystal structures of CmeR-bile acid complexes from Campylobacter jejuni. Prot Sci. 2011;20:712–723. doi: 10.1002/pro.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su CC, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, Yu EW. Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature. 2011;470:558–562. doi: 10.1038/nature09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su CC, Long F, Lei HT, Bolla JR, Do SV, Rajashankar KR, Yu EW. Charged amino acids (R83, E567, D617, E625, R669, and K678) of CusA are required for metal ion transport in the Cus efflux system. J Mol Biol. 2012;422:429–441. doi: 10.1016/j.jmb.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Pei H, Huang B, Zhu X, Zhang J, Zhou B, Zhu L, Zhang Y, Zhou FF. The expression of ABC efflux pump, Rv1217c-Rv1218c, and its association with multidrug resistance of Mycobacterium tuberculosis in China. Curr Microbiol. 2013;66:222–226. doi: 10.1007/s00284-012-0215-3. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor M. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The Phenix refinement framework. CCP4 Newsletter 42:contribution 8.
- 35.Otwinowski Z. MLPHARE, CCP4 Proc. 80. Warrington, UK: Daresbury Laboratory; 1991. [Google Scholar]
- 36.Collaborative Computational Project No. 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Terwilliger TC. Maximum-likelihood density modification using pattern recognition of structural motifs. Acta Crystallogr D Biol Crystallogr. 2001;57:1755–1762. doi: 10.1107/S0907444901013737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.