

Abstract
Synapsins are neuronal phosphoproteins crucial to regulating the processes required for normal neurotransmitter release. Synapsin II, in particular, has been implied as a candidate gene for schizophrenia. This study investigated synapsin II mRNA expression, using Real Time RT-PCR, in coded dorsolateral prefrontal cortical samples provided by the Stanley Foundation Neuropathology Consortium. Synapsin IIa was decreased in patients with schizophrenia when compared to both healthy subjects and patients with bipolar disorder, whereas the synapsin IIb was only significantly reduced in patients with schizophrenia when compared to healthy subjects, but not patients with bipolar disorder. Furthermore, lifetime antipsychotic drug use was positively associated with synapsin IIa expression in patients with schizophrenia. Results suggest that impairment of synaptic transmission by synapsin II reduction may contribute to dysregulated convergent molecular mechanisms which result in aberrant neural circuits that characterize schizophrenia, while implicating involvement of synapsin II in therapeutic mechanisms of currently prescribed antipsychotic drugs.
Keywords: Synapsin II Isoforms, Schizophrenia, Dorsolateral prefrontal cortex, Antipsychotic drugs, Postmortem Human Tissue, Real Time RT-PCR
INTRODUCTION
Synapsins are a family of neuronal phosphoproteins that are important for neurotransmitter release, synaptogenesis and the maintenance of synaptic vesicular pools (1–3). There is an increasing body of evidence to support a role for aberrant synaptic processes in the underlying pathophysiology of schizophrenia (2;4–8). In particular, abnormalities in synapsin II gene and protein expression have been shown to occur in patients with schizophrenia (8–13). A microarray study by Mirnics et al. (2000) demonstrated decreased mRNA concentrations of synapsin II in the prefrontal cortices of patients with schizophrenia (11). Additionally, research by Chen et al. (2004) reported positive associations between specific synapsin II gene polymorphisms and schizophrenia (14–16). The synapsin II gene is located on chromosome 3p25, which was suggested to be a region of vulnerability for schizophrenia, thus placing particular significance on the study of this gene (16;17).
Previous investigations from our laboratory have also reported that synapsin II concentrations are differentially regulated by the modulation of dopamine receptors (11;18;19). Dopamine receptors, in particular the dopamine D2 receptor, have been strongly implicated in the etiology of schizophrenia (20–24). Our cDNA study revealed that haloperidol, a typical antipsychotic drug and an antagonist of the inhibitory dopamine D2 receptor, increases mRNA expression of synapsin II in the rat striatum (11;18;19). Treatment with the atypical antipsychotic drug olanzapine, on the other hand, increases synapsin II expression in the medial prefrontal cortex (mPFC) of synapsin II knock-down rats (3). Furthermore, treatment with SCH23390, an antagonist of the excitatory dopamine D1 receptor, reduces concentrations of synapsin II, whereas treatment with SKF38393, an agonist of the dopamine D1 receptor, up-regulates expression of synapsin II in primary cell cultures (18). Given the importance of the dopamine D2 receptors in schizophrenia, studying the role of dopamine receptors and targeting therapeutics in the context of synapsin II and its expression is fundamental to understanding this debilitating mental disease.
Dysregulated glutamate neurotransmission in the prefrontal cortex has been a highly investigated theory of schizophrenia (25–27). Synapsins are located at the presynaptic site, and help tether the synaptic vesicle to the cytoskeleton at a reserve pool away from the active site of the synapse. In addition to synaptic proteins however, vesicular transporters are also present on synaptic vesicles for the uptake and storage of their respective neurotransmitters into the synaptic vesicles for subsequent release into the synaptic cleft (8;28). Evidence from previous studies have shown that synapsin II reduction in the prefrontal cortex is accompanied by reductions in VGAT (GABA vesicular transporter), VGLUT1 and VGLUT2 (vesicular glutamate transporters) (3). Altered prefrontal glutamatergic signaling, as a result of subnormal synapsin II expression, can subsequently disrupt local and long loop pathways and influence the release of neurotransmitters, including GABA and dopamine, in both the cortical and subcortical regions (2;3;29). This resembles the neurochemical changes which are characteristic of schizophrenia. Recent studies have now also taken the approach of studying schizophrenia as a disease of the synapse and connectivity, and presynaptic protein, synapsin II, has accordingly been uniquely positioned in a critical site of impact. Consequently, the study of the synapsin II and its isoforms is imperative to understanding the pathophysiology of this disease and the mechanisms involved in the therapeutic action of antipsychotic drugs (8).
In order to further assess synapsin II expression, the present study examined synapsin II mRNA expression in the dorsolateral prefrontal cortex of patients with schizophrenia, patients with bipolar disorder, and healthy subjects using coded RNA samples obtained from the Stanley Foundation Neuropathology Consortium (SFNC) array collection. This investigation utilized a large post-mortem sample size to accurately model the changes in genetic expression of synapsin II. The specific objectives of this study were to: (a) confirm reductions in synapsin II mRNA concentrations in the dorsolateral prefrontal cortex of patients with schizophrenia; (b) examine whether these reductions occur with both isoforms of synapsin II; (c) evaluate whether these decreases in synapsin II isoform expression occur in schizophrenia but not in bipolar disorder; and (d) ascertain whether lifetime use of antipsychotic drugs affect mRNA expression of synapsin II isoforms in the dorsolateral prefrontal cortex of patients with schizophrenia.
