

Abstract
Membrane proteins are essential for cell viability and are therefore important therapeutic targets1-3. Since they function in complexes4, methods to identify and characterize their interactions are necessary5. To this end, we developed the Membrane Strep-protein interaction experiment, called Membrane-SPINE6. This technique combines in vivo cross-linking using the reversible cross-linker formaldehyde with affinity purification of a Strep-tagged membrane bait protein. During the procedure, cross-linked prey proteins are co-purified with the membrane bait protein and subsequently separated by boiling. Hence, two major tasks can be executed when analyzing protein-protein interactions (PPIs) of membrane proteins using Membrane-SPINE: first, the confirmation of a proposed interaction partner by immunoblotting, and second, the identification of new interaction partners by mass spectrometry analysis. Moreover, even low affinity, transient PPIs are detectable by this technique. Finally, Membrane-SPINE is adaptable to almost any cell type, making it applicable as a powerful screening tool to identify PPIs of membrane proteins.
Keywords: Bioengineering, Issue 81, Membrane Proteins, in vivo protein-protein interaction, formaldehyde cross-linking, MS-analysis, Strep-tag
Introduction
To understand the function of a protein it is essential to know its interaction partners. Several classical techniques are available for the identification of interaction partners of soluble proteins. However, these techniques are not easily transferable to membrane proteins due to their hydrophobic nature4. To overcome this limitation, we have developed the Membrane Strep-protein interaction experiment (Membrane-SPINE)6. It is based on the SPINE method, which was only suitable for soluble proteins7.
Membrane-SPINE benefits from two advantages of the cross-linking agent formaldehyde: first, formaldehyde can easily penetrate membranes and therefore generates a precise snapshot of the interactome of a living cell8. Second, formaldehyde cross-links can be reversed by boiling9. Here, these two advantages are used to identify not only permanent but also transient PPIs of membrane proteins6.
In brief, a Strep-tag is fused to the C-terminus of the integral membrane bait protein. Cells expressing the membrane bait protein are incubated with formaldehyde which cross-links prey proteins to the membrane bait protein (Figure 1). Modifications of prey proteins are not needed. Next, the membrane fraction is prepared. Therefore, membrane proteins are solubilized by detergent treatment and bait proteins are co-purified with its prey proteins using affinity purification. Subsequently, the cross-link is reversed by boiling, and the bait and its co-eluted prey proteins are separated by SDS-PAGE. Finally, prey proteins can be identified by immunoblot analysis or mass spectrometry.
Protocol
Note: Detailed information regarding buffers indicated in the protocol is available in Table 1.
1. Fixation of Protein-protein Interactions by Formaldehyde Cross-linking in Living Cells
- To begin prepare growth media, stock solutions and buffer P1
- Prepare 500 ml medium: The medium should provide conditions that support the interaction between membrane bait protein and prey proteins. In addition, one experiment should be performed under conditions where no interaction is expected (e.g. without cross-linking). Note: We compare protein-protein interactions in stressed and non-stressed bacteria. Therefore, we prepare 500 ml Luria Bertani (LB) medium (Table 1) that we complement with a stress-inducing agent (e.g. 0.5 M NaCl) in a 2 L Erlenmeyer flask. For control, we use normal LB medium with 0.17 M NaCl (pH 7.0).
- Prepare Tris-buffer (20 mM Tris-HCl; pH 8.0) and 0.1 M ethylenediaminetetraacetic acid (EDTA), pH 8.0 stock solution (Table 1).
- Prepare buffer P1. Dissolve sucrose to a final concentration of 0.5 M in Tris-buffer. Sterilize buffer P1 by filtration and store it at 4 °C.
Grow cells expressing the membrane bait protein with a C-terminal Strep-tag fusion from a vector. Induce the expression of membrane bait proteins for a sufficient time. Note: We induce the expression of bacterial membrane bait proteins at early-log phase (A600 = 0.3) by addition of 250 µl 1 M isopropyl-β-D-thiogalactopyranoside (IPTG) to 500 ml medium (final concentration: 0.5 mM). To allow sufficient expression, we incubate the bacteria until the late-log phase (A600 = 1.2).
