

CONSPECTUS
The two principal components of biological membranes, the lipid bilayer and the proteins integrated within it, have coevolved for specific functions that mediate the interactions of cells with their environment. Molecular structures can provide very significant insights about protein function. In the case of membrane proteins, the physical and chemical properties of lipids and proteins are highly interdependent; therefore structure determination should include the membrane environment. Considering the membrane alongside the protein eliminates the possibility that crystal contacts or detergent molecules could distort protein structure, dynamics, and function and enables ligand binding studies to be performed in a natural setting.
Solid-state NMR spectroscopy is compatible with three-dimensional structure determination of membrane proteins in phospholipid bilayer membranes under physiological conditions and has played an important role in elucidating the physical and chemical properties of biological membranes, providing key information about the structure and dynamics of the phospholipid components. Recently, developments in the recombinant expression of membrane proteins, sample preparation, pulse sequences for high-resolution spectroscopy, radio frequency probes, high-field magnets, and computational methods have enabled a number of membrane protein structures to be determined in lipid bilayer membranes.
In this Account, we illustrate solid-state NMR methods with examples from two bacterial outer membrane proteins (OmpX and Ail) that form integral membrane β-barrels. The ability to measure orientation-dependent frequencies in the solid-state NMR spectra of membrane-embedded proteins provides the foundation for a powerful approach to structure determination based primarily on orientation restraints. Orientation restraints are particularly useful for NMR structural studies of membrane proteins because they provide information about both three-dimensional structure and the orientation of the protein within the membrane. When combined with dihedral angle restraints derived from analysis of isotropic chemical shifts, molecular fragment replacement, and de novo structure prediction, orientation restraints can yield high-quality three-dimensional structures with few or no distance restraints. Using complementary solid-state NMR methods based on oriented sample (OS) and magic angle spinning (MAS) approaches, one can resolve and assign multiple peaks through the use of 15N/13C labeled samples and measure precise restraints to determine structures.
Introduction
Biological membranes are essential for cellular life. These ancient structures evolved very early on before the split from the last universal common ancestor that led to the three branches of cellular life: bacteria, archaea, and eukarya. The chemical composition and structural organization of biological membranes indicate that they emerged through a process of coevolution of their two principal components: the lipid bilayer and the proteins integrated within it.1 The lipid bilayer provides an elastic yet ion-impermeable barrier for the effective compartmentalization of specialized cellular components, while integral membrane proteins mediate all interactions of cells with each other and with the outside world through their specific functions in transmembrane transport, signaling, adhesion, and much more. The coevolution model reflects the intimate relationship between membrane proteins and lipids. The lipid bilayer environment is anisotropic and heterogeneous, with large gradients in fluidity, water concentration, and dielectric constants from the bilayer core to the water–lipid interface. These features have profound effects on membrane protein structure, dynamics, and function,2 underscoring the importance of determining the structures of membrane proteins in lipid bilayers at or near physiological conditions of temperature, pH, and hydration.
X-ray diffraction and solution NMR structural studies of membrane proteins typically require proteins dissolved in detergents because lipid bilayers are incompatible with the requisite three-dimensional crystallization and isotropic motion. Lipid nanodiscs typically yield broader lines compared with micelles but can be useful solution NMR membrane mimics for some membrane proteins, as shown recently for the bacterial outer membrane protein OmpX.3 By contrast, solid-state NMR is compatible with structure determination of membrane proteins in a wide variety of phospholipid bilayer membranes under physiological conditions.
From the very beginning, solid-state NMR has played an important role in elucidating the physical and chemical properties of biological membranes. Early solid-state NMR studies provided key information about the structure and dynamics of the phospholipid components.4–7 More, recently, solid-state NMR studies have shifted attention to integral membrane proteins, facilitated by the use of recombinant DNA technology and automated PCR protocols, which enable a variety of proteins to be prepared biosynthetically and labeled isotopically. Recent developments in the areas of recombinant bacterial expression, sample preparation, pulse sequences for high-resolution spectroscopy, radio frequency probes, high-field magnets, and computational methods have enabled a number of membrane protein structures to be determined in phospholipid membranes (a few recent examples8–11 are shown in Figure 1). Furthermore, solid-state NMR is providing important structural information for membrane proteins and peptides in native cell membranes and for key cytoskeletal components of cell envelopes.12–15 These advances are described in recent excellent reviews.16–22
FIGURE 1.
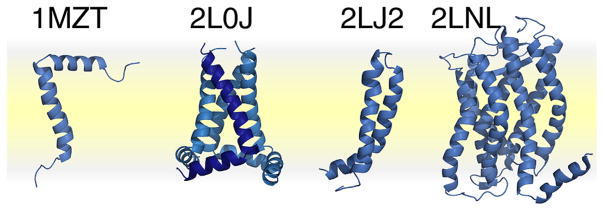
Recent structures of membrane proteins determined in phospholipid bilayers by solid-state NMR. PDB codes: 1MZT, membrane-bound bacteriophage fd coat protein;8 2L0J, pore-forming domain of influenza M2;9 2LJ2, mercury transporter MerF;10 2LNL, human chemokine receptor CXCR1.11 The phospholipid bilayer membrane is depicted in yellow.
