

Abstract
Duchenne and Becker muscular dystrophies (DMD/BMD) are X-linked inherited muscular disorders caused by mutations in the dystrophin gene. Two-thirds of DMD cases are thought to be caused by inheritance from carrier mothers and this study aimed to clarify and compare the carrier frequency of mothers of DMD and BMD patients according to the mutation type. We included 139 DMD and 19 BMD mothers. Of these, 113 patients (99 DMD and 14 BMD) and 13 patients (12 DMD and 1 BMD) had deletions and duplications of one or more exons, respectively. Small mutations, including nonsense mutations, small deletions/insertions and splice site mutations, were identified in 32 patients (28 DMD and four BMD). The overall carrier frequency for BMD mothers was significantly higher than for DMD (89.5% vs 57.6%, P<0.05), probably as BMD patients can leave descendants. The carrier frequency tended to be lower in mothers of DMD patients with deletion mutations than with duplications and small mutations (53.5%, 66.7% and 67.9%, respectively). It was suggested that de novo mutations are more prevalent for deletions than other mutations. This is the first report to analyze the carrier frequency according to mutation type.
Keywords: Becker muscular dystrophy, carrier frequency, Duchenne muscular dystrophy, dystrophin, mutations
Introduction
Duchenne muscular dystrophy (DMD; Online Mendelian Inheritance in Man (OMIM) no. 310200) and Becker muscular dystrophy (BMD; OMIM no. 300376) are allelic X-linked muscular disorders caused by mutations in the dystrophin gene (OMIM no. 300377). DMD is a serious disease in which muscle weakness becomes apparent at 4–5 years of age. Muscular weakness subsequently progresses until patients are unable to walk by the age of 12 years and die in their 20s or 30s from cardiac or respiratory failure. By contrast, BMD has a slower rate of progression and some affected individuals may lead near-normal lives.1
The dystrophin gene is a huge gene (2400 kb) composed of 79 exons that is located on the short arm of the X chromosome.2 Because of its size, it tends to mutate frequently and mutation types observed in DMD/BMD include the deletion or duplication of one or more exons, as well as small mutations such as nonsense mutations, small insertions/deletions and splice site mutations; deletion of one or more exons is the most common mutation type and accounts for 60% of mutations.3 It is known that reading frame rule explains the clinical difference between DMD and BMD at the molecular level; that is, deletions or duplications that shift the reading frame of the dystrophin mRNA (out-of-frame) lead to the more severe DMD phenotype, whereas the BMD phenotype occurs if the reading frame is preserved (in-frame).4 This rule was upheld for 93% of deletion and 66% of duplication mutation cases.3
Approximately one-third of DMD patients are thought to develop the disease because of de novo mutations, with the remaining two-thirds inheriting the mutation from carrier mothers. This can be interpreted from the theory based on the balance between mutation and selection in X-linked recessive mutations. For the incidence of the disease to be maintained, the mutant allele that is lost through selection must be replaced with de novo mutations. If it is assumed that the incidence of a severe X-linked disease does not change, then the mutation rate (μ) will equal the coefficient of selection (s) (percentage of mutant alleles not inherited by the next generation) multiplied by the allele frequency (q) multiplied by one-third as selection only acts on male alleles, which represent one-third of the population's alleles. Consequently, μ=sq/3. In X-linked lethal genetic diseases such as DMD, it is not possible to pass alleles to descendants; therefore, s=1. Thus, as it is supposed that one-third of mutated genes disappear in each generation, one-third must occur as new mutations in their place.5, 6, 7
It is also possible that the mechanism of mutation generation depends on the type of mutation8, 9 and that the percentages of de novo mutation and inheritance of the mutation from carrier mothers differ depending on the mutation type. However, thus far, no reports have examined a large number of cases for the carrier frequency of mothers of DMD/BMD patients for each mutation type, including small mutations. We have undertaken more than 400 genetic analyses of DMD and BMD patients including prenatal and carrier analysis. This report is based on our large cohort and examines the carrier frequency of the mothers of Japanese DMD and BMD patients according to each mutation type.
Materials and methods
Subjects
This study was approved by the Ethics Committee at Kobe University Hospital and informed consent was obtained from the patient or their family (approval No. 28:1998). It consisted of 158 mothers (139 DMD and 19 BMD) whose sons were Japanese DMD/BMD patients referred to the Department of Pediatrics, Kobe University Hospital, between 1995 and 2012. If there were more than two patients in the same family, only one was included in the present study. The definitive diagnosis of patients and diagnosis of the mother's carrier status were performed by dystrophin gene analysis.