MATERIALS AND METHODS
Post-mortem dorsolateral prefrontal cortical RNA samples
DNase-treated dorsolateral prefrontal cortical RNA samples from postmortem brain specimens, Brodmann’s area 46 (in the dorsolateral prefrontal cortex), from healthy subjects (n=35), patients with schizophrenia (n=35), and patients with bipolar disorder (n=35), were obtained from the Stanley Array Collection of the Stanley Foundation Brain Collection. The dorsolateral prefrontal cortical regions from which the RNA samples were extracted were independently identified in whole brain specimens by two neuropathologists employed at the Stanley Foundation Neuropathology Consortium (SFNC). Subjects were clinically assessed by two independent psychiatrists, and control subjects were confirmed to be free of psychiatric illness and substance abuse. Patient diagnosis with either schizophrenia or bipolar disorder was established using criteria from the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-V-TR). The demographics and patient histories of all the subjects were provided by the SFNC post-analysis and are summarized in Table 1. Analyses of RNA expression were performed with coded samples in order to keep experimenters blind to the diagnostic status of subjects, and the range of clinical variables and diagnostic status were only provided by the SFNC at the time of sample decoding, after results were deposited in the SFNC database.
Table 1.
Summary and Overview of Patient Profiles
| Variable | Normal control (n = 35) | Bipolar disorder (n = 35) | Schizophrenia (n = 35) |
|---|---|---|---|
| Age (mean ± SD, years) | 44.2 ± 7.6 (Range 31–60) | 45.3 ± 10.5 (Range 19–64) | 42.6 ± 8.5 (Range 19–59) |
| Sex (M/F) | 26/9 | 17/18 | 26/9 |
| Race (Caucasian/other) | 35/0 | 33/2 | 34/1 |
| Age of onset (mean ± SD, years) | N/A | 25.1 ± 9.1 (Range 14–48) | 21.3 ± 6.1 (Range 9–34) |
| Duration of illness (mean ± SD, years) | N/A | 20.1 ± 9.5 (Range 2–45) | 21.3 ± 10.2 (Range 1–45) |
| Time in hospital (mean ± SD, years) | N/A | 0.5 ± 1.4 (Range 0–8) | 1.2 ± 2.3 (Range 0–12) |
| Alcohol abuse at TOD (n) | 2 | 11 | 12 |
| Drug abuse at TOD (n) | 1 | 10 | 9 |
| Smoking at TOD (yes/no/unknown) | 9/9/17 | 16/6/13 | 23/4/8 |
| Psychotic feature (yes/no/unknown) | 0/35/0 | 21/12/2 | 35/0/0 |
| Lifetime antipsychotic use (FE ± SD, mg) | N/A | 10,035 ± 22,896 (Range 0– 130,000) | 85,004 ± 100,335 (Range 50– 400,000) |
| Relative brain mass (mean ± SD, g) | 1444 ± 148.4 (Range 1120– 1900) | 1394 ± 139.1 (Range 1120– 1670) | 1442 ± 107.5 (Range 1170–1630) |
| Right brain (n) | 19 | 15 | 18 |
| Left brain (n) | 16 | 20 | 17 |
| PMI (mean ± SD, h) | 29.4 ± 12.9 (Range 9–58) | 37.9 ± 18.4 (Range 12–81) | 31.4 ± 15.5 (Range 9–80) |
| Refrigerator interval (mean ± SD, h) | 3.6 ± 2.6 (Range 0–14) | 10.1 ± 10.4 (Range 1–54) | 6.0 ± 4.2 (Range 1–19) |
| Brain pH (mean ± SD) | 6.61 ± 0.27 (Range 6.00–7.03) | 6.43 ± 0.30 (Range 5.76–6.97) | 6.48 ± 0.24 (Range 5.90 ± 6.93) |
| 28S:18S rRNA ratio (mean ± SD) | 2.18 ± 0.50 (Range 0.90–3.74) | 2.21 ± 0.75 (Range 0.49–4.18) | 2.13 ± 0.55 (Range 1.18–3.81) |
Abbreviations: M, male; F, female; N/A, not applicable; TOD, time of death; PMI, postmortem interval; FE, fluphenazine equivalents.
Samples were shipped in 1.5 ml microcentrifuge tubes stored in dry ice, and each sample was matched for patient age, gender, RNA quality, and brain pH. The qualitative analysis of the RNA samples was provided by the SFNC. RNA purities were verified using A260/A280 ratio analyses and RNA integrities were confirmed by gel electrophoresis and ethidium bromide staining.