Using oriented sample (OS)23,24 and magic angle spinning (MAS)16,18 solid-state NMR approaches, it is possible to resolve and assign multiple peaks through the use of 15N/13C-labeled samples and to measure precise restraints for structure determination. OS solid-state NMR uses samples that are uniaxially aligned in the magnetic field (e.g., planar lipid bilayers) to yield orientation-dependent single line resonances. MAS solid-state NMR uses unaligned samples (e.g., proteoliposomes) and yields single line spectra by averaging the spin interactions to their isotropic values. In both cases, the uniaxial order inherent to membrane proteins undergoing rotational diffusion around the axis perpendicular to the membrane plane (i.e., the lipid bilayer normal) provides the foundation for a powerful approach to structure determination based primarily on orientation restraints.
In this Account, we illustrate the methods with examples from studies of the outer membrane protein Ail (attachment invasion locus) from Yersinia pestis, an extremely pathogenic organism with a long history of precipitating massive human pandemics. Ail belongs to the same protein family (pfam PF06316) as OmpX. However, unlike OmpX, Ail is a virulence factor essential for evading the human host’s immune system through its two principal functions: mediating the adhesion of bacteria to human cells and providing resistance to human innate immunity.25 Outer membrane proteins in this family share amino acid sequence homology in the membrane-spanning segments but vary widely in the sequences of the four extracellular loops, which are critical for function. Both Ail and OmpX adopt a transmembrane eight-stranded β-barrel structure.26,27 However, key functional loops of Ail are not visible in its crystal structure. Thus, structural studies in membranes will be needed to understand the molecular basis for protein functionality.
Sample Preparation
The efficient production of isotopically labeled, recombinant outer membrane proteins, including Ail and OmpX, can be typically obtained by cloning the genes corresponding to the mature proteins (i.e., without their signal sequence) in Escherichia coli, to drive protein expression into inclusion bodies (Figure 2a).26,28 The resulting inclusion bodies are highly enriched in recombinant protein, as evidenced by their white, fluffy appearance, enabling significant quantities of essentially pure protein to be readily obtained by performing a few centrifugation and detergent wash steps following bacterial cell lysis (Figure 2b) and subsequent purification by chromatography yields highly purified protein (Figure 2c). The purification strategy depends on the specific chemical properties of each protein. For example, the isolelectric points of OmpX (pI = 5.0) and Ail (pI = 7.8) dictate the use of anion and cation exchange chromatography, respectively. Notably, additional purification by size exclusion chromatography is needed to obtain samples of Ail suitable for high-resolution spectroscopy, even though SDS polyacrylamide gel electrophoresis (PAGE) shows no evidence of impurities prior to this step. This is likely due to the presence of lipids or other E. coli components that copurify with the protein during ion exchange.
FIGURE 2.
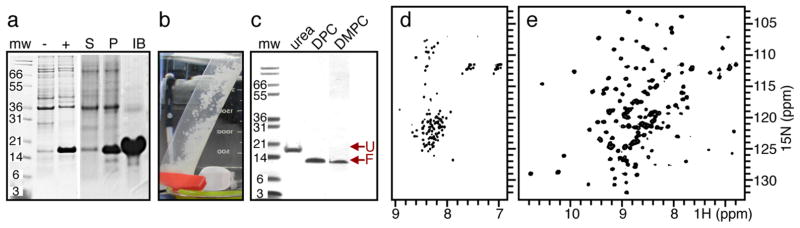
Expression, purification, and refolding of Y. pestis Ail. (a) SDS PAGE analysis. Whole cells isolated before (−) or after (+) induction with IPTG show that induction of Ail yields a band near the predicted molecular weight (mw). Cell lysis and separation into supernatant (S), pellet (P), and inclusion bodies (IB) fractions show that inclusion bodies are enriched in Ail. (b) Isolated Ail inclusion bodies are white and fluffy. (c) SDS-PAGE analysis of Ail folded from urea into DPC micelles or DMPC bilayers. Unfolded Ail (U) migrates at an apparent molecular weight (mw) of 18 kDa; folded Ail (F) migrates near 12 kD. (d, e) Solution NMR 1H/15N HSQC spectra of Ail unfolded in urea (d) or folded in DPC (e). Tris-tricine gels were stained with Coomassie blue.