Prepare for each culture two centrifuge beakers with a minimum volume of 300 ml each and place them on ice.
- Perform formaldehyde cross-linking. CAUTION: Formaldehyde is highly toxic. We recommend working under a safety fume hood.
- Transfer the culture vessel under a safety fume hood. Split each sample into two samples, one omitting cross-linking (- X) and one including the formaldehyde cross-linking (+ X). Add 4 ml 37% formaldehyde solution to 250 ml culture to reach a final concentration of 0.6%.
Transfer the culture vessels back and grow cells for further 20 min as before.
Fill your cultures into the centrifuge tubes under a safety fume hood. Collect cells by centrifugation at 3,000 x g for 30 min.
Discard supernatant by carefully pipetting it up under a safety fume hood. Collect toxic formaldehyde-containing supernatant and dispose of it properly.
- In order to reach optimal yield during membrane protein preparation, prepare spheroplasts first. Here, we provide a protocol for spheroplast formation of Gram-negative bacteria:
- Prepare buffer P2 (Table 1) from lyophilized powder. Gently dissolve 2 mg lysozyme in 0.1 M EDTA, pH 8, in a 1.5 ml tube. Always prepare buffer P2 freshly for the day of the experiment.
- Prepare Tris-buffer with protease inhibitor (buffer P3). To 10 ml of Tris-buffer, add 0.1 ml 1 M phenylmethylsulfonylfluoride (PMSF) (Table 1) to a final concentration of 10 mM. Use the buffer immediately, to ensure proper function of PMSF as protease inhibitor.
- Resuspend cell pellets in 10 ml Tris-buffer with protease inhibitor and transfer solution to a 15 ml conical tube.
- Add 1 ml buffer P2 and incubate on ice for 30 min.
- Collect spheroplasts by centrifugation at 3,000 x g for 30 min and discard supernatant carefully.
Incubate the spheroplast pellets overnight at -20°C.
2. Purification of Strep-tag Membrane Protein (Bait)
Place spheroplast pellet on ice.
During the time the spheroplast pellets thaw on ice, prepare 10 ml buffer P3 for each spheroplast preparation. Gently dissolve 1 mg DNaseI in 10 ml Tris-buffer. Add 0.1 ml 1 M PMSF just before use.
Resuspend the pellet in 6 ml freshly prepared P3. Sonicate the sample four times for 1 min continuously on ice with 1 min pause between each burst, in order to disrupt spheroplasts.
Centrifuge the sample at 10,000 x g for 10 min to harvest cell debris. Transfer the supernatant using a pipette to an ultracentrifuge tube.
Pellet the membrane fraction at 100,000 x g for 30 min. Wash the pellet carefully in Tris-buffer without dissolving it. Dry the tube with a paper tissue (e.g. Kleenex). Avoid disturbing the pellet at any time.
Resuspend the pellet (= membranes) carefully in 1 ml Tris-buffer. Use a 20 µl aliquot to determine the protein concentration by e.g. BCA assay. At this step, the membranes can be shock frozen in liquid nitrogen and stored at -80 °C.
3. Purification of Strep-tag Membrane Bait Protein and SDS-PAGE
Normalize the protein concentration of the membrane fraction to 5 mg/ml with Tris-buffer. Take 2.5 ml membrane fraction (5 mg/ml) in an ultracentrifuge tube and add 0.25 ml of 20% Triton X-100 to reach a final concentration of 2%, in order to solubilize the membrane proteins. Add a micro-magnetic rod and stir on ice for 1 hr. Note: Although in general we have good results using Triton X-100 as detergent, it might be useful to change the detergent for optimization. In this respect, we have best results with those detergents used for functional purification of the respective membrane bait protein.