DMD and BMD were diagnosed from muscle pathological findings or clinical findings. Patients who were unable to walk by the age of 12 years and those in whom dystrophin protein was absent from muscle biopsy, pathology specimens were classified as DMD, whereas patients who were able to walk at 13 years of age or older were classified as BMD.10
Genetic analysis of the patient
Genomic DNA (gDNA) was extracted from peripheral blood using standard phenol-chloroform extraction methods. gDNA analysis was performed as previously reported.3 Briefly, Southern blotting or multiplex ligation-dependent probe amplification (MLPA; SALSA MLPA KIT P034/035 DMD/Becker, MRC-Holland, Amsterdam, The Netherlands) was used to identify one or more exon deletions or duplications. In cases where the deletion of a single exon was detected by MLPA, the analysis was confirmed by PCR or direct sequencing to determine whether one exon was indeed deleted or the MLPA result was modified by a small mutation.11 In cases where deletion or duplication mutations were not detected, a small mutation was identified by direct sequence analysis using lymphocyte gDNA or analysis of mRNA extracted from muscles or lymphocytes by the reverse-transcription PCR method.3 Mutations that were identified in complementary DNA were also analyzed and confirmed in the corresponding gDNA region.
Genetic analysis of the mother
If the mutation in patient was a deletion or duplication of one or more exons, the gDNA of mother was analyzed by Southern blotting or MLPA. In cases where the patient's mutation was small, PCR and direct sequencing of gDNA were used to analyze the presence of heterozygous mutations and to determine whether the mother was a carrier.
Statistical analysis
Statistical analysis was performed using the Fisher's exact test or the Chi-square test as appropriate. The differences were deemed statistically significant at P<0.05.
Results
Distribution of mutations in DMD/BMD patients
Of the DMD mutations, 99 were deletions of one or more exons, 12 were duplications of one or more exons and 28 were small mutations (13 nonsense mutations, 9 small insertions/deletions and 6 splice site mutations) (Table 1). The BMD mutations included 14 deletions of one or more exons, 1 duplication and 4 small mutations (1 small insertion/deletion and 3 splice site mutations) (Table 1).
Table 1. Distribution of mutations in DMD and BMD patients.
| Clinical entity | Exon deletion | Exon duplication | Small mutation | Total |
|---|---|---|---|---|
| DMD | 99 | 12 | 28 | 139 |
| BMD | 14 | 1 | 4 | 19 |
Abbreviations: BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy.
Mutation site and carrier frequency of the mothers of DMD patients
First, we examined the relationship between the mutation site and the carrier status of the DMD mothers. We investigated the exons deleted in patients with causative exon deletions, and initially found no clear tendency in the deleted exon distribution between patients whose mothers were carriers or non-carriers (Figure 1a). As previously reported, many deletions were clustered in exons 2–20, which is a proximal hot spot, and exons 45–55, which is a distal hot spot.3, 12
Figure 1.

The location of mutations in Duchenne muscular dystrophy patients from carrier and non-carrier mothers. (a) Exon deletions identified in 99 patients. Horizontal bars represent deleted exon regions. Black and gray bars represent patients from carrier and non-carrier mothers, respectively. Exons 1–79 are numbered on the bottom line. Deletions are clustered in two hot spots. No tendency was identified based on the carrier status. (b) Exon duplications identified in 12 patients. Horizontal bars represent duplicated exon regions. Black and gray bars represent patients from carrier and non-carrier mothers, respectively. On the 5th line from the top, dotted lines indicate normal exons between separated duplications (duplication of exons 19–31 and 45–51). No tendency was identified based on the carrier status. (c) Twenty-eight patients were identified with small mutations, including thirteen with nonsense mutations, nine with small deletions/insertions and six with splice site mutations. Diamonds, triangles and circles represent nonsense mutations, small deletions/insertions and splice site mutations, respectively. Black and gray marks represent patients from carrier mothers and non-carrier mothers, respectively. No tendency was identified based on the carrier status.