SDS-PAGE analysis is very useful for monitor folding and unfolding of outer membrane proteins: unfolded and folded species migrate at distinctly different apparent molecular weights, and the protein fold, once established, is resistant to SDS as well as significant concentrations of urea and other denaturing conditions. For example, folded Ail is clearly detected at lower molecular weight by SDS-PAGE (Figure 2c). Fold is confirmed by solution NMR spectroscopy, where the 1H/15N HSQC spectra obtained for Ail in DPC (decylphosphocholine) micelles have dramatically greater chemical shift dispersion, consistent with β-barrel formation, compared with those obtained in urea where the protein is unfolded (Figure 2d,e).
Samples of folded Ail and OmpX can be obtained in a variety of phospholipid samples suitable for OS and MAS solid-state NMR studies,28–30 including uniaxially oriented bilayers and unoriented liposomes (Figures 3 and 5). Efficient refolding in relatively short-chain phospholipids, such as DMPC(1,2-dimyristoyl-sn-glycero-3-phosphocholine) and DMPG (1,2-dimyristoyl-sn-glycero-3-phosphoglycerol), seems to be a common feature shared by many outer membrane proteins,31 including Ail and OmpX. This observation suggests that the hydrophobic thickness of bacterial outer membranes may be smaller than that of the inner membranes, and is in agreement with the relatively thin belt of hydrophobic residues surrounding the β-barrel of OmpX (see below).
FIGURE 3.

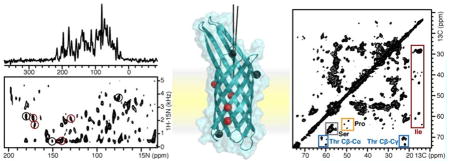
OS solid-state NMR spectra of 15N labeled OmpX in DMPC phospolipid bilayers. (a, b) One- and two-dimensional 1H/15N SLF spectra of OmpX in magnetically aligned DMPC/DHPC lipid bilayers with the bilayer normal parallel to the magnetic field. Peaks from phenylalanine sites were assigned as described.30 Peaks labeled in red resist 2H exchange. Spectra were obtained on a Bruker Avance 700 MHz spectrometer using a home-built radio frequency probe. (c) Crystal structure of OmpX26 aligned in the membrane on the basis of orientation restraints derived from 15N phenylalanine signals. Amide N atoms from phenylalanine are shown as spheres. Phenylalanine residues labeled in red define a 16 Å band of membrane-integrated, nonexchangeable H bonds.
FIGURE 5.

Uniaxially ordered samples yield orientation-dependent solid-state NMR spectra. (a) A membrane protein undergoing rotational diffusion around an axis (n) normal to the membrane plane. The angle θB defines the membrane orientation relative to the magnetic field (Bo); the angle θn defines the orientation of an amide NH bond (blue) relative to the axis n. Rotational diffusion of the protein around n averages the angle θ between the NH bond and Bo. (b) Predicted solid-state NMR 1H/15N spectra. A static powder sample yields a butterfly-shaped two-dimensional SLF spectrum (gray) whose edges correspond to the maximum values of the 15N CS tensor (σ11, σ22, σ33) and 1H–15N DC (νmax). Rotational diffusion around n produces motionally averaged, axially symmetric powder spectra (black), whose edges correspond to parallel (σ||, ν||, θB = 0°) and perpendicular (σ⊥, ν⊥, θB = 90°) orientations of n relative to Bo. The two-dimensional SLF spectrum has a characteristic cross-like pattern, as observed for gramicidin.49 Uniaxial alignment of the sample (e.g., with n parallel to Bo) yields single line spectra (red) with 15N CS and 1H–15N DC frequencies that correspond to the parallel edges of rotationally averaged powder patterns.
NMR Restraints for Membrane Protein Structure Determination
Modern NMR methods for protein structure determination rely increasingly on orientation restraints derived from dipolar coupling (DC) and chemical shift anisotropy (CSA) measurements and on dihedral angle (ϕ,ψ) restraints derived from isotropic chemical shift (CS) analysis. Orientation restraints can be used very effectively both in the early stages of structure determination to guide the generation of structural models and in the final stages of structure refinement, and they are especially powerful when coupled with de novo structure prediction and molecular fragment replacement. Examples of this approach include solid-state NMR structures of membrane proteins in lipid bilayers,8–11 as well as solution NMR structures of membrane proteins in detergent micelles32 and globular proteins in water.33,34 Importantly, they help shift the burden away from time-consuming measurements of multiple long-range distances between side chain sites, thus facilitating the determination of high-quality three-dimensional structures with very few or no distance restraints, derived from spin diffusion,35 rotational resonance,36 and paramagnetic relaxation enhancement.37
The orientation-dependent DC and CSA signals from 1H/15N/13C-labeled protein sites contain information about molecular structure, orientation, and dynamics. They are particularly useful in solid-state NMR structural studies of membrane proteins incorporated in liquid crystalline lipid bilayers; since the membrane-integrated regions of membrane proteins typically exhibit uniform dynamics dominated by uniaxial diffusion around the membrane normal, their corresponding DC and CSA signals can be readily interpreted in terms of orientation restraints. The extra-membrane, globular regions can exhibit different modes and time scales of internal motions, with significant scaling of the observed DC and CSA values that must be considered in their interpretation as orientation restraints.
