

Abstract
OCT2 is the entry step for organic cation (OC) secretion by renal proximal tubules. Although many drugs inhibit OCT2 activity, neither the mechanistic basis of their inhibition nor their transport status is generally known. Using representatives of several structural classes of OCT2-inhibitory ligands described recently (Kido Y, Matsson P, Giacomini KM. J Med Chem 54: 4548–4558, 2011), we determined the kinetic basis of their inhibition of 1-methyl-4-phenylpyridinium (MPP) transport into Chinese hamster ovary cells that stably expressed hOCT2. The “cluster II” inhibitors (which contain known OCT2 substrates) metformin and cimetidine interacted competitively with MPP. However, other cluster II compounds, including tetraethylammonium (TEA), diphenidol and phenyltoloxamine, were mixed-type inhibitors of MPP transport (i.e., decreasing Jmax and increasing Kt). A cluster III (neutral steroid) representative, adrenosterone, and a cluster I (large, flexible cation) representative, carvedilol, displayed noncompetitive inhibitory profiles. Competitive counterflow (CCF) was used to determine whether the inhibitory ligands served as substrates of hOCT2. Carvedilol (cluster I) and adrenosterone (cluster III) did not support CCF, consistent with the prediction that members of these structural classes are likely to be nontransported inhibitors of OCT2. The cluster II representatives MPP, metformin, cimetidine, and TEA all supported CCF, consistent with independent assessments of their OCT2-mediated transport. However, the other cluster II representatives, diphenidol and phenyltoloxamine, failed to support CCF, suggesting that neither compound is transported by OCT2. An independent assessment of diphenidol transport (using liquid chromatography with tandem mass spectroscopy) confirmed this observation. The results underscore the caution required for development of predictive models of ligand interaction with multidrug transporters.
Keywords: transport, proximal tubule, organic cation, multidrug
in a recent assessment of the physicochemical determinants of renal drug clearance (51), the kidney was found to contribute >50% of total body clearance for >30% of the ∼400 compounds surveyed. Renal secretion, mediated principally by proximal tubule cells, plays a central role in this process, a hallmark of which is the structurally diverse array of both exogenous (xenobiotic), and endogenous, organic compounds it handles (43). A consequence of the structural diversity of ligand interaction that is a functional hallmark of a comparatively small set of “multidrug” transport processes is the high probability of unwanted drug-drug-interactions (DDIs) at the point of renal secretion, interactions that can significantly influence a drug's clinical or toxicological profile. In addition, inhibition of renal drug transport can influence drug accumulation within the kidney, thereby contributing to the nephrotoxic impact associated with use of some pharmaceutical agents. Unwanted DDIs are typically detrimental, so predicting and, ideally, preempting their occurrence is a major focus of current work on renal drug transport.
Our focus is on the role of organic cation transporter 2 (OCT2; SLC22A2) in renal drug secretion. Approximately 40% of prescribed drugs carry a net positive charge at or near physiological pH (1) and, so, are collectively referred to as “organic cations” (OCs), and the mechanism of OC secretion by renal proximal tubule cells has received considerable attention. The basic cellular model of renal OC secretion in renal proximal tubule (RPT) cells, proposed first by Holohan and Ross (45), includes the sequential activity of 1) a basolateral “entry step,” from blood to cell, that involves an electrogenic organic cation transporter; and 2) an apical “exit step,” from cell to tubular filtrate mediated by electroneutral OC/H+ exchange. Following the cloning of a series of electrogenic organic cation transporters (19, 23, 40), there is now broad consensus that, in the human kidney, the basolateral step in this process is dominated by activity of OCT2 (36, 43).
The increasing attention given to the clinical impact of unwanted DDIs has lead to development of several predictive models of ligand interaction with OCT2 (24, 49, 56) and its hepatic homolog, OCT1 (1, 6, 35). All of these studies correlated inhibition of OCT-mediated transport produced by sets of test ligands with various structural and/or physicochemical descriptors of the inhibitory agents, as the basis for development of two dimensional (2D)- or 3D-quantitative structure activity relationships (QSARs) of ligand/transporter interaction. Although details vary, 2D-QSARs for both OCTs consistently show that inhibitory effectiveness is correlated with 1) cationic charge and 2) various measures of ligand lipophilicity; and 3D-QSARs (“pharmacophores”) generally include a “cationic feature,” one or more “hydrophobes,” and, frequently, one or more hydrogen bond donor/acceptor features. In the most extensive study of ligand interaction with OCT2 to date, Kido et al. (24) screened 900 compounds and the effective inhibitors of transport (244 compounds) were distributed into 3 structural classes, referred to as “clusters,” based on selected molecular properties. Cluster I inhibitors are generally long, flexible cations; cluster II inhibitors are smaller, globular, cations and, interestingly, included the known substrates of OCT2; and cluster III inhibitors are noncharged heterotypic ring structures (including many sterols).