Further analysis was carried out after the patients were divided into two groups. The number of patients in which one of the ends of the deleted exon was located in a proximal or distal hot spot was 25 and 65, respectively. Within these groups, the number of patients whose mother was a carrier was 12 (48.0%) and 33 (50.8%), respectively. Thus, with respect to the carrier frequency of the mother, there was no significant difference between the cases with deletion mutations in the proximal or distal hot spots (Table 2). Regarding duplications, three of the four patients from non-carrier mothers had mutations in the proximal side with duplications at exons 3–7, 3–9 and 11. The remaining patients had a distal side mutation with duplication at exons 41–69 (Figure 1b). The small mutations (nonsense mutations, small insertions/deletions and splice site mutations) were distributed across the entire exon region, and no clear bias was identified in the mutation distribution according to whether the mother was a carrier (Figure 1c).
Table 2. Number of DMD patients with deletion mutations located in the two deletion hot spots.
| Proximal hot spot | Distal hot spot | Total | |
|---|---|---|---|
| From carrier mothers | 12 | 33 | 45 |
| From non-carrier mothers | 13 | 32 | 45 |
| Total | 25 | 65 | 90 |
Abbreviation: DMD, Duchenne muscular dystrophy.
Mutation site and carrier frequency of the mothers of BMD patients
Next, we examined the site of mutation and relation to carrier status in BMD. The carrier frequency of the mothers was 12 of 14 deletion cases, and no clear bias was identified in the mutation distribution according to whether the mother was a carrier (Figure 2). The mothers were carriers in the cases of the single patient with a duplication (of exon 7) and all four patients with small mutations, including a four-nucleotide deletion in exon 38 and three splice site mutations (introns 20, 25 and 42). No relationship was observed between carrier status of the mothers and the mutation site.
Figure 2.
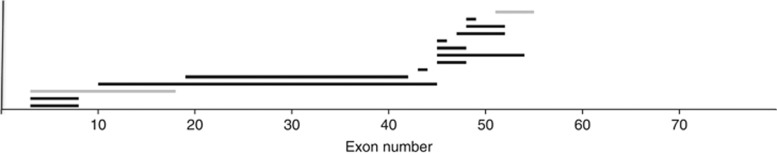
The location of deletion mutations in Becker muscular dystrophy cases from carrier and non-carrier mothers. Exons 1–79 are numbered on the bottom line. Exon deletions were identified in 14 cases. Horizontal bars represent deleted exon regions. Black and gray bars represent cases from carrier mothers and non-carrier mothers, respectively.
Comparison of carrier frequency in DMD and BMD
We examined the difference in carrier frequency of the mothers in DMD and BMD. In DMD, the overall carrier frequency including all mutation types was 80 of 139 mothers (57.6%). In BMD, this carrier frequency was significantly higher at 89.5% (P<0.05; Figure 3).
Figure 3.

The carrier frequency of mothers of Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) patients. Based on all types of mutations, the carrier frequency was significantly lower in mothers of DMD patients compared with BMD patients using the Fisher's exact test (57.6% vs 89.5% P<0.05).
The carrier frequency of mothers of DMD patients according to mutation type
Finally, we examined the difference in the carrier frequency of the mothers of DMD patients according to mutation types. The carrier frequency was 53 of 99 mothers of DMD patients with deletions of one or more exons (53.5%), 8 of 12 with duplications (66.7%) and 19 of 28 with small mutations (67.9% Figure 4). The carrier frequency was lower in the mothers of DMD patients with deletions compared with duplications or small mutations, but this difference was not statistically significant.
Figure 4.

The carrier frequency of mothers of Duchenne muscular dystrophy patients based on the type of mutation. The carrier frequency was 53.5% in the mothers of patients with deletion mutations, which was lower than that with duplication mutations (66.7%) and small mutations (67.9%), although the difference was not statistically significant using the χ2-test.
Discussion
In this study, we investigated the difference in carrier frequency between DMD and BMD, and the carrier frequency of DMD mothers according to the mutation type. We observed an overall carrier frequency of mothers in DMD of 57.6%, which is lower than the theoretical value of two-thirds predicted as the balance between mutation and selection in X-linked recessive mutations. One possible reason for this is that DMD diagnosis has improved in recent years because of advances in genetic testing. Because carrier diagnosis and prenatal diagnosis have become possible, the number of children born with the disease from carrier mothers is declining. In fact, in the Netherlands, the percentage of first-affected boys was 62% between 1961 and 1974, and this significantly increased to 88% between 1993 and 2002. This is thought to be because carrier diagnosis and prenatal diagnosis caused a decline in the number of births of disease-affected children.13 As this trend is likely to become more obvious in the future, it is possible that the measured carrier frequency will decrease further.