Many DC and CS tensors are well characterized for 1H/15N/13C-labeled protein sites, and CS tensors themselves can be used as effective structural restraints.38 Their dependence on secondary structure and amino acid type can be significant39 However, CS tensor variations can be factored into the structure calculation protocol. For example, the 15N CSA restraints of glycine sites are typically computed separately from those of other residues to account for their significantly different tensors.
Isotropic CSs are important sources of structural information in both solid-state NMR and solution NMR. Furthermore, the ability to predict isotropic CS from structural models could be useful for guiding MAS solid-state NMR assignments, which can be complicated by the lack of 1H signals and by broader or overlapped lines. Because proteins are intrinsically dynamic, experimentally measured isotropic CS represent an ensemble and time average over all conformational states explored by the protein up to the millisecond time scale. Thus, improvements in isotropic CS prediction can be obtained by averaging predicted CS values over extended molecular dynamics trajectories, as well as over an ensemble of predicted structural models.40,41 In two recent examples, the integral membrane protein MerF and the soluble protein MerP, the agreement between predicted and experimentally observed CS was shown to improve significantly by averaging the predicted CS over 10–20 structural models.41 Since the models were “blind” predictions from Rosetta42 (i.e., obtained by excluding known structures from the database search), this approach could be used to guide MAS solid-state NMR assignment during de novo structure determination and provide confidence in the selection of a starting structural model for structural refinement.
Orientation Restraints from Uniaxially Oriented Lipid Bilayers
Uniaxial alignment of planar lipid bilayers in the magnetic field yields spectra with orientation-dependent, single line resonances, and high-resolution separated local field (SLF) spectroscopy of membrane proteins enables multiple orientation restraints to be measured directly in a single spectrum from uniformly labeled protein.8,43 For example, the one-and two-dimensional 1H/15N spectra of OmpX in magnetically aligned lipid bicelles show very high resolution (Figure 3). Each peak in the SLF spectrum provides a unique set of DC and CSA orientation restraints for protein structure determination.
The OS solid-state NMR spectra of uniaxially oriented samples exhibit distinctive wheel-like patterns of single line resonances, PISA wheels, that reflect protein structure and orientation.44–46 Well-defined PISA wheels are observed in the spectra of many integral membrane protein domains, indicating that the structure-dependent features of the spectra are not overshadowed by any residue-dependent 15N CS tensor variations. PISA wheels are useful for guiding resonance assignment performed with traditional spectroscopic approaches and for obtaining resonance assignments through methods that have been developed for simultaneous assignment and structure refinement (SASR). They also help reduce or eliminate the degeneracy of orientation solutions associated with DC and CSA measurements, allowing structures to be built by linking consecutive peptide planes or fragments through their common CA atom.8
The SASR approach is based on minimizing the difference between the experimentally observed spectral frequencies and the frequencies back-calculated from a structural model.8,30 It relieves the burden of having to obtain near-complete resonance assignments prior to structure determination: assignments are obtained as a side product of fitting a structural model to the NMR data, but it is not a prerequisite for structure determination.
Several recently developed bioinformatics methods enable the generation of structural models of proteins, including membrane proteins, based on their amino acid sequence. Of these, Rosetta42 has been used extensively in conjunction with NMR structure determination. Rosetta can provide very effective starting structural models for SASR and for the refinement of atomic resolution structures with high precision. Furthermore, even a few DC and CSA measurements can provide effective orientation restraints enabling structural information to be obtained prior to complete resonance assignment.
Typically, a SASR calculation cycle starts with an initial structural model (e.g., an ideal α-helix) and a set of unassigned DC and CSA frequencies for a particular residue type (e.g., from a selectively labeled sample). Each SASR cycle consists of generating optimal residue-specific assignments for the input data consistent with the structural model, and then using the assigned DC and CSA restraints to refine the model, which provides the input for the next SASR cycle where a new set of DC and CSA are assigned. The cycles are continued until all DC and CSA restraints are assigned and the resulting structure is consistent with the entire data set.
The program AssignFit,47 facilitates SASR by computing and scoring for best fit all of the multiple residue-specific assignment possibilities for a given NMR data set and structural model. This type of analysis can quickly evolve into a complicated problem when the number of assignment permutations to be tested is very large, since for n number of peaks there are n! possible assignment sets. The quality of the final result can be assessed in terms of RMSD between the experimental NMR data and the frequencies that are back-calculated from the refined structure (Figure 4).
FIGURE 4.