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) quantification of synapsin IIa and IIb mRNA expression
Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) was performed in triplicates for each sample using a MX-3000P Real-Time RT-PCR machine (Stratagene La Jolla, CA, USA). The following primers were used to examine synapsin IIa and IIb mRNA concentrations: 5′-CCCACAGCTCAACAAGTC-3′ (synapsin IIa forward); 5′-ATCTGAAAAGAGGCTGGC-3′ (synapsin IIa reverse); 5′-AGACCCCTAACAACCCAG-3′ (synapsin IIb forward); 5′-CTTGTTTTGGCCCTACTG-3′ (synapsin IIb reverse). Respective forward and reverse primers for housekeeping cyclophilin are as follows: 5′ GCA AGA CCA GCA AGA AGA 3′; and 5′ CAG CGA GAG CAC AAA GAT 3′. cDNA was reverse transcribed from the RNA specimens. Prior to real-time RT-PCR analysis, standard curves were established for primer sets. These curves were then used to standardize synapsin IIa and IIb mRNA concentrations amongst different patient groups, and template copy numbers were calculated at the end of each reaction. Melting temperature analysis of resulting amplicons confirmed the presence of a single product for each reaction. Real-time RT-PCR conditions were optimized to ensure that the amplification efficiencies remained constant throughout all reactions. Additional controls included the use of no-reverse transcriptase and no-cDNA template. Human cyclophyllin mRNA concentrations were also quantified to show no significant alterations from sample to sample in the synapsin IIa and IIb mRNA concentrations obtained for each sample. The following components were used in the reactions described above: 10 μL of Invitrogen Platinum SyberGreenqPCR 2X Supermix; 0.4 μL of Rox; 375 nM of forward primer; 375 nM of reverse primer; 5.6 μL of DEPC water; 50ng of cDNA prepared from the RNA samples. The real-time RT-PCR conditions were as follows: 50°C for 2 min (1 cycle); 94°C for 2 min (1 cycle); 94°C for 30 sec, 54°C for 30 sec, and 72°C for 1 min (40 cycles).
Statistical Analyses
Data were entered and analyzed with SAS, Version 9.2 (SAS Institute, United States). The differences in synapsin IIa and IIb mRNA copy number between each patient group (control, bipolar disorder or schizophrenia) were determined using analysis of variance (ANOVA), and pairwise comparisons were conducted post hoc with the Scheffé test, as this is the most conservative approach (30). Multiple regression was used to examine the impact of lifetime antipsychotic drug use on synapsin IIa and IIb mRNA expression within the schizophrenia patient profile, while controlling for the potential confounding effects of age, brain pH, refrigerator interval, time spent in hospital, post-mortem interval cause of death, history of substance abuse, alcohol use, and smoking status. Diagnostic analyses for the regression analysis indicated that no outliers were influential; as such, all samples were retained in the analysis to maximize statistical power. As the distribution of the synapsin IIa and IIb isoform mRNA expression was negatively skewed, the data were log-transformed. A log-transformation resulted in normally distributed synapsin IIa and IIb data – a requirement for the application of the statistical analyses utilized in this study in order to draw valid inferences from the data. All statistical tests were two-tailed using a confidence level of α=0.05.
RESULTS
Patient data and study variables
Post-mortem dorsolateral prefrontal cortex RNA samples extracted from 35 healthy subjects, 35 patients with schizophrenia, and 35 patients with bipolar disorder obtained from the SFNC were evaluated. A summary of the relevant patient information is provided in Table 1 and stratified according to patient diagnosis (control, bipolar disorder, or schizophrenia).
Synapsin II mRNA expression is significantly decreased in patients with schizophrenia
Representative real-time RT-PCR amplicons are shown in Figures 1 and 2. Results from Figure 1 display reductions in synapsin II mRNA in dorsolateral prefrontal cortex of a representative patient with schizophrenia, without any changes in the housekeeping gene cyclophilin. This same reduction was not seen in a representative patient with bipolar disorder (Figure 2).
Figure 1.



Representative amplification plot of Real Time RT-PCR data for (A) synapsin IIa, (B) synapsin IIb, and (C) housekeeping gene cyclophilin in the dorsolateral prefrontal cortex of normal controls and patients with schizophrenia. Forward primer for synapsin IIa - 5′ ATCTGAAAAGAGGCTGGC 3′; reverse primer for synapsin IIa - 5′ GACTTGTTGAGCTGTGGG 3′. Forward primer for synapsin IIb - 5′ AGACCCCTAACAACCCAG 3′; reverse primer for synapsin IIb - 5′ CTTGTTTTGGCCCTACTG 3′. Forward and reverse primers for housekeeping cyclophilin: 5′ GCAAGACCAGCAAGAAGA 3′; and 5′ CAGCGAGAGCACAAAGAT 3′ respectively.
Figure 2.



Representative amplification plot of Real Time RT-PCR data for (A) synapsin IIa, (B) synapsin IIb, and (C) housekeeping gene cyclophilin in the dorsolateral prefrontal cortex of normal controls and patients with bipolar disorder. Forward primer for synapsin IIa - 5′ ATCTGAAAAGAGGCTGGC 3′; reverse primer for synapsin IIa - 5′ GACTTGTTGAGCTGTGGG 3′. Forward primer for synapsin IIb - 5′ AGACCCCTAACAACCCAG 3′; reverse primer for synapsin IIb - 5′ CTTGTTTTGGCCCTACTG 3′. Forward and reverse primers for housekeeping cyclophilin: 5′ GCAAGACCAGCAAGAAGA 3′; and 5′ CAGCGAGAGCACAAAGAT 3′ respectively.
Box plots in Figures 3 and 4 show a comparison of synapsin mRNA expression among different patient groups. Analysis of variance followed by pairwise comparisons using the post hoc Scheffé test revealed that synapsin IIa mRNA concentrations were significantly decreased in patients with schizophrenia when compared to both controls and patients with bipolar disorder (F = 15.625, p< 0.002) (Figure 3). However, analysis of synapsin IIb mRNA data revealed that expression levels of this isoform were significantly reduced in patients with schizophrenia only when compared to normal controls (F = 3.65, p< 0.030) (Figures 4). As patients with schizophrenia and patients with bipolar disorder display similar clinical features (31), we examined whether bipolar disorder individuals also exhibit the same reductions in synapsin II gene expression observed among individuals with schizophrenia. No differences in synapsin II levels in the dorsolateral prefrontal cortex were observed in patients with bipolar disorder when compared to normal control subjects (p>0.05) (Figures 2, 3 and 4).
