

Iron-sulfur clusters are widespread in metalloproteins, where they most often function to transfer electrons, but also can act as sites for catalysis.[1] In known iron sulfide clusters, the iron ions are in the +2 and +3 oxidation states.[2,3] Even in synthetic chemistry, with a much broader range of supporting ligands, the iron ions in iron sulfide complexes are always Fe2+ or Fe3+. Synthetic all-Fe2+ clusters using cyanide or N-heterocyclic carbene ligands are a recent advance.[4] However, there are no reports of iron-sulfide compounds in which iron ions are reduced to the Fe1+ level.[5] In this contribution, we describe the first examples of isolable iron(I)-sulfide compounds, which establishes that iron(I) is a feasible oxidation state in iron sulfide chemistry.
The progenitor of the new compounds is the previously reported μ-sulfidodiiron(II) compound [LMeFe]2(μ-S) (1-H), (LMe = HC[C(Me)N(2,6-iPr2C6H3)]2).[6] This molecule is the only literature example of an iron-sulfide with a three-coordinate iron atom. The work reported here used a close variant of this compound, [MeLMeFe]2(μ-S) (1-Me), (MeLMe = MeC[C(Me)N(2,6-iPr2C6H3)]2, in which the supporting ligand contains an additional methyl group. [MeLMeFe]2(μ-S) (1-Me) was spectroscopically similar to its LMe analogue (1-H).
We also developed a novel organometallic route to the μ-sulfidodiiron(II) complexes. This strategy takes advantage of rapid, clean β-hydride elimination from low-coordinate alkyl complexes,[7] and the ability of low-coordinate iron(II) hydride complexes to reductively eliminate H2 upon addition of coordinating ligands.[8] Thus, LMeFe(iso-butyl) or MeLMeFe(iso-butyl) were mixed with PMe3S and heated to 100 °C in toluene overnight to give the diiron(II) sulfides 1-H or 1-Me (Scheme 1 shows 1-Me). All of the byproducts PMe3, H2, and isobutylene were conveniently removed by evaporation. The products were isolated in 65% and 73% yield, respectively.
Scheme 1.
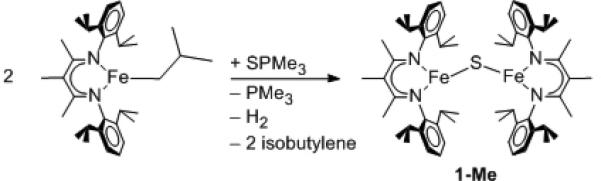
Synthesis of the diiron(II) sulfide complex 1-Me.
A red solution of 1-Me in diethyl ether reacted with two molar equivalents of potassium graphite (KC8) to give a color change to green. The product, [KMeLMeFe]2(μ-S) (2-Me) (Scheme 2), was isolated in 62% yield and crystallographically characterized. 1-Me can instead be reacted with excess metallic sodium in THF to give [NaMeLMeFe]2(μ-S) (3-Me) in 56% yield. Compounds 2-Me and 3-Me had similar 1H NMR spectra, and had half-lives of ca. 80 hrs at 60 °C in C6D6 (Figures S-6 and S-7, Supporting Information).
Scheme 2.
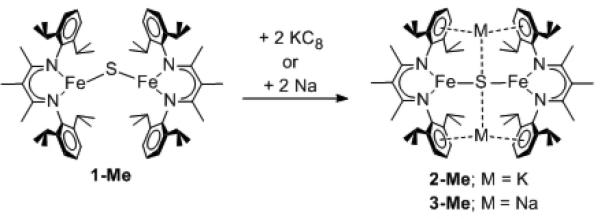
Synthesis of the diiron(I) sulfide complexes.
X-ray diffraction studies showed the solid-state structures of 2-Me and 3-Me (Figure 1 and Supporting Information). The Fe-S bond lengths in 2-Me and 3-Me are 2.1745(13) Å and 2.1957(3) Å, respectively. These Fe-S bond distances are typical for μ2-S atoms in diiron compounds (2.22(3) Å).[9] However, the Fe-S distances in 2-Me and 3-Me are significantly longer than the 2.102(2) Å for a three-coordinate iron(II) atom to a bridging sulfide in LMeFe(μ-S)Fe(NCCH3)LMe.[6] The longer Fe-S bonds suggest that the iron is in a lower oxidation state, and charge counting in the structure suggests a diiron(I) formulation. This hypothesis is addressed below using spectroscopic and computational evidence.
Figure 1.

Thermal-ellipsoid plot of [KMeLMeFe]2(μ-S), 2-Me, using 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. The sodium analogue 3-Me has also been crystallographically characterized, and is shown in the Supporting Information.