Despite the increase in understanding of physical and structural factors that influence ligand interaction with the OCTs, the work to date has largely overlooked two issues we suggest are central to understanding the molecular basis of OCT activity and successful development of predictive models of their influence on OC clearance. Specifically, existing studies have largely ignored 1) the mechanistic/kinetic basis of observed inhibition of OCT transport activity and 2) whether an inhibitory ligand is itself a transported substrate. The first of these issues is central to development of pharmacophores that reflect the possibility, frequently alluded to but not directly addressed, that OCTs may have multiple binding sites and that substrates and inhibitors may interact with one or more of these sites, perhaps simultaneously (e.g., Refs. 15 and 34). There is a large extant literature on ligand interaction with OCTs (see Ref. 39 for a thorough review of these data). Unfortunately, large variability in quantitative data, even from studies examining the same substrates/inhibitors using similar (if not identical) experimental systems, precludes any effort to draw broad conclusions about the kinetic basis of OCT/ligand interactions.
The second issue noted above, i.e., the need to understand the structural basis of ligand interaction that can result in successful transport, is critical to development of “physiologically based pharmacokinetic models.” Whereas hundreds of compounds have been shown to inhibit OCT transport activity (e.g., Refs. 1 and 24), evidence for transport is limited to fewer than 30 compounds (39, 46).
The present study provides an initial examination of both of these issues for human OCT2. Within the structurally diverse set of compounds studied, which included representatives of the three structural classes of OCT2 inhibitors defined by Kido et al. (24), examples of “competitive,” “noncompetitive,” and “mixed-type” inhibition were noted. We also describe a method, “competitive counterflow” (CCF), that distinguished between nontransported inhibitors and those that also serve as transported substrates of OCT2.
MATERIALS AND METHODS
Reagents
Chinese hamster ovary (CHO) cells containing a single integrated Flp recombination target site (CHO Flp-In), Flp recombinase expression plasmid (pOG44), platinum high-fidelity DNA polymerase, hygromyosin, zeocin, and mammalian expression vector pcDNA5/FRT/V5-His TOPO were acquired from Invitrogen (Carlsbad, CA). Ham's F-12 Nutrient Mixture, 1-methyl-4-phenylpyridinium (MPP), cimetidine, tetraethyl ammonium chloride (TEA), imipramine hydrochloride, pyrimethamine, adrenosterone, carvedilol, and phenyltoloxamine were obtained from Sigma-Aldrich (St. Louis, MO). Metformin was obtained from AK Scientific (Mountain View, CA). Tacrine hydrochloride was obtained from Cayman Chemical (Ann Arbor, MI). Diphenidol hydrochloride was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][l,2,5]oxadiazol-4-yl)amino]ethanaminium iodide (NBD-MTMA) and N1-ethylacridinium were synthesized by the Synthesis Core of the Southwest Environmental Health Sciences Center/Department of Chemistry at the University of Arizona (Tucson, AZ).
Cell Culture and Stable Expression of hOCT2
CHO cells containing the Flp recombination target site were grown in Ham's F-12 Nutrient Mixture with 10% fetal bovine serum and supplemented with 100 μg/ml zeocin. Cells that stably expressed hOCT2 were prepared using methods described previously (42). Briefly, cells (5 × 106) were transfected by electroporation (BTX ECM 630, San Diego; 260 V with time constant of ∼25 ms) with 10 μg of salmon sperm, 18 μg of pOG44, and 2 μg of pcDNA5/FRT/V5-His TOPO containing an OCT2 (hOCT2) construct. The CHOhOCT2 cells were grown and maintained under hygromycin pressure (100 μg/ml) in plastic cell culture flasks at 37°C in a humidified atmosphere with 5% CO2. Cells were passed every 3–4 days and seeded into 48-well plates at 550,000 cells/ml (24-h confluence) or 275,000 cells/ml (48-h confluence).