Figure 3.
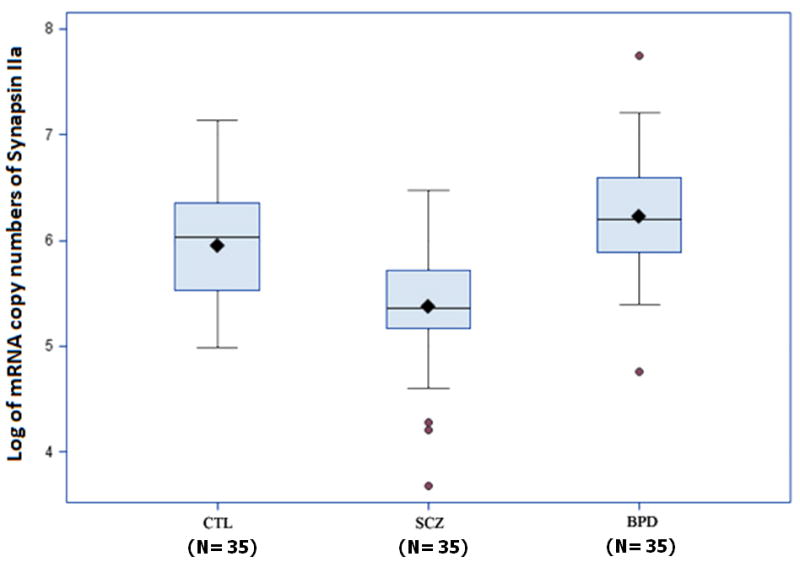
Synapsin IIa mRNA in post-mortem dorsolateral prefrontal cortex brain specimens. Real Time RT-PCR was performed on 105 specimens obtained from the SFNC. Synapsin IIa was significantly reduced in patients with schizophrenia (SCZ) (P<0.0001) compared to normal controls (CTL) and patients with bipolar disorder (BPD). Data are presented as box plots of log-transformed mean synapsin IIa mRNA expressions with shaded circles identifying outliers.
Figure 4.
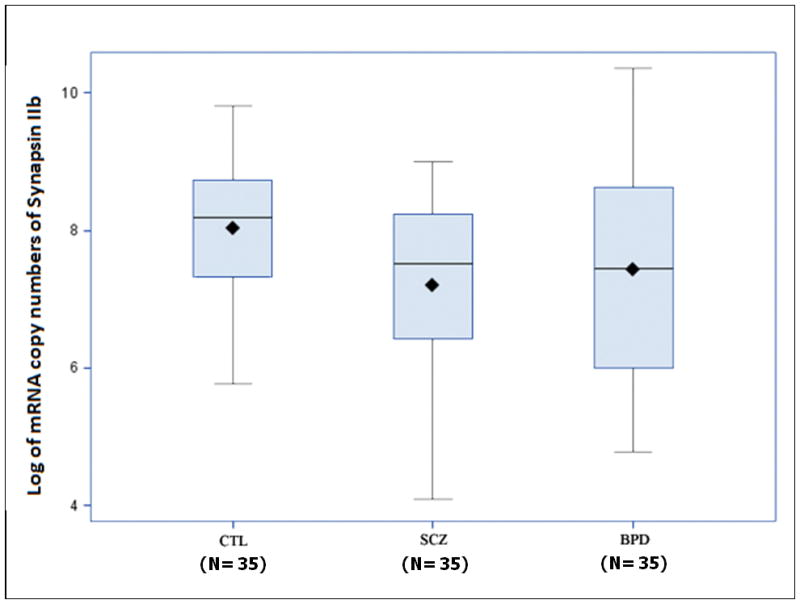
Synapsin IIb mRNA expression in post-mortem dorsolateral prefrontal cortex brain specimens. Real Time RT-PCR was performed on 105 specimens obtained from the SFNC. Synapsin IIb was significantly decreased in patients with schizophrenia (SCZ) compared to normal controls (CTL) (P<0.030). However, no significant differences were observed between patients with schizophrenia (SCZ) and bipolar disorder (BPD). Data are presented as box plots of log-transformed mean synapsin IIb mRNA expressions.
Effects of antipsychotic drug use on synapsin II mRNA expression
In the present study, multiple regression analysis showed that lifetime antipsychotic drug use was positively associated with synapsin IIa mRNA expression in patients with schizophrenia (beta 5.9×10−6, p< 0.001) (Figure 5). The confounding factor effects of age, brain pH, refrigerator interval, time spent in hospital, post-mortem interval, cause of death, history of substance abuse, alcohol use, and smoking status were controlled for in this analysis. However, analysis of the synapsin IIb isoforms revealed no association between lifetime antipsychotic drug use and mRNA expression levels (beta 3.0×10−6, p < 0.411) (Figure 6). Furthermore, multiple regression analysis revealed no evidence to suggest a correlation between antipsychotic drug use and mRNA expression in patients with bipolar disorder (data not shown).
Figure 5.

Graph depicting a direct linear relationship between expression levels of synapsin IIa mRNA (expressed as copy numbers) and lifetime antipsychotic drug use (expressed as fluphenazine equivalents) in patients with schizophrenia. A solid line represents a fitted regression line, a dotted line represents the probability (0.95) that the “true” fitted line (in the population) falls between the bands, while the shaded region depicts the 95% confidence interval.