In [KMeLMeFe]2(μ-S) (2-Me) and [NaMeLMeFe]2(μ-S) (3-Me), the μ-sulfido bridges are linear (Fe-S-Fe angles of 179.70(4)° and 180°, respectively). Linear sulfide bridges are uncommon, and the average Fe-S-Fe bond angle for diiron complexes with a single bridging sulfur atom is 126(24)°.[9] The next most linear Fe-S-Fe bond angle is 167.0(2)°, in a 5-coordinate iron complex with a bulky salen ligand.[10] Linear sulfido bridges have been seen in complexes of other transition metals such as V, Mo, Co, Ni, and Cu.[11,12,13,14]
In the crystal structures of 2-Me and 3-Me, the alkali metal cations are sandwiched between the aryl groups of the β-diketiminate ligands, as found in formally iron(I) hydride and dinitrogen complexes.[15] Geometric restraints from the cation-π interactions may play a role in enforcing the linear sulfide bridge, though there are literature examples of linear sulfide bridges without such restraints.[11-13] The K-S distances in 2-Me of 2.932(2) Å and 2.936(2) Å are the shortest known.[9] The next shortest K-S bond is 3.039(2) Å in a compound where the K+ ion also has a cation-π interaction.[16] The Na-S distance in 3-Me is 2.6994(7) Å which is only slightly shorter than the average Na-S bond of 2.9(2) Å.[9] Other parameters from the crystal structures of 2-Me and 3-Me are similar, and thus no major structural differences arise from the choice of alkali metal cation.
The ability to exchange the alkali metals was evaluated using NMR spectroscopy. The 1H NMR spectra of 2-Me and 3-Me were consistent with D2d or D2h symmetry in solution, with seven paramagnetically shifted resonances. Mixing 2-Me with 3-Me resulted in the growth of a third set of resonances in the 1H NMR spectrum with a shift pattern similar to the reactants (Supporting Information). We assign the new peaks to the mixed-cation complex [NaKMeLMeFe]2(μ-S) (4-Me). This reaction reached an equilibrium in which all 3 species (2-Me, 3-Me, 4-Me) were present, requiring 24 h in C6D6 and 6 h in Et2O. To further support the exchange of cations, Na+ and K+ sources (1 equiv of NaBArF4 or KOTf) were added to 2-Me and to 3-Me in Et2O solution. When the alkali salts matched (e.g. addition of KOTf to 2-Me), no reaction was seen, but the mixed alkali-metal experiments produced 4-Me, as shown by 1H NMR spectroscopy. These results demonstrate that the potassium and sodium cations can exchange between the aryl rings on the β-diketiminate ligands. However, the compounds are not stable without the alkali metal cations (see below).
We next turned to spectroscopic studies to support the oxidation state assignment as iron(I). The Mössbauer spectrum of solid 2-Me showed a single quadrupole doublet for the two equivalent iron atoms, with isomer shift δ = 0.67 mm s-1 and quadrupole splitting |ΔEQ| = 2.17 mm s-1 that was temperature-independent from 4.2 to 80 K (Figure 2a). The zero-field Mössbauer spectrum of 3-Me was similar with an isomer shift δ = 0.64 mm s-1 and quadrupole splitting of |ΔEQ| = 2.28 mm s-1 at 80 K. For comparison, the Mössbauer spectrum of the diiron(II) sulfide complex 1-Me had distinctly different parameters of δ = 0.59 mm s-1 and |ΔEQ| = 0.89 mm s-1. The increase in isomer shift upon reduction supports the hypothesis that reduction has occurred at the iron centers. The isomer shifts observed for 2-Me and 3-Me also resemble those for thioether-supported iron(I) complexes (δ = 0.62 – 0.76 mm s-1),[17] and a phosphine-supported iron(I) complex (δ = 0.57).[18]
Figure 2.


(a) Mössbauer spectrum of 2-Me at 80 K (top) and at 4.2 K with a 4 T field perpendicular to the gamma rays (bottom). The solid lines are fits for δ = 0.67 mm s-1 and |ΔEQ| = 2.17 mm s-1. The magnetic simulation reveals S = 0 at 4.2 K, Vzz = -2.17 mm s-1 and η = 0.3. (b) Solid-state variable temperature magnetic susceptibility of 2-Me. The solid line is a fit where both iron centers have a spin state of Si = 3/2 and antiferromagnetic coupling with J = -123 cm-1 (H = -2JS1·S2 + gμB(S1+S2)·B). The dashed line represents a 1.4% paramagnetic impurity (PI) with S = 5/2, which was necessary to account for the offset below 50 K.