SASR refinement of the transmembrane helix of fd coat protein in lipid bilayers aligned with the bilayer normal (n) parallel to the magnetic field.8,47 (a) Peaks in the 1H/15N SLF spectrum are first assigned to residue types leucine, alanine, and valine (red dots) by comparison with spectra from selectively 15N-Leu-, 15N-Ala-, and 15N-Val-labeled protein. Then AssignFit is used to specifically assign each peak to a residue number based on best fit of the observed signals (black) to the signals back-calculated from an ideal helix starting model (yellow circles). (b) Refinement of the ideal helix with the assigned DC and CSA restraints extracted from the experimental spectrum yields back-calculated signals (pink circles) that agree closely with the observed data (black). (c) The refined helix (pink) differs from the starting ideal model (yellow) and changes direction after Lys40.
Additional assignments of OS solid-state NMR spectra can be made through the implementation of multidimensional triple-resonance experiments.23 Assignments can also be made through comparisons with isotropic NMR data48 obtained by MAS for similar, albeit unoriented, samples, or even obtained by solution NMR for micelle samples, provided that the protein structure is maintained in detergent.
Orientation Restraints from Unoriented Proteoliposome Samples
Accurate orientation restraints can also be measured from the solid-state NMR powder patterns that are observed for unoriented samples in which uniaxial order is established by rapid rotational diffusion around a unique axis (Figure 5). This principle was first demonstrated with solid-state 31P NMR spectroscopy for phospholipids assembled in liquid crystalline bilayer membranes: the lipids undergo rapid rotational diffusion around the axis perpendicular to the membrane plane (i.e., the bilayer normal) and, thus, yield narrowed, axially symmetric powder patterns whose frequency edges reflect the orientation of the phosphate group relative to the membrane.4
Membrane-integrated proteins also undergo uniaxial rotational diffusion around the bilayer normal and thus yield rotationally narrowed powder patterns that can be used to extract orientation restraints. Solid-state 13C NMR spectroscopy provided the first demonstration of this effect in a protein, by showing that rotationally narrowed 13CO powder patterns of 13CO-Leu-labeled bacteriorhodopsin incorporated in membranes can provide CSA orientation restraints useful for determining protein secondary structure and overall orientation within the membrane.50 Uniaxial protein rotation was further shown to yield collinear 15N CS and 1H–15N DC tensors, resulting in two-dimensional SLF spectra with distinctive cross-like patterns whose end points provide precise DC and CSA values (Figure 6, black).49 This property was used to measure a pair of 1H–15N DC and 15N CSA orientation restraints for the single 15N-Trp-labeled site membrane-integrated gramicidin, without the need for physically aligning the sample relative to the magnet.49 More recently, useful solid-state NMR orientation restraints have been measured from unoriented samples of membrane peptides and proteins labeled with 13C, 15N, or 2H at a single or a few specific sites by chemical synthesis.19 In these cases, isotopic labeling at one or a few sites was necessary because the observation of powder patterns cannot provide single-site resolution.
FIGURE 6.

Solid-state NMR spectroscopy of 15N/13C-labeled Ail in DMPC proteoliposomes. Two-dimensional 13C/13C correlation MAS solid-state NMR spectrum of 15N/13C-labeled Ail in DMPC liposomes. Peaks from 4 threonine (blue), 4 proline (gold), 16 serine (gray), and 7 isoleucine (red), are highlighted. Spectra were obtained on a Bruker Avance 700 MHz spectrometer using a Bruker MAS probe and controller.
To address this issue, a method has been developed for structure determination of uniformly 15N/13C-labeled membrane proteins reconstituted in proteoliposomes.10,11,51 Multiple peaks are resolved and assigned by MAS through the use of triple-resonance 1H/13C/15N experiments,16,18–22,52,53 and MAS also provides isotropic CS frequencies that can be interpreted as dihedral angle restraints for structure determination. Rotationally averaged DC and CSA powder patterns, associated with individually resolved protein sites from uniformly labeled protein, are recoupled and measured under MAS; since the frequency measured from the edge of a rotationally averaged powder pattern is equivalent to that measured from OS NMR spectra (Figure 6, red), the same analytical methods developed for data analysis and structure determination are applicable. This approach has been used to determine the structures of two α-helical integral membrane proteins in DMPC liposomes: the bacterial mercury transporter MerF and the G-protein coupled receptor CXCR1.10,11
As for OmpX, Ail can also be incorporated in phospholipid bilayers for high-resolution solid-state NMR spectroscopy. As shown in Figure 6, the two-dimensional MAS 13C–13C correlation spectra of uniformly 15N/13C-labeled Ail incorporated in DMPC proteoliposomes show several resolved signals (Figure 6a). Peaks from alanine, isoleucine, serine, and threonine residues populate the regions expected for β-sheet conformation, as described.54 Four signals are observed with 13C shifts in the expected region corresponding to the four threonine residues of Ail. Furthermore, seven signals are resolved in the region expected for isoleucine signals, including two signals with significantly greater intensity, as might be expected from residues in the extracellular loops of Ail (Figure 6b). Additional experiments demonstrating rotational diffusion of the β-barrel in the membrane will be needed before the implementation of MAS experiments that recouple DC and CSA similar to those described for helical proteins.