Figure 6.

Graph depicting a direct linear relationship between expression levels of synapsin IIb mRNA (expressed as copy numbers) and lifetime antipsychotic drug use (expressed as fluphenazine equivalents) in patients with schizophrenia. A solid line represents a fitted regression line, a dotted line represents the probability (0.95) that the “true” fitted line (in the population) falls between the bands, while the shaded region depicts the 95% confidence interval.
DISCUSSION
Patients with schizophrenia consistently display deficits in cognitive abilities, including attention, executive functioning, memory, and language (32–34). These cognitive deficiencies have primarily been associated with a dysfunction of the prefrontal cortex. Accordingly, evidence gathered from various neuropsychological, neuroimaging, histopathological, and neurochemical investigations have implicated the dorsolateral prefrontal cortex in the pathophysiology of schizophrenia (35). However, the physiological mechanisms underlying such disturbances remain unclear (32–34). Since the synapsin family of phosphoproteins are involved in neurotransmitter release, synapse formation and synapse maintenance, dysfunction of these molecules in the prefrontal cortex may cause aberrant neurotransmitter release, and has been suggested to play a role in the pathology of schizophrenia (2;3;8;9;28;36–39). Isoforms of synapsins interact with multiple kinases, and converge on several signaling pathways. As such, isoforms of synapsins can have similar, differing, or overlapping roles at the synapse (40). Synapsin II, in particular, is particularly essential for synaptic trafficking during repetitive stimulation and long-term regulation of neurotransmitter release (40).
Synapsin IIb can have an effect on the number of core vesicles, as well as the presence on all of the other isoforms of synapsins and synaptophysin (40). Synapsin IIa, on the other hand, has been primarily shown to regulate the kinetics of synaptic depression by controlling the size of the glutamatergic vesicular reserve pool. Thus, results from this study showing a reduction in synapsin IIa suggest an altered size of the glutamatergic vesicle reserve pools in the dorsolateral prefrontal cortex in schizophrenia (28). Given the active role of the synapsin IIa isoform in the regulation of glutamatergic vesicles, and the role glutamatergic neurotransmission in the dorsolateral prefrontal cortex, our results support the hypothesis that lifetime antipsyhotic drug use would have a greater effect on synapsin isoform IIa over IIb in the dorsolateral prefrontal cortex (Figures 5 and 6).
Furthermore, the present investigation further showed that these reductions are specific to schizophrenia, as patients with bipolar disorder exhibit prefrontal cortical synapsin II mRNA expression levels comparable to those observed in control subjects. Although potential variables, such as age, brain pH, post-mortem interval, cause of death, history of substance abuse, alcohol use, and smoking status, can affect the integrity of RNA and gene expression, these were not found to be confounding factors via multiple regression analysis in our study of synapsin II expression.
The present study also provides evidence that the synapsin isoform IIa is positively correlated with lifetime cumulative antipsychotic drug intake in patients with schizophrenia. Conversely, Mirnics et al. (2000) demonstrated that antipsychotic agents had no effect on the prefrontal cortical mRNA concentrations of these phosphoproteins, contradicting the findings of this study and of previously published preclinical and human experiments (3;10;11;18;19). In the Mirnics study, synapsin II gene expression remained unchanged in the prefrontal cortices of monkeys chronically treated with haloperidol. However, the sample size (n=2) used to obtain those results was small (41). Furthermore, haloperidol plasma concentrations administered were in the lower range (5ng/ml) of its therapeutic window (5–12ng/ml), and analyses of individual synapsin II isoforms were not distinguished in these monkeys, which may explain why these results contradict the present observations.
Another study by Imai et al. (2001) also reported that prefrontal cortical synapsin II mRNA concentrations were similar between patients with schizophrenia and healthy controls (11;42). However, a number of technical differences may also account for the discrepant observations. Firstly, these previous investigations evaluated prefrontal cortical synapsin II expression in only six patients with schizophrenia, and such a small sample size may not be sensitive enough to detect underlying differences. Our current investigation utilizes a much larger population of patients with schizophrenia, and may consequently provide a more accurate representation of the general population. Secondly, the investigators did not provide patient information, such as antipsychotic drug treatment and dosage, both of which have shown to play significant roles in our findings. Thirdly, the study by Imai et al. (2001) did not distinguish between the synapsin IIa and IIb isoforms. Our observations support a differential effect on synapsin IIa and IIb isoforms, both in the disease of schizophrenia and in the treatment with antipsychotic drugs, distinctions which were paramount in our findings. Results in this extensive study have taken into account various confounding factors and have utilized a significantly larger sample size of the population affected with schizophrenia. As such, we contend that results from this study may depict a more accurate representation of the role of synapsin II in this patient population.
Previous studies from other investigators have utilized these same specimens for analysis of expression and alterations of various other genes in schizophrenia, including catecholamine-regulated protein/mortalin 2, carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase (CAPON), tryptophan hydroxylase 2, Sprouty2, and the human endogenous retrovirsus-K10, (35;43–46). Despite the apparent strengths in this investigation, there are, regrettably, some unavoidable shortcomings. While appropriate statistical analyses have been performed in this study to assess the effect of antipsychotic drugs in schizophrenia and bipolar disorder, it is not feasible to account for the potential roles of individual antipsychotic drug use and other additional types of medications used to treat these disorders. Due to the complexity of the disorder, patients are often treated with multiple types and/or classes of drugs. Further, while this study utilized a relatively large sample size, future investigations can further strengthen the results of this study in larger clinical populations to better address the influence of various treatments prescribed for schizophrenia. A characteristic shortcoming of human post-mortem studies in schizophrenia is the lack of availability of patient tissue which has not been exposed to antipsychotic drugs or any other medications. As such, a direct association cannot be made for synapsin II expression levels in untreated patients with schizophrenia. Finally, the phosphorylated state of synapsin II also plays an important role in its regulation of synaptic vesicles, and is a fundamentally important question that should be addressed. However, at present time no specific antibodies are available for the direct measure of phosphorylated synapsin II in these human post-mortem samples. Until such tools and specimens are available, a direct measure of phosphorylation in synapsin II cannot be performed.