- While performing step 3.1, prepare 50 ml buffer W and 5 ml buffer E from (Table 1). Fill up 10 ml 5x buffer W concentrate to 50 ml and add 150 µl 20% Triton X-100.
- Fill up 1 ml 5x buffer E concentrate to 10 ml and add 30 µl 20% Triton X-100. Equilibrate a 1 ml Strep-Tactin superflow gravity flow column with 8 ml buffer W. Note: It is essential to add the detergent used for solubilization in a concentration 10-fold above the critical micelle concentration (cmc) in any buffer during the purification procedure.
Remove the micro-magnetic rod from the solubilization sample and ultracentrifuge at 100,000 x g for 30 min, to pellet the insoluble membrane fraction. Remove the supernatant using a pipette for further purification.
Load the supernatant to the column. Run the column only with gravity flow. Wash the column with 5 ml buffer W. Repeat this washing step 5x.
Elute membrane bait proteins with 1 ml buffer E. Repeat this elution step 4x.
Concentrate elution fractions 2, 3, and 4 to 300 µl with a centrifugal filter unit.
Mix 200 µl of each sample with 50 µl 5x SDS-PAGE loading dye. Split each preparation in 125 µl aliquots. Boil one aliquot of each preparation for 20 min at 95 °C, to reverse formaldehyde cross-links. Let samples cool down to RT for at least 10 min on bench top.
Load 30 µl from each sample to a single lane of a polyacrylamide-gel suitable for immunoblotting. Use a prestained molecular weight marker for best orientation and run SDS-PAGE10.
4. Immunoblot Analysis to Confirm Interaction Partners
Once step 3.8 is completed, transfer proteins to a nitrocellulose membrane by e.g. semidry.
Block the membrane to prevent unspecific labeling. Prepare blocking buffer by weighing bovine serum albumin (BSA) to achieve 3% weight per volume in Tris-buffered saline supplemented with final a concentration of 0.05% Tween-20 (TBS-T). Use a sufficient volume of blocking buffer to cover the membrane. Block the membrane for 1 hr at RT.
Dilute and incubate the prey protein specific first antibody as usual. Use an HRP-linked antibody as the second antibody.
Develop the immunoblot using a chemiluminescent detection kit with high sensitivity. Monitor the signal using a classical film processing procedure or digital imaging equipment.
5. NanoLC-ESI-MS/MS High Resolution Experiments to Identify Interaction Partners
In case that no specific antibody is available for the prey protein or unknown interaction partners should be identified, use mass spectrometry (MS) for identification. In order to prevent keratin contamination, use precasted gels for protein separation.
Silver stain the SDS-PAGE using a MS-compatible staining kit according to the manufacturer's protocol. Perform all staining and washing steps in glass tanks.
Excise respective bands and analyze these by high resolution LC/MS9.
Representative Results
Membrane SPINE analysis allows the co-purification of membrane proteins and transiently interacting protein partners. The co-purification is achieved by using the cross-linking agent formaldehyde. Two parameters are critical to prevent unspecific cross-links: the formaldehyde concentration and the cross-linking time. The sufficient, but not excessive use of formaldehyde can easily be controlled by immunoblotting. Formaldehyde cross-linked protein complexes can be separated by boiling but not by SDS treatment. Hence, they can be visualized after blotting of the Strep-tagged membrane bait protein as smear in the upper section of the immune-blot of an unboiled sample.
A representative result of a Membrane SPINE assay, including all required controls, is presented in Figure 2. As bait protein the integral membrane protein CpxA of Escherichia coli was used6,11. CpxA is a sensor kinase and consists of an N-terminal sensor domain with two transmembrane domains (TMD) integrating a large extracytoplasmic sensor domain and a C-terminal highly conserved cytoplasmic catalytic domain12. After stimulation, CpxA activates its cognate response regulator CpxR. Activated CpxR diffuses off to mediate the response. For Membrane SPINE, the Strep-tag was fused to the C-terminus of CpxA (CpxA-Strep). CpxA-Strep cross-linked to other proteins or protein complexes is detected as a smear in the unboiled formaldehyde-treated sample (Figure 2, line 1 versus line 3) indicating sufficient cross-linking. Moreover, a direct protein-protein interaction of CpxA-Strep with its cognate response regulator protein CpxR as the prey protein is only detectable in the presence of formaldehyde (Figure 2, line 2 versus line 4) supporting the specificity of formaldehyde cross-linking.