Applied-field Mössbauer measurements on 2-Me revealed an energetically well-isolated diamagnetic (Stotal = 0) ground state for the dimer, and a positive sign of the electric field gradient with small asymmetry η = 0.3. Solid-state magnetic susceptibility studies (Figure 2b) also indicated antiferromagnetic coupling of two paramagnetic iron subsites to give a regular spin ladder with an Stotal = 0 ground state, as expected for strong exchange interaction that dominates the single-ion zero-field splitting (zfs). The data fit to a fundamental model where each iron(I) ion is high-spin (SFe = 3/2) and J = -123±8 cm-1 quantifies the antiferromagnetic coupling. Interestingly, this system does not have the strong first-order orbital moment that was observed for a related mononuclear iron(I) complex.[19] We estimate that D = 0±30 cm-1; the simulations are not particularly sensitive to zfs in such a dinuclear system where a spin singlet is the ground state.
The spectroscopic studies were supplemented with calculations on the full molecule using density-functional theory (DFT) using the crystallographic coordinates. The functional and basis set were varied to find the best match to the geometry, the Mössbauer parameters and the J value.[15b] The electronic structure description of 2-Me derived from the best-fit (broken-symmetry calculations with TPSSh functional and TZVP basis set) calculations showed two antiferromagnetically coupled high-spin iron(I) centers with J = -170 cm-1 (Figure 3). The β-diketiminates showed no compelling evidence for “redox non-innocent” sharing of spin density from the metals. There is slight π-backbonding from the iron(I) centers to the unoccupied β-diketiminate orbitals, suggesting that the electronic properties of the supporting ligand may play a role in stabilizing the low oxidation state of iron(I). However, the interaction between the iron atoms and the π-system of the β-diketiminates is small.
Figure 3.

(Top) d-Orbitals from the computational model of 2-Me. The Fe character is shown for each orbital, and the S values give the overlap between pairs of corresponding orbitals. This picture indicates two high-spin d7 centers, with antiferromagnetic coupling mediated by sulfur p orbitals. (Bottom) Spin-density plot for 2-Me; since there is little spin on the supporting ligands, they are “redox innocent.”
The role of the alkali metal in stabilizing the low iron oxidation state was evaluated experimentally by studying the diiron(I) sulfide in the presence of solvents and additives that have the ability to remove the alkali metal cation. In C6D6, 2-Me and 3-Me were stable for about 5 days at 60 °C, while under the same conditions in THF-d8 they were stable for less than 2 hours. This result suggests that THF may pull the alkali metal cations away from the Fe/S core, destabilizing 2-Me and 3-Me. In a more muscular test of this hypothesis, the potassium chelators 18-crown-6 or cryptand-222 were added to solutions of 2-Me under argon or N2, which led to immediate decomposition. Electrochemical reduction of 1-Me in Et2O indicated a one-electron wave at -2.7 V (vs. Fc+/0), also supporting the idea that two-electron reduction is not possible without the alkali metal cations. All of these results indicate that the cation plays a significant role in stabilizing the iron(I) complexes, most likely by the close interactions between cations and the negatively charged core of the molecule.
In conclusion, we have shown that diiron(I) sulfide compounds can be isolated, and their characterization as bona fide iron(I) complexes is supported by crystallography, spectroscopy, magnetism, and computations. The unprecedented stability of an iron-sulfide complex in this oxidation state is enabled by steric contributions (bulky ligands that protect the Fe-S-Fe core) and electronic contributions (especially close interactions between cations and the anionic core).
Supplementary Material
Footnotes
Funding was provided by the National Institutes of Health (GM-065313). P.L.H. was also supported by a Fulbright Scholar grant.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Meghan M. Rodriguez, Department of Chemistry University of Rochester Rochester, NY 14627 (USA)
Bryan D. Stubbert, Department of Chemistry University of Rochester Rochester, NY 14627 (USA)
Christopher C. Scarborough, Department of Chemistry Emory University Atlanta, GA 30322 (USA) scarborough@emory.edu
William W. Brennessel, Department of Chemistry University of Rochester Rochester, NY 14627 (USA)
Eckhard Bill, Max-Planck-Institut für Bioanorganische Chemie 45470 Mülheim an der Ruhr (Germany).
Patrick L. Holland, Department of Chemistry University of Rochester Rochester, NY 14627 (USA).