Clinical potency of existing antipsychotic drugs has been directly correlated to the drug’s ability to antagonize dopamine D2 receptors. However, antagonism of a receptor is achieved instantaneously, while the beneficial effect of antipsychotic drugs shows a delayed therapeutic effect (47;48). As such, the therapeutic effect of antipsychotic drugs must be explained by a more complicated and gradual mechanism, which can account for the delayed therapeutic effects (47;48). Recent evidence suggests modification of synaptic connections and synaptic proteins by antipsychotic drugs as a more likely explanation for the therapeutic effects seen in antipsychotic drug use. Previous studies from our laboratory have shown that antipsychotic drug use induces increases in synapsin II levels in neuronal cells, through the cyclic adenosine-3′,5′-monophosphate/PKA-dependent mechanism involving the AP-2α transcription factor (18;49). In addition to changes in levels of synapsin II, other studies have observed synaptic plasticity and neuronal remodeling in various regions of the brain, including the prefrontal cortex, nucleus accumbens, as a result of antipsychotic drug use. Both classes of antipsychotic drugs have been found to reverse the markers of pathology in schizophrenia, by increasing brain volume, and inducing neuroplasticity by affecting synaptic rearrangements (ie. increasing synaptic connections, activity, and density) and altering gene expression (ie. increasing synthesis of synaptic proteins, such as synapsin II) in the respective brain regions (47;48).
Antipsychotic drugs do not induce change in synaptic mechanisms directly, but rather modulate other neurotransmitter systems to do so, thus suggesting indirect modulation of synaptic activity (47;48). Mechanistically, antagonism of dopamine D2 receptors (in addition to other receptor types, depending on the antipsychotic drug administered), can lead to subsequent glutamatergic signaling through local circuits or long-loop neurotransmitter pathways. Glutamate receptor activity plays an important role in synapse formation and stabilization (47;48). A reduced number of glutamate receptors and activity leads to the ‘synaptic disorder’ in schizophrenia, whereas an increase in this glutamatergic activity induced (directly or indirectly) by antipsychotic drugs can lead to synaptic plasticity, and thus an attenuation of the behavioural symptoms and biochemical markers of schizophrenia (47;48;50).
In conclusion, results indicate that dorsolateral prefrontal cortical synapsin IIa and IIb gene expression is specifically reduced in patients with schizophrenia, and that prefrontal cortical synapsin IIa is preferentially up-regulated by antipsychotic agents. Given that dorsolateral prefrontal cortical synaptic aberrations are also associated with the deficits in attention, executive function and other cognitive abilities seen in schizophrenia, our results may contribute to a better understanding of the role of synapsin II in the development of hypo-frontal activity in patients with schizophrenia. Clinical studies examining the relationship between reductions in prefrontal cortical synapsin II gene expression and deficiencies in the cognitive functions observed in schizophrenia are, however, clearly warranted. The present study also provides insight into the therapeutic and potentially adverse mechanisms of action with use of antipsychotic drugs at the synaptic level. Additional roles played by other synapsin isoforms have, however, yet to be established. While this study alludes to a fundamental role of synapsin II in the underlying pathophysiology of schizophrenia, the exact mechanistic pathways underlying remain to be identified in future studies. The understanding of synapsin II in mediating the dynamics of synaptogenesis and synaptic vesicle regulation remains critical to unraveling the enigma of schizophrenia. However, given our understanding of abnormal synaptic function and projections in schizophrenia, and the importance of synaptic mechanisms in the treatment of schizophrenia, the development of novel antipsychotic drugs which target synaptic remodeling will be particularly beneficial (47;48;50). Results from our manuscript support the notion that modulation of synapsin II, and thus neuroplasticity, in specific brain regions, may be relevant to the design of an effective antipsychotic drug for schizophrenia. The further understanding of synapsin II in these synaptic mechanisms, at both the levels of disease production and treatment design, will facilitate the production of more specific targets and potentially quicker-acting drugs in comparison to existing antipsychotic drugs in the market (47;48).
Acknowledgments
This work was supported by the Canadian Institutes of Health Research.
We are thankful to Dr. Victor Chong for his helpful discussion in this study.
Footnotes
CONFLICT OF INTEREST
All authors report neither competing financial interests nor potential conflicts of interest in relation to the work described.