A representative result of a Membrane SPINE analysis is presented in Figure 3. Samples corresponding to those of Figure 2 were silver stained. The arrow marks a band that is specific for the boiled sample. Due to the background, MS analysis also identified other proteins besides CpxR6. Therefore, the non-formaldehyde treated sample should always be analyzed, to assign background noise and specific interaction partner.
Table 1: Buffers and reagents required for membrane-SPINE.
| Buffer / Reagent / Medium | Working concentration | comment | |
|---|---|---|---|
| LB | 10 g Tryptone 5 g yeast extract 10 g NaCl to 1 L, adjust pH to 7.0 | Luria Broth medium | |
| IPTG | 1 M Isopropyl-β-D-thiogalactopyranoside | 0.5 mM | |
| Tris-buffer | 20 mM Tris-HCl, pH 8 | Adjust pH to 8.0 using NaOH | |
| 0,1 M EDTA, pH 8 | 0.1 M EDTA | Adjust pH to 8.0 using NaOH | |
| PMSF | 1 M Phenylmethylsulfonylfluoride in 100% Isopropanol | 10 mM | PMSF is stable in 100% isopropanol but not in water! Stock solution can be stored at -20 °C; PMSF has to be adapted to room temperature before diluting in buffer; PMSF containing buffers should be used in within 10 min of preparation. |
| P1 | 20 mM Tris-HCl, pH 8.0 0.5 M Sucrose | ||
| P2 | 2 mg/ml lysozyme in 0.1 M EDTA, pH 7.5 | ||
| P3 | 20 mM Tris-HCl, pH 8.0 10 mM PMSF | Use immediately after preparation | |
| Detergent | 20% Triton X-100 | 2% for solubilization | |
| Buffer W | Fill up 10 ml 5x concentrate to 50 ml add 150 µl 20% Triton X-100 | 100 mM Tris-HCl, pH 8.0 150 mM NaCl 1 mM EDTA 0.06% Triton X-100 | 5x concentrate is part of Strep-tag protein purification buffer set (IBA) |
| Buffer E | Fill up 1 ml 5x concentrate to 10 ml add 30 µl 20% Triton X-100 | 100 mM Tris-HCl, pH 8.0 150 mM NaCl 1 mM EDTA 2.5 mM Dethiobiotin 0.06% Triton X-100 | 5x concentrate is part of Strep-tag protein purification buffer set (IBA) |
| 5x SDS-PAGE loading dye | 0.3125 M Tris-HCl, pH 6.8 10% SDS 0.5 M DTT 50% glycerol 0.05% bromophenol blue |
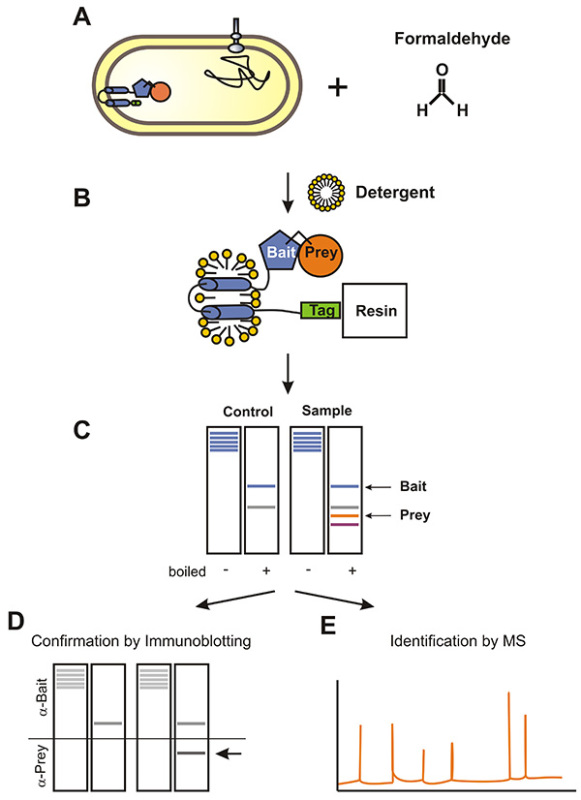 Figure 1. Flow chart of the Membrane-SPINE procedure using an Escherichia coli membrane protein as bait protein.A) Bacteria expressing the Strep-tagged membrane bait protein are treated with formaldehyde. Formaldehyde penetrates membranes and cross-links prey proteins to the membrane bait protein. B) The membrane