Transport Studies
CHO cells grown to confluence in 48-well plates were rinsed twice with Waymouth buffer (WB; 135 mM NaCl, 13 mM HEPES, 2.5 mM CaCl2·2H2O, 1.2 mM MgCl2, 0.8 mM MgSO4·7H2O, 5 mM KCl, and 28 mM d-glucose). A loading buffer containing [3H]MPP (80 Ci/mmol; typically 10–20 nM), plus the desired concentration of a test compound, was added to each well, removed after a set amount of time, and washed three times with cold WB to stop transport. The cells were then solubilized in 200 μl of 0.5 N NaOH with 1% SDS/well and shaken for 15 min. The solubilized cells were neutralized with 100 μl of 1 N HCl, and 250 μl were placed in a scintillation vial. The radioactivity in each sample was determined using scintillation spectroscopy (Beckman model LS6000IC). Individual transport experiments were conducted in triplicate and typically repeated at least three times. Experiments were conducted using cells between passages 4 and 35 with no consistent difference in transport rates between earlier and later passages.
Mass Spectroscopy
Analyses were performed using a TSQ Quantum Ultra triple-quadrupole mass spectrometer in conjunction with a Finnigan Surveyor Autosampler and Finnigan Surveyor quaternary HPLC pump (Thermo Finnigan, San Jose, CA).
Diphenidol.
The method was adapted from that of Marcelin-Jimanez et al. (32). The mass spectrometer was operated in the positive ionization mode using electrospray (ESI) ionization under the following conditions: spray voltage of 4,800 V, sheath gas (nitrogen) flow of 35 (arbitrary units), capillary temperature of 370°C with argon as the collision gas at a pressure of 0.8 mtorr. Detection used selective reaction monitoring (SRM), the parent mass/charge ratio for diphenidol was 310.2, and the fragment monitored was 292.3. The collision energy was 24 eV. HPLC separation was achieved using a Kinetex 2.6μ C-18 50 × 2.1-mm column (Phenomenex, Torrance, CA). Mobile phase A was 5:95 acetonitrile:water with 0.1% formic acid (vol/vol); mobile phase B was 95:5, acetonitrile:water with 0.1% formic acid (vol/vol). Mobile phase gradient was 80% A from 0 to 4 min, increasing to 100% B by 4.10 min, and maintaining 100% B until 8 min, followed by reequilibration at 80% A from 8.10 min until 12 min. Flow rate was 200 μl/min, and injection volume was 5 μl. Total analysis time was 12 min; diphenidol eluted at ∼2.4 min.
Metformin.
The mass spectrometer was operated in the positive ionization mode using atmospheric pressure chemical ionization (APCI) under the following conditions: spray voltage of 3,500 V, sheath gas (nitrogen) flow of 21 (arbitrary units), auxiliary gas (nitrogen) flow of 5, vaporizer temperature of 395°C, and capillary temperature of 200°C with argon as the collision gas at a pressure of 0.8 mtorr. Detection used SRM. The parent mass/charge ratio for metformin was 130.1, and the fragment monitored was 71.1. The collision energy was 33 eV. HPLC separation was achieved using a BDS HypersilCyano3μ 50 × 4.6-mm column (Thermo Scientific, Bellefonte, PA). The mobile phase was 60:20:20, 10 mM ammonium acetate:acetonitrile:methanol (vol/vol), at a flow rate of 650 μl/min, and injection volume was 20 μl. Total analysis time was 5 min; metformin eluted at ∼3.6 min.
Statistical Analysis
Data are expressed as means ± SE, with calculations of SEs based on the number of separate experiments conducted on cells at a different passage number. One-way ANOVA was used to test the effect of multiple treatments and was followed by the Student-Newman-Keuls test for pairwise comparisons. Comparison of sample means was done using an unpaired Student's t-test. All statistical analyses were performed with Prism 5 (GraphPad Software, La Jolla, CA) or Excel 2007 (Microsoft, Redmond, WA) and deemed significant when P < 0.05.