REFERENCE LIST
- 1.Dyck BA, Skoblenick KJ, Castellano JM, Ki K, Thomas N, Mishra RK. Behavioral abnormalities in synapsin II knockout mice implicate a causal factor in schizophrenia. Synapse. 2009;63(8):662–672. doi: 10.1002/syn.20643. [DOI] [PubMed] [Google Scholar]
- 2.Dyck BA, Mishra RK. Regulation of Synapsin II by Dopaminergic Mechanisms. Dopamine: Function, Regulation and Health Effects. New York: Nova Science Publishers; 2011. [Google Scholar]
- 3.Dyck BA, Beyaert MG, Ferro MA, Mishra RK. Medial prefrontal cortical synapsin II knock-down induces behavioral abnormalities in the rat: Examining synapsin II in the pathophysiology of schizophrenia. Schizophr Res. 2011;130(1–3):250–259. doi: 10.1016/j.schres.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 4.Kasai K, Iwanami A, Yamasue H, Kuroki N, Nakagome K, Fukuda M. Neuroanatomy and neurophysiology in schizophrenia. Neurosci Res. 2002;43(2):93–110. doi: 10.1016/s0168-0102(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 5.Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6(4):348–357. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- 6.Weinberger DR, Berman KF, Zec RF. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry. 1986;43(2):114–124. doi: 10.1001/archpsyc.1986.01800020020004. [DOI] [PubMed] [Google Scholar]
- 7.Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. 2004;27(11):683–690. doi: 10.1016/j.tins.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91(4):313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci. 2002;59(4):589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guest KA, Dyck BA, Shethwala S, Mishra RK. Atypical antipsychotic drugs upregulate synapsin II in the prefrontal cortex of post-mortem samples obtained from patients with schizophrenia. Schizophr Res. 2010;120(1–3):229–231. doi: 10.1016/j.schres.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 11.Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28(1):53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 12.Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, et al. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004;24(14):3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, et al. A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci. 2002;5(5):431–437. doi: 10.1038/nn840. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, He G, Qin W, Chen QY, Zhao XZ, Duan SW, et al. Family-based association study of synapsin II and schizophrenia. Am J Hum Genet. 2004;75(5):873–877. doi: 10.1086/425588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Q, He G, Wang XY, Chen QY, Liu XM, Gu ZZ, et al. Positive association between synapsin II and schizophrenia. Biol Psychiatry. 2004;56(3):177–181. doi: 10.1016/j.biopsych.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Saviouk V, Moreau MP, Tereshchenko IV, Brzustowicz LM. Association of synapsin 2 with schizophrenia in families of Northern European ancestry. Schizophr Res. 2007;96(1–3):100–111. doi: 10.1016/j.schres.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pulver AE, Lasseter VK, Kasch L, Wolyniec P, Nestadt G, Blouin JL, et al. Schizophrenia: a genome scan targets chromosomes 3p and 8p as potential sites of susceptibility genes. Am J Med Genet. 1995;60(3):252–260. doi: 10.1002/ajmg.1320600316. [DOI] [PubMed] [Google Scholar]
- 18.Chong VZ, Skoblenick K, Morin F, Xu Y, Mishra RK. Dopamine-D1 and -D2 receptors differentially regulate synapsin II expression in the rat brain. Neuroscience. 2006;138(2):587–599. doi: 10.1016/j.neuroscience.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 19.Chong VZ, Young LT, Mishra RK. cDNA array reveals differential gene expression following chronic neuroleptic administration: implications of synapsin II in haloperidol treatment. J Neurochem. 2002;82(6):1533–1539. doi: 10.1046/j.1471-4159.2002.01104.x. [DOI] [PubMed] [Google Scholar]
- 20.Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci U S A. 2000;97(14):7673–7675. doi: 10.1073/pnas.97.14.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, et al. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc Natl Acad Sci U S A. 2005;102(9):3513–3518. doi: 10.1073/pnas.0409766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeman P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin Ther Targets. 2006;10(4):515–531. doi: 10.1517/14728222.10.4.515. [DOI] [PubMed] [Google Scholar]
- 23.Seeman P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin Schizophr Relat Psychoses. 2010;4(1):56–73. doi: 10.3371/CSRP.4.1.5. [DOI] [PubMed] [Google Scholar]
- 24.Seeman P. All Roads to Schizophrenia Lead to Dopamine Supersensitivity and Elevated Dopamine D2 Receptors. CNS Neurosci Ther. 2011;17(2):118–132. doi: 10.1111/j.1755-5949.2010.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51(10):775–787. doi: 10.1016/s0006-3223(01)01362-2. [DOI] [PubMed] [Google Scholar]
- 26.Stone JM, Morrison PD, Pilowsky LS. Glutamate and dopamine dysregulation in schizophrenia--a synthesis and selective review. J Psychopharmacol. 2007;21(4):440–452. doi: 10.1177/0269881106073126. [DOI] [PubMed] [Google Scholar]
- 27.Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr Bull. 2013;39(1):120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gitler D, Cheng Q, Greengard P, Augustine GJ. Synapsin IIa controls the reserve pool of glutamatergic synaptic vesicles. J Neurosci. 2008;28(43):10835–10843. doi: 10.1523/JNEUROSCI.0924-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, et al. Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem. 2006;96(5):1458–1466. doi: 10.1111/j.1471-4159.2005.03636.x. [DOI] [PubMed] [Google Scholar]