fraction is prepared and membrane proteins are solubilized by detergent treatment. Subsequently, prey proteins are co-purified with the bait protein. C) Formaldehyde cross-links are reversed by boiling and proteins are separated by SDS-PAGE. Finally, prey proteins are either monitored by immunoblotting (D) or identified by MS-analysis (E). Click here to view larger image.
Figure 1. Flow chart of the Membrane-SPINE procedure using an Escherichia coli membrane protein as bait protein.A) Bacteria expressing the Strep-tagged membrane bait protein are treated with formaldehyde. Formaldehyde penetrates membranes and cross-links prey proteins to the membrane bait protein. B) The membrane fraction is prepared and membrane proteins are solubilized by detergent treatment. Subsequently, prey proteins are co-purified with the bait protein. C) Formaldehyde cross-links are reversed by boiling and proteins are separated by SDS-PAGE. Finally, prey proteins are either monitored by immunoblotting (D) or identified by MS-analysis (E). Click here to view larger image.
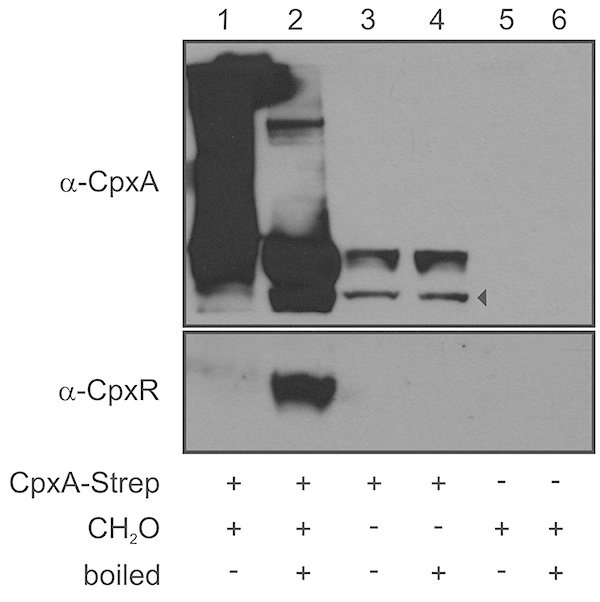 Figure 2. Representative immunoblot used to monitor a PPI of a membrane protein. Bacteria producing CpxA-Strep from a plasmid as membrane bait protein, were grown in LB medium to an OD600 of 1 and exposed to formaldehyde (CH2O) for 20 min. The inner membrane fraction was prepared, membrane proteins were solubilized by detergent treatment and CpxA-Strep was purified (lanes 1 and 2). Bacteria producing either CpxA-Strep without formaldehyde treatment (lanes 3 and 4) or carrying the empty vector with formaldehyde treatment (lanes 5 and 5) served as controls. Aliquots of each sample were boiled at 95 °C for 20 min (lanes 2, 4, and 6) to separate cross-linked proteins from CpxA-Strep. Proteins were separated in a 12.5% SDS-PAGE. Immunoblotting was performed and the blot was separated according to the size of CpxA-Strep (51 kDa) and the respective prey proteins CpxR (26 kDa). The two parts of the immunoblot were incubated with polyclonal antibodies raised against CpxA and CpxR, respectively. Subsequently, the blots were further treated with an anti-rabbit horse (HRP) antibody and developed using the SuperSignal West Pico Chemiluminescent substrate. The arrowhead marks degradation products of CpxA. Click here to view larger image.