The degree of disease severity differs between DMD and BMD, so it is conceivable that the frequency of de novo mutations will also differ. However, thus far, no studies have investigated a large number of cases in which DMD and BMD were differentiated. The present study found that the carrier frequency of mothers was significantly higher for BMD than for DMD. This can be explained by the difference in disease severity as DMD patients often die by their 20s or 30s without having children, whereas some patients with mild cases of BMD first develop symptoms during young adulthood or later, and many of these patients have children. Thus, it can be supposed that there are cases in which the mutation has been passed on by a male patient to his daughter, and the carrier daughter passes it on to the next generation. The present study included one such family in which the patient was diagnosed with BMD, his mother as a carrier, and his grandfather was suspected to have BMD because of a walking disability, increased leaking enzymes and dilated cardiomyopathy. When this is applied to the above-mentioned mutation rate calculation formula (μ=sq/3), the rate of new mutations will be less than one-third because s is less than 1 in BMD. The results of our study are consistent with this theory.
DMD mutations include the most commonly observed mutation type, the deletion of one or more exons, as well as duplications and small mutations. It is possible that the percentages of de novo mutations and inheritance of the mutation from carrier mothers differ depending on the mutation type. In previous reports of comparatively large sample numbers, the carrier frequency of the mothers of DMD and BMD patients was reported as 1102 of 1472 cases (74.9%),12 which compares with 98 of 189 cases (51.9%) in another study of DMD sporadic cases.13 However, neither of these studies examined the carrier frequency according to the mutation type, instead including all mutations in their analyses. In a report of a large number of cases in Japan, the carrier frequency of the mothers in 108 cases of DMD/BMD was 63.9% however, this study only included cases with deletion and duplication mutations, so the carrier frequency for small mutations was not reported.14 Our report is the first to examine the carrier frequency of the mothers in a large number of cases according to the type of mutation.
The carrier frequencies of the mothers of DMD patients were 53.5% for deletions, 66.7% for duplications and 67.9% for small mutations. These frequencies did not differ significantly from each other, although the frequency was lower for deletions. This might be related to the nature of the respective mutations; many large-scale deletions tend to occur because of unequal crossover in the oogenesis process,8, 15 whereas many simple point mutations occur because of DNA replication or repair errors during reproductive cell division. As spermatogenesis undergoes far more replication cycles than oogenesis, point mutations are usually inherited from the father rather than the mother. In fact, although almost all de novo large-scale deletions in the dystrophin gene are maternally derived, point mutations tend to be paternally derived.9, 16 Thus, sperm-derived mutations in DMD are handed down to the next generation via a carrier mother so carrier frequencies of mothers for small mutations are higher than for other mutation types. With deletions, even if the mother is not a carrier, the frequency of a mutation occurring during the mother's oogenesis process and the child developing the disease is high. Our findings support this theory.
Point mutations, which tend to be sperm-derived, make up a smaller proportion of BMD cases than DMD cases,3 whereas carrier frequency of mother was higher in BMD than in DMD. This discrepancy is also explained by the difference in the severity of DMD and BMD. In BMD cases who can leave descendants deletion mutations can be passed on by a male patient to his daughter, and the carrier daughter passes it on to her son, which is similar to sperm-derived mutations. Therefore, the carrier frequency of mothers in BMD is higher than that in DMD.
Although it is envisaged that the mechanism that generates duplication mutations may, as for deletion mutations, be oogenesis recombination and other processes, the details have not been fully revealed. In previous reports, the carrier frequency of mothers for duplications was high,14 and our findings support this. These results suggest that duplications do not occur easily at oogenesis-like deletions. However, because only 15 cases were identified with duplication mutations in the present study, it is necessary to perform further studies on larger numbers of cases in the future.
Passos-Bueno et al. previously observed that in familial cases with deletion mutations, 47% were found in the proximal region and 53% in the distal region, whereas in sporadic cases 28% were found in the proximal region and 72% in the distal region. Furthermore, in cases with germinal mosaicism, an important factor in the cause of de novo mutations, the frequency of deletion mutations in the proximal region was significantly higher than in the distal region.17 We observed no clear site bias in the examination of the deleted exon site and whether the mother was a carrier. In our study, 26.7% and 73.3% of deletions were in proximal and distal regions, respectively, in DMD cases with carrier mothers, and 28.9% and 71.1% were found in proximal and distal regions, respectively, in cases with non-carrier mothers. However, our study did not include cases thought to be caused by germinal mosaicism, which may explain why no significant difference was seen between cases with carrier and non-carrier mothers.