RESULTS
Kinetics of MPP Transport
The hOCT2-mediated uptake of [3H]MPP was linear for at least 90 s (data not shown), so 1-min uptakes were used for all subsequent studies of transport kinetics. To establish the kinetics of hOCT2-mediated MPP transport, the rate of transport of 12 nM [3H]MPP into CHO cells that stably expressed the transporter was measured in the presence of increasing concentrations of unlabeled MPP (Fig. 1). The relationship was adequately described by the Michaelis-Menten equation for competitive interaction of labeled and unlabeled substrate (31)
| 1 |
where J* is the rate of transport of the radiolabeled substrate (in this case, [3H]MPP) from a concentration of the labeled substrate equal to [S*]; Jmax is the maximal rate of mediated substrate transport; Kt is the Michaelis constant of the transported substrate; [S] is the concentration of unlabeled substrate; and Dns is a first-order rate constant that describes the nonsaturable component of labeled substrate accumulation (reflecting the combined influence of diffusion, nonspecific binding, and incomplete rinsing of labeled substrate from the cell culture well). In 22 separate experiments Kt and Jmax values for hOCT2-mediated MPP transport were 11.1 ± 1.2 μM and 16.0 ± 1.4 pmol·cm−2·min−1, respectively. Expressed per milligram of membrane protein, Jmax was (approximately) 400 pmol·mg−1·min−1.
Fig. 1.
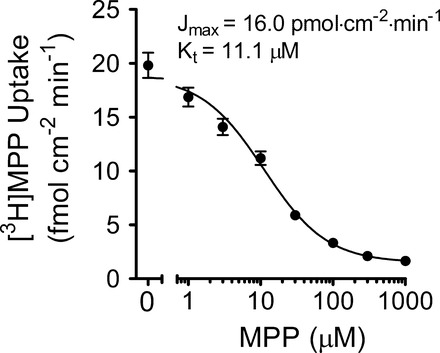
Kinetics of 1-methyl-4-phenylpyridinium (MPP) transport into Chinese hamster ovary (CHO) cells that stably express human organic cation transporter 2 (hOCT2). Each point is the mean (±SE) of the 1-min uptake of 12 nM [3H]MPP, measured in the presence of increasing concentrations of unlabeled MPP, determined in 22 separate experiments (each in triplicate). Kinetic values were determined using the relationship outlined in Eq. 1. Jmax, maximal rate of mediated substrate transport; Kt, Michaelis constant of the transported substrate.
Ligand Inhibition of hOCT2
Thirteen structurally diverse OCs (Fig. 2) were tested for their inhibition of OCT2-mediated [3H]MPP transport (Table 1). The rationale for selection of these compounds varied: cimetidine, imipramine, and tacrine IC50 values were reported by Kido et al. (24) in their study on ligand interaction with hOCT2; and carvedilol, diphenidol, phenyltoloxamine, and adrenosterone were cited as representatives of the three categories of OC structures they defined as OCT2 inhibitors. The remaining compounds were chosen either because of their use, either as substrates (metformin, TEA) (6, 25, 49) or inhibitors (pyrimethamine, corticosterone) (15, 27) in studies of OCT activity; or their use by us in previous studies of renal OC transport (N1-ethylacridinium, and NBD-MTMA) (7, 53). Figure 3 shows the inhibitory kinetic profiles of several representative test agents: N1-ethylacridinium, tacrine, diphenidol, and metformin. For these (and all the other) compounds tested, uptake of [3H]MPP was inhibited by increasing concentrations of the test agent, although the concentrations that inhibited 50% of the mediated (i.e., blockable) component of total MPP uptake (IC50) varied from <<1 μM to >>100 μM (Table 1), according to the relationship
| 2 |
where IC50 is the concentration of the inhibitor [I] that reduced mediated (i.e., blockable) [3H]MPP transport by 50%; Jmapp is the product of the maximum rate of S* (i.e., [3H]MPP) uptake (Jmax) and the ratio of the IC50 of the test agent and Kt for MPP transport. Table 1 presents the average IC50 values for these and the other test agents included in this study.
Fig. 2.
Structural formulas of the test compounds used in this study.
Table 1.