Figure 2. Representative immunoblot used to monitor a PPI of a membrane protein. Bacteria producing CpxA-Strep from a plasmid as membrane bait protein, were grown in LB medium to an OD600 of 1 and exposed to formaldehyde (CH2O) for 20 min. The inner membrane fraction was prepared, membrane proteins were solubilized by detergent treatment and CpxA-Strep was purified (lanes 1 and 2). Bacteria producing either CpxA-Strep without formaldehyde treatment (lanes 3 and 4) or carrying the empty vector with formaldehyde treatment (lanes 5 and 5) served as controls. Aliquots of each sample were boiled at 95 °C for 20 min (lanes 2, 4, and 6) to separate cross-linked proteins from CpxA-Strep. Proteins were separated in a 12.5% SDS-PAGE. Immunoblotting was performed and the blot was separated according to the size of CpxA-Strep (51 kDa) and the respective prey proteins CpxR (26 kDa). The two parts of the immunoblot were incubated with polyclonal antibodies raised against CpxA and CpxR, respectively. Subsequently, the blots were further treated with an anti-rabbit horse (HRP) antibody and developed using the SuperSignal West Pico Chemiluminescent substrate. The arrowhead marks degradation products of CpxA. Click here to view larger image.
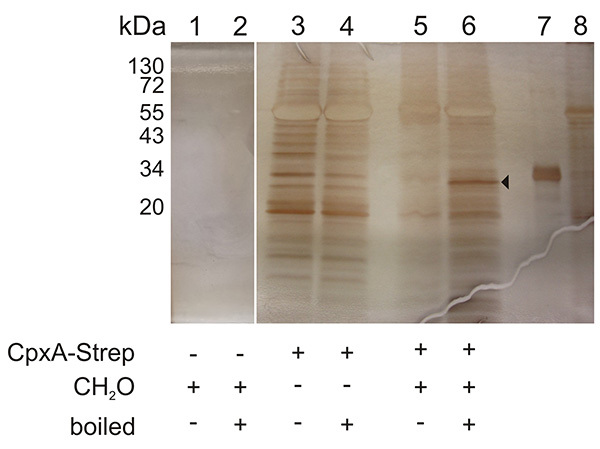 Figure 3. Representative silver-stained SDS-PAGE used for the identification of an interaction partner of a membrane protein by MS analysis. Samples corresponding to those of Figure 2 were silver stained. The arrow marks a band which was analyzed by MS analysis and which confirmed CpxR as the interaction partner of CpxA5. Lane 7 shows purified CpxR-His6 and lane 8 shows purified CpxA-His6 protein. Click here to view larger image.
Figure 3. Representative silver-stained SDS-PAGE used for the identification of an interaction partner of a membrane protein by MS analysis. Samples corresponding to those of Figure 2 were silver stained. The arrow marks a band which was analyzed by MS analysis and which confirmed CpxR as the interaction partner of CpxA5. Lane 7 shows purified CpxR-His6 and lane 8 shows purified CpxA-His6 protein. Click here to view larger image.
Discussion
Membrane SPINE analysis is a biochemical approach that enables one to confirm and to identify to this point unknown interaction partners of membrane proteins. Membrane SPINE combines in vivo cross-linking by formaldehyde with purification of a Strep-tagged membrane bait protein. The combination with immunoblotting facilitates the confirmation of predicted interaction partners (Figure 2). Additionally, the combination with MS analysis permits the identification of unknown interaction partners (Figure 3). For both applications, there is no requirement for modifying your prey protein. Moreover, Membrane SPINE is sufficiently sensitive to allow the detection of endogenous prey proteins6.