References
- 1.Beinert H, Holm RH, Münck E. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.a Angove HC, Yoo SJ, Burgess BK, Münck E. J. Am. Chem. Soc. 1997;119:8730–8731. [Google Scholar]; b Angove HC, Yoo SJ, Münck E, Burgess BK. J. Biol. Chem. 1998;273:26330–26337. doi: 10.1074/jbc.273.41.26330. [DOI] [PubMed] [Google Scholar]; c Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Münck E. J. Am. Chem. Soc. 1999;121:2534–2545. [Google Scholar]; d Musgrave KB, Angove HC, Burgess BK, Hedman B, Hodgson KO. J. Am. Chem. Soc. 1998;120:5325–5326. [Google Scholar]
- 3.All-ferrous biological clusters: Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC. Biochemistry. 1997;36:1181–1187. doi: 10.1021/bi9626665.Mayer SM, Lawson DM, Gormal CA, Roe SM, Smith BE. J. Mol. Biol. 1999;292:871–891. doi: 10.1006/jmbi.1999.3107.
- 4.a Scott TA, Berlinguette CP, Holm RH, Zhou H-C. Proc. Natl. Acad. Sci. USA. 2005;102:9741–9744. doi: 10.1073/pnas.0504258102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deng L, Holm RH. J. Am. Chem. Soc. 2008;130:9878–9886. doi: 10.1021/ja802111w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL. Inorg. Chem. 2009;48:2735–2747. doi: 10.1021/ic802192w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.There are some nitrosyl-containing iron-sulfur clusters that could be described as iron(I), but the iron oxidation state assignment depends on the formalism used for the NO ligand. For example: Lu T-T, Huang H-W, Liaw W-F. Inorg. Chem. 2009;48:9027–9035. doi: 10.1021/ic9012679.
- 6.Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J. Am. Chem. Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]
- 7.Vela J, Vaddadi S, Cundari TR, Smith JM, Gregory EA, Lachicotte RJ, Flaschenriem CJ, Holland PL. Organometallics. 2004;23:5226–5239. [Google Scholar]
- 8.Yu Y, Sadique AR, Smith JM, Dugan TR, Cowley RE, Brennessel WW, Flaschenriem CJ, Bill E, Cundari TR, Holland PL. J. Am. Chem. Soc. 2008;130:6624–6638. doi: 10.1021/ja710669w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cambridge Structural Database. Allen FH. Acta Crystallogr. 2011;2002:B58, 380–388. doi: 10.1107/s0108768102003890. update. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee RN, Stack TDP, Holm RH. J. Am. Chem. Soc. 1988;110:1850–1861. [Google Scholar]
- 11.Vanadium: Schiemann J, Hübener P, Weiss E. Angew. Chem. Int. Ed. 1983;22:980–981.Angew. Chem. 1983;95:1021.
- 12.Molybdenum: Lincoln S, Soong SL, Koch SA, Sato M, Enemark JE. Inorg. Chem. 1985;24:1355–1359.Yoshida T, Adachi T, Matsumura K, Baba K. Angew. Chem. Int. Ed. 1993;32:1621–1623.McDonough JE, Mendiratta A, Curley JJ, Fortman GC, Fantasia S, Cummins CC, Rybak-Akimova EV, Nolan SP, Hoff CD. Inorg. Chem. 2008;47:2133–2141. doi: 10.1021/ic701611p.Davies SC, Hughes DL, Richards RL, Sanders JR. Dalton Trans. 2000:719–725.
- 13.Cobalt, nickel: Mealli C, Midollini S, Sacconi L. Inorg. Chem. 1978;17:632–637.Jaeheung C, Van Heuvelen KM, Yap GPA, Brunold TC, Riordan CG. Inorg. Chem. 2008;47:3931–3933. doi: 10.1021/ic800321x.
- 14.Copper: Delgado S, Miguel PJS, Priego JL, Jiménez-Aparicio R, Gómez-García CJ, Zamora F. Inorg. Chem. 2008;47:9128–9130. doi: 10.1021/ic801314s.
- 15.a Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J. Am. Chem. Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]; b Chiang KP, Scarborough CC, Horitani M, Lees NS, Ding K, Dugan TR, Brennessel WW, Bill E, Hoffman BM, Holland PL. Angew. Chem. Int. Ed. 2012;51:3658–3662. doi: 10.1002/anie.201109204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niemeyer M, Power PP. Inorg. Chem. 1996;35:7264–7272. doi: 10.1021/ic960570t. [DOI] [PubMed] [Google Scholar]
- 17.Mock MT, Popescu CV, Yap GPA, Dougherty WG, Riordan CG. Inorg. Chem. 2008;47:1889–1891. doi: 10.1021/ic7023378. [DOI] [PubMed] [Google Scholar]
- 18.Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc. Natl. Acad. Sci. USA. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoian SA, Yu Y, Smith JM, Holland PL, Bominaar EL, Münck E. Inorg. Chem. 2005;44:4915–4922. doi: 10.1021/ic050321h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.