Embedding Proteins in Membranes
OS solid-state NMR provided an early view of the orientation of OmpX within the lipid bilayer membrane (Figure 3c).30 The seven pairs of DC and CSA restraints in the 1H/15N SLF spectrum of 15N-Phe-labeled OmpX were assigned by SASR and 2H exchange experiments, performed by acquiring the data after equilibrating the sample in D2O, provided information about the depth of membrane integration. Four of the seven phenylalanines (F24, F43, F115, F125) form strong backbone hydrogen bonds that resist water exchange after overnight equilibration in D2O at pH 7. They are located within a relatively narrow, 16 Å, band of hydrophobic residues on the exterior of the β-barrel. The other three phenylalanines (F90, F107, F148) exchange readily. They are located just outside the 16 Å HD-exchange-resistant band and, therefore, help to define its boundaries, which include about half of the total 32 Å length of the OmpX β-barrel. This is significantly narrower than the approximately 27 Å thickness predicted to form the membrane-spanning region of the barrel solely on the basis of residue hydrophobicity.26 It is also narrower than the 27 Å hydrophobic thickness estimated on the basis of NOEs observed between backbone amide or side chain methyl groups of OmpX and the hydrocarbon chain of DHPC (dihexanoyl-phosphatidylcholine), which provided the micelle environment for solution NMR studies.55
The preference of β-barrels for thinner membranes has been noted previously and has been proposed to provide a mechanism for targeting outer membrane proteins to the outer membranes of Gram-negative bacteria rather than to the inner membranes.31 Indeed, most outer membrane proteins have relatively short hydrophobic, membrane-spanning regions (~20–24 Å), compared with the longer transmembrane regions of α-helical membrane proteins (~30–32 Å) that are found predominantly in inner membranes.
Conclusions
Structure determination in a membrane environment provides the most biologically relevant view of an integral membrane protein; it eliminates the potential for distorting protein structure, dynamics, and function due to crystal contacts or detergent molecules and enables ligand binding studies to be performed in a setting as close as possible to native. The solid-state NMR spectra of both α-helical and β-barrel membrane proteins have very high-resolution, enabling measurements of isotropic CS, anisotropic DC and CSA and strategic distances that provide restraints for structure determination. A number of solid-state NMR structures have already been determined for membrane proteins in phospholipid bilayers, and many more will be forthcoming with recent progress in sample preparation, instrumentation, NMR experiments, and computational methods.
Acknowledgments
This research was supported by grants from the National Institutes of Health (Grants GM075917; GM094727; GM100265; and AI074805). The NMR studies utilized the NMR Facility at Sanford-Burnham Medical Research Institute and the Resource for Molecular Imaging of Proteins at UCSD, each supported by grants from the National Institutes of Health (Grants P30 CA030199; P41 EB002031).
Biographies
Yi Ding obtained her B.Sc. degree in Chemistry from the University of Science and Technology of China where her research project focused on understanding the antitumor activity of trans-coordinated platinum compounds. She joined the graduate program at the Sanford-Burnham Medical Research Institute in 2011. Her thesis research focuses on the structure determination and functional characterization of the outer membrane protein Ail, a major virulence factor of Yersinia pestis.
Yong Yao obtained his B.Sc. and Ph.D. degrees in Chemistry from Nanjing University in China. His thesis research focused on solution NMR structural studies of heme proteins. He was postdoctoral associate with Jane Dyson at the Scripps Research Institute from 2003 to 2006. He joined the Sanford-Burnham Medical Research Institute in 2006 where he is currently staff scientist. His research focuses on solution and solid-state NMR structural studies of integral membrane proteins.
Francesca M. Marassi obtained her Ph.D. degree in Chemistry from the University of Toronto, where she studied NMR of lipids in membranes with Peter MacDonald. From 1993 to 1998, she was a postdoctoral fellow of the Natural Sciences and Engineering Research Council of Canada and of the Medical Research Council of Canada at the University of Pennsylvania. She was subsequently an Assistant Professor at the Wistar Institute in Philadelphia from 1998 to 2000 and is currently on the faculty of the Sanford-Burnham Medical Research Institute in La Jolla, CA. Her research interests are focused on developing methods for the structure determination of membrane proteins within their natural environment of the phospholipid bilayer membrane.
Footnotes
The authors declare no competing financial interest.