IC50 values for inhibition of hOCT2-mediated transport
| IC50, μM |
||
|---|---|---|
| Compound | Means ± SE; n = 3 | Other studies |
| N1-ethylacridinium | 0.26 ± 0.16 | 0.09TEA (49) |
| Tacrine | 3.1 ± 1.5 | 0.7ASP (24) |
| Imiprimine | 3.3 ± 1.3 | 6MPP (56) |
| 0.6ASP (24) | ||
| Phenyltoloxamine | 4.2 ± 2.8 | |
| Adrenosterone | 7.0 ± 2 | |
| Corticosterone | 17 ± 2.9 | 34MPP (20) |
| Pyrimethamine | 24 ± 5.4 | 10Met (27) |
| Diphenidol | 35 ± 12 | |
| Carvedilol | 55 ± 7.3 | 63MPP (56) |
| NBD-MTMA | 63 ± 12 | |
| Cimetidine | 110 ± 32 | 126MPP (56) |
| 23ASP (24) | ||
| 70TEA (49) | ||
| TEA | 269 ± 190 | 189TEA (33) |
| Metformin | 545 ± 45 | 398MPP (56) |
| 336TEA (49) | ||
Means ± SE values were determined in the present study using the methods outlined for the studies in Fig. 3 (each value is the mean of 3 separate experiments). TEA, tetraethylammonium. The IC50 values for inhibition of hOCT2 activity presented in the right-hand column were reported in the indicated references (in parentheses), and the superscripts refer to the human organic cation transporter 2 (hOCT2) substrate used to generate each value (note: all measurements reflect transport into cultured mammalian cells transfected with hOCT2).
Fig. 3.
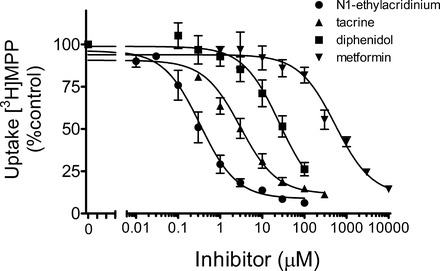
Kinetics of inhibition of hOCT2-mediated [3H]MPP transport produced by increasing concentrations of the indicated test agents. Data points are mean 1-min uptakes (±SE) of 12 nM [3H]MPP, normalized to the rate of transport measured in the absence of inhibitor, determined in 3 separate experiments (for diphenidol, n = 4), each performed in triplicate.
N1-ethylacridinium was the most potent inhibitor of hOCT2-mediated MPP transport (IC50 of 0.26 ± 0.16 μM); metformin was the least effective inhibitor (IC50 of 545 ± 45 μM).
As we are about to discuss a more detailed view of the kinetic basis of ligand interaction with hOCT2, it is relevant to compare, where available, our results to those obtained by others using similar methods. Figure 4 plots the data presented in Table 1, comparing the IC50 values we obtained for inhibition of hOCT2-mediated MPP transport in CHO cells to those reported in other studies. We restricted the comparison to studies of expression of human OCT2 in cultured mammalian cells (typically HEK 293 cells) and indicate the identity of the transported substrate used as the measure of OCT2 activity. Using these selection criteria, we found comparable IC50 data for 9 of our 13 test ligands, including 5 studies that used MPP as the transported substrate. In general, there was a strong correlation between the values we measured and those reported by others. We found it particularly intriguing that the correlation appeared strongest when limited to the IC50 values that used MPP as the transported probe; the several IC50 values obtained using ASP as the test probe were consistently lower than the values obtained using MPP. The correlation between our results, and the technical methods we employed, with those obtained by others lends credence, we suggest, to the data obtained in the more detailed kinetic analyses described below.
Fig. 4.
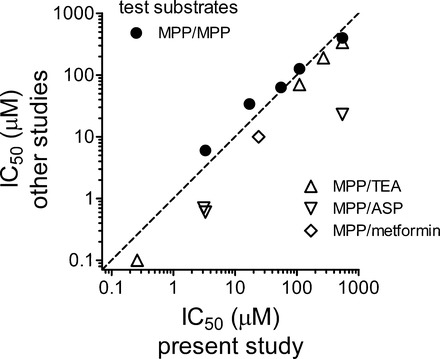
Comparison of IC50 values, listed in Table 1, for the inhibition of hOCT2-mediated transport produced by selected test agents. Filled circles compare values determined in the present study, using [3H]MPP as the test substrate, with those determined in other studies that also used radiolabeled MPP as the test substrate. Open symbols compare values determined in the present study (using [3H]MPP) with those determined in other studies using the indicated compound (TEA, ASP, or metformin) as the test substrate. The dashed line is the line of identity (equal IC50 values).