- 30.Kleinbaum DG, Kupper LL, Muller KE, Nizam A. Applied regression analysis and other multivariable methods. Pacific Grove: Duxbury Press; 1998. [Google Scholar]
- 31.van Os J, Verdoux H, Maurice-Tison S, Gay B, Liraud F, Salamon R, et al. Self-reported psychosis-like symptoms and the continuum of psychosis. Soc Psychiatry Psychiatr Epidemiol. 1999;34(9):459–463. doi: 10.1007/s001270050220. [DOI] [PubMed] [Google Scholar]
- 32.Andreasen NC, Rezai K, Alliger R, Swayze VW, Flaum M, Kirchner P, et al. Hypofrontality in neuroleptic-naive patients and in patients with chronic schizophrenia. Assessment with xenon 133 single-photon emission computed tomography and the Tower of London. Arch Gen Psychiatry. 1992;49(12):943–958. doi: 10.1001/archpsyc.1992.01820120031006. [DOI] [PubMed] [Google Scholar]
- 33.Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, et al. Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry. 2001;50(11):825–844. doi: 10.1016/s0006-3223(01)01252-5. [DOI] [PubMed] [Google Scholar]
- 34.Liddle PF. Inner connections within domain of dementia praecox: role of supervisory mental processes in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 1995;245(4–5):210–215. doi: 10.1007/BF02191799. [DOI] [PubMed] [Google Scholar]
- 35.Pillai A. Decreased expression of Sprouty2 in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder: a correlation with BDNF expression. PLoS One. 2008;3(3):e1784. doi: 10.1371/journal.pone.0001784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller HK, Wegener G, Popoli M, Elfving B. Differential expression of synaptic proteins after chronic restraint stress in rat prefrontal cortex and hippocampus. Brain Res. 2011;1385:26–37. doi: 10.1016/j.brainres.2011.02.048. [DOI] [PubMed] [Google Scholar]
- 37.Ferreira AF, Real CC, Rodrigues AC, Alves AS, Britto LR. Moderate exercise changes synaptic and cytoskeletal proteins in motor regions of the rat brain. Brain Res. 2010;1361:31–42. doi: 10.1016/j.brainres.2010.09.045. [DOI] [PubMed] [Google Scholar]
- 38.Fdez E, Hilfiker S. Vesicle pools and synapsins: new insights into old enigmas. Brain Cell Biol. 2006;35(2–3):107–115. doi: 10.1007/s11068-007-9013-4. [DOI] [PubMed] [Google Scholar]
- 39.Fornasiero EF, Bonanomi D, Benfenati F, Valtorta F. The role of synapsins in neuronal development. Cell Mol Life Sci. 2010;67(9):1383–1396. doi: 10.1007/s00018-009-0227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matus-Leibovitch N, Nevo I, Vogel Z. Differential distribution of synapsin IIa and IIb mRNAs in various brain structures and the effect of chronic morphine administration on the regional expression of these isoforms. Molecular Brain Research. 1997;45(2):301–316. doi: 10.1016/s0169-328x(96)00265-3. [DOI] [PubMed] [Google Scholar]
- 41.Van Putten T, Marder SR, Mintz J, Poland RE. Haloperidol plasma levels and clinical response: a therapeutic window relationship. Am J Psychiatry. 1992;149(4):500–505. doi: 10.1176/ajp.149.4.500. [DOI] [PubMed] [Google Scholar]
- 42.Imai C, Sugai T, Iritani S, Niizato K, Nakamura R, Makifuchi T, et al. A quantitative study on the expression of synapsin II and N-ethylmaleimide-sensitive fusion protein in schizophrenic patients. Neurosci Lett. 2001;305(3):185–188. doi: 10.1016/s0304-3940(01)01844-4. [DOI] [PubMed] [Google Scholar]
- 43.Gabriele JP, Chong VZ, Pontoriero GF, Mishra RK. Decreased expression of a 40-kDa catecholamine-regulated protein in the ventral striatum of schizophrenic brain specimens from the Stanley Foundation Neuropathology Consortium. Schizophr Res. 2005;74(1):111–119. doi: 10.1016/j.schres.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 44.De LV, Likhodi O, Van Tol HH, Kennedy JL, Wong AH. Tryptophan hydroxylase 2 gene expression and promoter polymorphisms in bipolar disorder and schizophrenia. Psychopharmacology (Berl) 2005;183(3):378–382. doi: 10.1007/s00213-005-0191-4. [DOI] [PubMed] [Google Scholar]
- 45.Xu B, Wratten N, Charych EI, Buyske S, Firestein BL, Brzustowicz LM. Increased expression in dorsolateral prefrontal cortex of CAPON in schizophrenia and bipolar disorder. PLoS Med. 2005;2(10):e263. doi: 10.1371/journal.pmed.0020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank O, Giehl M, Zheng C, Hehlmann R, Leib-Mosch C, Seifarth W. Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders. J Virol. 2005;79(17):10890–10901. doi: 10.1128/JVI.79.17.10890-10901.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konradi C, Heckers S. Antipsychotic drugs and neuroplasticity: insights into the treatment and neurobiology of schizophrenia. Biol Psychiatry. 2001;50(10):729–742. doi: 10.1016/s0006-3223(01)01267-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horacek J, Bubenikova-Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, et al. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs. 2006;20(5):389–409. doi: 10.2165/00023210-200620050-00004. [DOI] [PubMed] [Google Scholar]
- 49.Skoblenick KJ, Argintaru N, Xu Y, Dyck BA, Basu D, Tan ML, et al. Role of AP-2alpha transcription factor in the regulation of synapsin II gene expression by dopamine D1 and D2 receptors. J Mol Neurosci. 2010;41(2):267–277. doi: 10.1007/s12031-009-9299-z. [DOI] [PubMed] [Google Scholar]
- 50.Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr Res. 2006;81(1):47–63. doi: 10.1016/j.schres.2005.08.014. [DOI] [PubMed] [Google Scholar]