To conclude, our study clearly showed that the carrier frequency of the mothers was significantly higher in BMD than in DMD. We also found that the carrier frequency of mothers in DMD tends to vary among types of mutation, and envisage that this is related to the nature of the mutation. To our knowledge, this is the first report to analyze the carrier frequency according to the mutation type, including small mutations.
Acknowledgments
We thank Ms Kanako Yokoyama for her secretarial help and Ms Naomi Okahisa for her technical assistance. This work was supported by a Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Emery A. E. The muscular dystrophies. Lancet. 2002;23:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Ahn A. H., Kunkel L. M. The structural and functional diversity of dystrophin. Nat. Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- Takeshima Y., Yagi M., Okizuka Y., Awano H., Zhang Z., Yamauchi Y., et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010;55:379–388. doi: 10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]
- Monaco A. P., Bertelson C. J., Liechti-Gallati S., Moser H., Kunhekel L. M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- Haldane J. B. The rate of spontaneous mutation of a human gene. J. Genet. 1935;31:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- Moser H. Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum. Genet. 1984;66:17–40. doi: 10.1007/BF00275183. [DOI] [PubMed] [Google Scholar]
- Nussbaum R. L., McInnes R. R., Willard H. F.Genetics in Medicine7th edneds Thompson J., Thompson M.,198–199.Saunders Elsevier: Philadelphia; 2007 [Google Scholar]
- Winter R. M., Pembrey M. E. Does unequal crossing over contribute to the mutation rate in Duchenne muscular dystrophy. Am. J. Med. Genet. 1982;12:437–441. doi: 10.1002/ajmg.1320120407. [DOI] [PubMed] [Google Scholar]
- Grimm T., Meng G., Liechti-Gallati S., Bettecken T., Müller C. R., Müller B. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J. Med. Genet. 1994;31:183–186. doi: 10.1136/jmg.31.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman E. P., Kunkel L. M., Angelini C., Clarke A., Johnson M., Harris J. B. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39:1011–1017. doi: 10.1212/wnl.39.8.1011. [DOI] [PubMed] [Google Scholar]
- Okizuka Y., Takeshima Y., Awano H., Zhang Z., Yagi M., Matsuo M. Small mutations detected by multiplex ligation-dependent probe amplification of the dystrophin gene. Genet. Test Mol. Biomarkers. 2009;13:427–431. doi: 10.1089/gtmb.2009.0002. [DOI] [PubMed] [Google Scholar]
- Tuffery-Giraud S., Béroud C., Leturcq F., Yaou R. B., Hamroun D., Michel-Calemard L., et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 2009;30:934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- Helderman-van den Enden A. T., Madan K., Breuning M. H., van der Hout A. H., Bakker E., de Die-Smulders C., et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. Eur. J. Hum. Genet. 2013;21:21–26. doi: 10.1038/ejhg.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura J., Kato S., Ishihara T., Hiraishi Y., Kawashiro T. Difference of new mutation rates in dystrophin gene between deletion and duplication mutation in Duchenne and Becker muscular dystrophy. Rinsho. Shinkeigaku. 1997;37:212–217. [PubMed] [Google Scholar]
- Strachan T., Read A. P.Human Molecular Genetics4th edn329–332.Garland Science: New York; 2004 [Google Scholar]
- Helderman-van den Enden A. T., de Jong R., den Dunnen J. T., Houwing-Duistermaat J. J., Kneppers A. L., Ginjaar H. B., et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin. Genet. 2009;75:465–472. doi: 10.1111/j.1399-0004.2009.01173.x. [DOI] [PubMed] [Google Scholar]
- Passos-Bueno M. R., Bakker E., Kneppers A. L., Takata R. I., Rapaport D., den Dunnen J. T., et al. Different mosaicism frequencies for proximal and distal Duchenne muscular dystrophy (DMD) mutations indicate difference in etiology and recurrence risk. Am. J. Hum. Genet. 1992;51:1150–1155. [PMC free article] [PubMed] [Google Scholar]