Kinetic Basis of Inhibition
The inhibitory profiles evident in Fig. 3, and the IC50 values reported in Table 1, provided no insight by themselves into the mechanistic basis of the decreases in OCT2-mediated MPP transport produced by the test ligands; although consistent with “competition” between [3H]MPP and the test agent, qualitatively similar profiles are expected if the interactions were noncompetitive, uncompetitive, or a “mixed type” (47). To establish the kinetic basis of observed inhibition of hOCT2-mediated transport, we measured the kinetics of MPP uptake in the absence and presence of a fixed concentration of inhibitor equal to approximately two times its IC50 value (Fig. 5). Inhibition was considered competitive if its presence resulted in an increase in the apparent Kt for MPP transport but no change in Jmax. Moreover, because the inhibitor concentrations used were each about twice the measured IC50 value, apparent Kt values were expected to increase about threefold (47). Based on these criteria, cimetidine's inhibition of OCT2-mediated MPP transport was competitive: in three experiments, the presence of 200 μM cimetidine increased Kt from 7.4 ± 2.5 to 24.9 ± 7.9 μM (P < 0.01, n = 6), while Jmax remained unchanged (8.6 ± 4.9 vs. 11.4 ± 4.1 pmol·cm−2·min−1; Table 2). Metformin's inhibition of OCT2-mediated MPP transport also displayed a competitive profile (Table 2). Carvedilol, in contrast, was a noncompetitive inhibitor of MPP transport, reducing the Jmax of transport from 16.7 ± 4.3 to 3.8 ± 3.7 pmol·cm−2·min−1 (P < 0.05, n = 3), without significantly influencing the Kt (13.0 ± 4.0 vs. 8.5 ± 2.4 μM; Fig. 5, Table 2). Adrenosterone produced a similar, noncompetitive inhibitory profile (Table 2). Interestingly, TEA, a well-characterized substrate of OCT2 (e.g., Refs. 5 and 40), proved to be a mixed-type, rather than competitive, inhibitor of MPP transport; 500 μM TEA produced a decrease in Jmax (from 10.6 ± 2.5 to 5.8 ± 1.1 pmol·cm−2·min−1; P < 0.05, n = 3) and an increase in apparent Kt (from 7.6 ± 1.0 to 18.5 ± 0.6 μM; P < 0.01) (Fig. 5, Table 2). Diphenidol and phenyltoloxamine also produced mixed-type inhibitory profiles (Table 2).
Fig. 5.
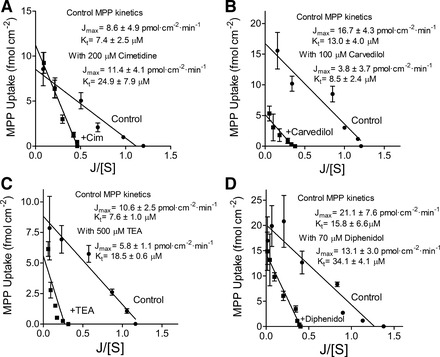
Kinetics of [3H]MPP uptake in CHOhOCT2 cells in the absence (control) and presence of the indicated inhibitory ligands: 200 μM cimetidine (n = 6; A); 100 μM carvedilol (n = 3; B); 500 μM TEA (n = 3; C); 70 μM diphenidol (n = 3; D). Data points represent mean uptakes (1 min; ±SE) under the indicated condition from several separate experiments (as indicated by n values), each performed in triplicate. Uptakes were corrected for the nonsaturable component of transport as determined by the calculated value of Dns for each experiment (see Eq. 1).
Table 2.