Here, we present a protocol optimized for membrane proteins of gram-negative bacteria. The envelope of gram-negative bacteria separates the cytoplasm from the environment and posses, in addition to the cytoplasmic membrane, an outer membrane and a murein sacculus. Hence, our protocol includes the removal of the outer membrane and the murein sacculus, resulting in so-called spheroplasts. Because such protocols are available for most cell types, our protocol should be adaptable for most membrane proteins. In addition, the following points should be considered.
First of all, the copy number of the vector used to overproduce the membrane bait protein has to be balanced between a high level to allow sufficient membrane protein purification and a low level to prevent unspecific interactions
Second, for some membrane proteins an N-terminal fusion might be more optimal than a C-terminal fusion. In both cases, the functionality of the fusion should be confirmed by trans-complementation.
Third, other affinity purification protocols are also compatible with subsequent MS analysis, such as FLAG purification and tandem affinity purification (TAP). We prefer the Strep-tag II because of its small size with only 8 amino acids (WSHPQFEK).
Fourth, the concentration of formaldehyde used and the cross-linking time should be optimized to prevent unspecific cross-linking. A sufficient but not excessive use of formaldehyde can be monitored by immunoblotting of unboiled samples as the ratio between cross-linked and nonlinked membrane bait protein (Figure 2). Cross-linked proteins migrate as a smear in the upper part of the immunoblot. Un-linked proteins migrate as untreated protein. The ratio between cross-linked and unlinked protein should be about 3:1.
Fifth, there is no "general detergent" for all membrane proteins for solubilization. Hence, in some cases the appropriate detergent for solubilization of the membrane bait protein has to be determined. Thereby, the chosen detergent must be compatible with the Strep-tag column.
Finally, the silver staining procedure has to be compatible with subsequent MS analysis. MS compatible silver staining kits are available from different suppliers. We used different ones with comparable good results.
Disclosures
We have nothing to disclose.
Acknowledgments
This research was supported by grants of the Deutsche Forschungsgemeinschaft GraKo1121, Hu1011/2-1 and SFB940 to S.H.
References
- Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25:451–457. doi: 10.1093/bioinformatics/btp002. [DOI] [PubMed] [Google Scholar]
- Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat. Biotechnol. 2007;25 doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- Daley DO. The assembly of membrane proteins into complexes. Curr. Opin. Struct. Biol. 2008;18:420–424. doi: 10.1016/j.sbi.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Jung K, Fried L, Behr S, Heermann R. Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 2012;15:118–124. doi: 10.1016/j.mib.2011.11.009. [DOI] [PubMed] [Google Scholar]
- Müller VS, Jungblut PR, Meyer TF, Hunke S. Membrane-SPINE: An improved method to identify protein-protein interaction partners of membrane proteins in vivo. Proteomics. 2011;11:2124–2128. doi: 10.1002/pmic.201000558. [DOI] [PubMed] [Google Scholar]
- Herzberg C, et al. SPINE: A method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics. 2007;7:4032–4035. doi: 10.1002/pmic.200700491. [DOI] [PubMed] [Google Scholar]
- Sutherland BW, Toews J, Kast J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 2008;43:699–715. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- Hernandez P, Müller M, Appel RD. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrom. Rev. 2006;25:235–254. doi: 10.1002/mas.20068. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Hunke S, Keller R, Müller VS. Signal integration by the Cpx-envelope stress system. FEMS Microbiol. Lett. 2012;326:12–22. doi: 10.1111/j.1574-6968.2011.02436.x. [DOI] [PubMed] [Google Scholar]
- Weber RF, Silverman PM. The Cpx proteins of Escherichia coli K12 : Structure of the CpxA polypeptide as an inner membrane component. J. Mol. Biol. 1988;203:467–478. doi: 10.1016/0022-2836(88)90013-7. [DOI] [PubMed] [Google Scholar]