References
- 1.Mulkidjanian AY, Galperin MY, Koonin EV. Co-evolution of primordial membranes and membrane proteins. Trends Biochem Sci. 2009;34:206–215. doi: 10.1016/j.tibs.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem Sci. 2011;36:117–125. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaughlin AC, Cullis PR, Hemminga MA, Hoult DI, Radda GK, Ritchie GA, Seeley PJ, Richards RE. Application of 31P NMR to model and biological membrane systems. FEBS Lett. 1975;57:213–218. doi: 10.1016/0014-5793(75)80719-8. [DOI] [PubMed] [Google Scholar]
- 5.Griffin RG. Letter: Observation of the effect of water on the 31P nuclear magnetic resonance spectra of dipalmitoyllecithin. J Am Chem Soc. 1976;98:851–853. doi: 10.1021/ja00419a044. [DOI] [PubMed] [Google Scholar]
- 6.Seelig J. Deuterium magnetic resonance: theory and application to lipid membranes. Q Rev Biophys. 1977;10:353–418. doi: 10.1017/s0033583500002948. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald PM. Deuterium NMR and the topography of surface electrostatic charge. Acc Chem Res. 1997;30:196–203. [Google Scholar]
- 8.Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12:403–411. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, Marassi FM, Opella SJ. Structure determination of a membrane protein in proteoliposomes. J Am Chem Soc. 2012;134:2047–2056. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis A, Marassi FM, Opella SJ. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–783. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharif S, Kim SJ, Labischinski H, Schaefer J. Characterization of peptidoglycan in fem-deletion mutants of methicillin-resistant Staphylococcus aureus by solid-state NMR. Biochemistry. 2009;48:3100–3108. doi: 10.1021/bi801750u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koch K, Afonin S, Ieronimo M, Berditsch M, Ulrich A. In: Solid State NMR. Chan JCC, editor. Vol. 306. Springer; Berlin Heidelberg: 2012. pp. 89–118. [DOI] [PubMed] [Google Scholar]
- 14.Miao Y, Qin H, Fu R, Sharma M, Can TV, Hung I, Luca S, Gor’kov PL, Brey WW, Cross TA. M2 proton channel structural validation from full-length protein samples in synthetic bilayers and E. coli membranes. Angew Chem, Int Ed. 2012;51:8383–8386. doi: 10.1002/anie.201204666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renault M, Tommassen-van Boxtel R, Bos MP, Post JA, Tommassen J, Baldus M. Cellular solid-state nuclear magnetic resonance spectroscopy. Proc Natl Acad Sci U S A. 2012 doi: 10.1073/pnas.1116478109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDermott A. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 17.Judge PJ, Watts A. Recent contributions from solid-state NMR to the understanding of membrane protein structure and function. Curr Opin Chem Biol. 2011;15:690–695. doi: 10.1016/j.cbpa.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Franks WT, Linden AH, Kunert B, van Rossum BJ, Oschkinat H. Solid-state magic-angle spinning NMR of membrane proteins and protein-ligand interactions. Eur J Cell Biol. 2012;91:340–348. doi: 10.1016/j.ejcb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Hong M, Zhang Y, Hu F. Membrane protein structure and dynamics from NMR spectroscopy. Annu Rev Phys Chem. 2012;63:1–24. doi: 10.1146/annurev-physchem-032511-143731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reif B. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: implications for structure and dynamics. J Magn Reson. 2012;216:1–12. doi: 10.1016/j.jmr.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Shi L, Ladizhansky V. Magic angle spinning solid-state NMR experiments for structural characterization of proteins. Methods Mol Biol. 2012;895:153–165. doi: 10.1007/978-1-61779-927-3_12. [DOI] [PubMed] [Google Scholar]
- 22.Goncalves J, Eilers M, South K, Opefi CA, Laissue P, Reeves PJ, Smith SO. Magic angle spinning nuclear magnetic resonance spectroscopy of G protein-coupled receptors. Methods Enzymol. 2013;522:365–389. doi: 10.1016/B978-0-12-407865-9.00017-0. [DOI] [PubMed] [Google Scholar]
- 23.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray DT, Das N, Cross TA. Solid state NMR strategy for characterizing native membrane protein structures. Acc Chem Res. 2013 doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartra SS, Styer KL, O’Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect Immun. 2008;76:612–622. doi: 10.1128/IAI.01125-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogt J, Schulz GE. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure. 1999;7:1301–1309. doi: 10.1016/s0969-2126(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita S, Lukacik P, Barnard TJ, Noinaj N, Felek S, Tsang TM, Krukonis ES, Hinnebusch BJ, Buchanan SK. Structural insights into Ail-mediated adhesion in Yersinia pestis. Structure. 2011;19:1672–1682. doi: 10.1016/j.str.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plesniak LA, Mahalakshmi R, Rypien C, Yang Y, Racic J, Marassi FM. Expression, refolding, and initial structural characterization of the Y. pestis Ail outer membrane protein in lipids. Biochim Biophys Acta. 2011;1808:482–489. doi: 10.1016/j.bbamem.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahalakshmi R, Franzin CM, Choi J, Marassi FM. NMR structural studies of the bacterial outer membrane protein OmpX in oriented lipid bilayer membranes. Biochim Biophys Acta. 2007;1768:3216–3224. doi: 10.1016/j.bbamem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahalakshmi R, Marassi FM. Orientation of the Escherichia coli outer membrane protein OmpX in phospholipid bilayer membranes determined by solid-state NMR. Biochemistry. 2008;47:6531–6538. doi: 10.1021/bi800362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleinschmidt JH, Tamm LK. Secondary and tertiary structure formation of the beta-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J Mol Biol. 2002;324:319–330. doi: 10.1016/s0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 32.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci U S A. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raman S, Lange OF, Rossi P, Tyka M, Wang X, Aramini J, Liu G, Ramelot TA, Eletsky A, Szyperski T, Kennedy MA, Prestegard J, Montelione GT, Baker D. NMR structure determination for larger proteins using backbone-only data. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veshtort M, Griffin RG. Proton-driven spin diffusion in rotating solids via reversible and irreversible quantum dynamics. J Chem Phys. 2011;135(134509) doi: 10.1063/1.3635374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaefer J. “Development of REDOR rotational-echo double-resonance NMR” by Terry Gullion and Jacob Schaefer [J. Magn. Reson. 81 (1989) 196–200] J Magn Reson. 2011;213:421–422. doi: 10.1016/j.jmr.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Sengupta I, Nadaud PS, Helmus JJ, Schwieters CD, Jaroniec CP. Protein fold determined by paramagnetic magic-angle spinning solid-state NMR spectroscopy. Nat Chem. 2012;4:410–417. doi: 10.1038/nchem.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wylie BJ, Sperling LJ, Nieuwkoop AJ, Franks WT, Oldfield E, Rienstra CM. Ultrahigh resolution protein structures using NMR chemical shift tensors. Proc Natl Acad Sci U S A. 2011;108:16974–16979. doi: 10.1073/pnas.1103728108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito H, Ando I, Ramamoorthy A. Chemical shift tensor - the heart of NMR: Insights into biological aspects of proteins. Prog Nucl Magn Reson Spectrosc. 2010;57:181–228. doi: 10.1016/j.pnmrs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li DW, Bruschweiler R. Certification of molecular dynamics trajectories with NMR chemical shifts. J Phys Chem Lett. 2010;1:246–248. [Google Scholar]
- 41.Tian Y, Opella SJ, Marassi FM. Improved chemical shift prediction by Rosetta conformational sampling. J Biomol NMR. 2012;54:237–243. doi: 10.1007/s10858-012-9677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 43.Marassi FM, Ramamoorthy A, Opella SJ. Complete resolution of the solid-state NMR spectrum of a uniformly 15N-labeled membrane protein in phospholipid bilayers. Proc Natl Acad Sci U S A. 1997;94:8551–8556. doi: 10.1073/pnas.94.16.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 46.Marassi FM. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys J. 2001;80:994–1003. doi: 10.1016/S0006-3495(01)76078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian Y, Schwieters CD, Opella SJ, Marassi FM. AssignFit: A program for simultaneous assignment and structure refinement from solid-state NMR spectra. J Magn Reson. 2012;214:42–50. doi: 10.1016/j.jmr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Angelis AA, Howell SC, Opella SJ. Assigning solid-state NMR spectra of aligned proteins using isotropic chemical shifts. J Magn Reson. 2006;183:329–332. doi: 10.1016/j.jmr.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 49.Tian F, Song Z, Cross TA. Orientational constraints derived from hydrated powder samples by two-dimensional PISEMA. J Magn Reson. 1998;135:227–231. doi: 10.1006/jmre.1998.1544. [DOI] [PubMed] [Google Scholar]
- 50.Lewis BA, Harbison GS, Herzfeld J, Griffin RG. NMR structural analysis of a membrane protein: Bacteriorhodopsin peptide backbone orientation and motion. Biochemistry. 1985;24:4671–4679. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- 51.Marassi FM, Das BB, Lu GJ, Nothnagel HJ, Park SH, Son WS, Tian Y, Opella SJ. Structure determination of membrane proteins in five easy pieces. Methods. 2011;55:363–369. doi: 10.1016/j.ymeth.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffin RG. Dipolar recoupling in MAS spectra of biological solids. Nat Struct Biol. 1998;5(Suppl):508–512. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 53.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bauer AJ, Gieschler S, Lemberg KM, McDermott AE, Stockwell BR. Functional model of metabolite gating by human voltage-dependent anion channel 2. Biochemistry. 2011;50:3408–3410. doi: 10.1021/bi2003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez C, Hilty C, Wider G, Wuthrich K. Lipid-protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]