Kinetic impact of inhibitory ligands on hOCT2-mediated MPP transport
| Inhibitor | MPP Transport, Jmax, pmol·cm−2·min−1 | Jmax in the Presence of Inhibitor, pmol·cm−2·min−1 | MPP Transport Kt, μM | Kt in the Presence of Inhibitor, μM |
|---|---|---|---|---|
| Competitive | ||||
| Metformin (1 mM) | 12.8 ± 1.8 | 14.0 ± 3.5 | 8.1 ± 1.9 | 22.9 ± 4.3† |
| Cimetidine (200 μM) | 8.6 ± 4.9 | 11.4 ± 4.1 | 7.4 ± 2.5 | 24.9 ± 7.9† |
| Noncompetitive | ||||
| Adrenosterone (15 μM) | 13.9 ± 3.9 | 2.6 ± 0.7† | 11.6 ± 2.6 | 7.3 ± 1.7 |
| Carvedilol (100 μM) | 16.7 ± 4.3 | 3.8 ± 3.7* | 13.0 ± 4.0 | 8.5 ± 2.4 |
| Mixed Type | ||||
| TEA (500 μM) | 10.6 ± 2.5 | 5.8 ± 1.1* | 7.6 ± 1.0 | 18.5 ± 0.6† |
| Diphenidol (70 μM) | 21.1 ± 7.6 | 13.1 ± 3.0* | 15.8 ± 6.6 | 34.1 ± 4.1† |
| Phenyltoloxamine (15 μM) | 19.3 ± 4.6 | 8.0 ± 3.7* | 17.6 ± 2.4 | 30.1 ± 6.9* |
Values are means ± SE of 3–6 separate, paired experiments (each performed in triplicate). MPP, 1-methyl-4-phenylpyridinium. The maximal rate of mediated substrate transport (jmax) and the Michaelis constant of the transported substrate (Kt) values were measured in the absence (control) and presence of the indicated concentration of each inhibitory ligand. *,†: Differences at the P < 0.05 (
) or 0.01 (
) level.
Indirect Assessment of Inhibitor Transport: Competitive Counterflow
Knowledge of the kinetic basis of ligand interaction with OCT2 cannot support conclusions on whether an inhibitor of transport is itself a transported substrate of OCT2. Addressing the issue of transport directly can be a challenge; unfortunately, most of the inhibitory ligands used in this (and other) studies are not routinely available in radiolabeled form (and custom syntheses can be prohibitively expensive). Furthermore, although the transport of most of these compounds, if not all, can be determined employing various other analytic methods (e.g., HPLC and/or mass spectroscopy), their use in a study attempting to screen transport of a large number of ligands can also be prohibitively expensive (in both time and money). In light of these issues, we sought to develop a method that is both cost effective and yet still capable of providing information concerning the “transportability” of inhibitory ligands.
The approach we developed is a modification of methods introduced for the study of glucose transport in red cells by Rosenberg and Wilbrandt (“counterflow”) (44) and Lacko and Burger (“competitive exchange diffusion”) (28), as discussed by Stein (48). We refer to the method as CCF as it combined elements of the aforementioned studies to determine whether two compounds share the same transport process. Briefly, cells expressing the transporter of interest (e.g., hOCT2) are allowed to accumulate a probe substrate to steady state. The cells are then rinsed of the extracellular solution and immediately exposed to an experimental solution containing either 1) the same concentration of the labeled substrate or 2) the same concentration of labeled substrate plus a test compound to determine whether the presence of the test compound stimulates efflux of the intracellular compound. In the first condition, cell content of the radiolabeled substrate should remain unchanged because the cells have already achieved a steady-state distribution arising from equal rates of substrate entry and exit. However, under the second condition, the presence of the extracellular inhibitory ligand reduces the rate of influx of the labeled substrate. If binding of the test ligand still permits turnover of the transporter (i.e., if the inhibitor is a substrate), then the intracellular labeled substrate can still exit and cell content of the radiolabel will decrease with time. Importantly, if binding of an inhibitory ligand prevents effective turnover of the transporter, then mediated efflux of the label is eliminated, indicating that the test compound is a nontransported inhibitor.
As a “proof-of-concept” for our modified CCF protocol, we determined the influence of extracellular unlabeled MPP on the efflux of preloaded [3H]MPP. Figure 6 shows experiments that established the time and concentration dependence of [3H]MPP transport using the CCF protocol. Figure 6A shows that steady-state accumulation of [3H]MPP (∼12 nM) into hOCT2-expressing cells was achieved within ∼30 min. In the experiment summarized in Fig. 6B, cells allowed to accumulate [3H]MPP to steady state (30 min) were exposed to media containing either 1) 12 nM [3H]MPP (identical to the loading solution) or 2) 12 nM [3H]MPP plus 100 μM unlabeled MPP. When the solution contained only [3H]MPP (the “control” condition), cell content of labeled MPP remained unchanged over 1 min, reflecting maintenance of the steady-state condition. The addition of unlabeled MPP in the extracellular solution was associated with a rapid net loss of label from the cells that approached a new steady state after 60 s. This was expected because OCTs support mediated exchange of an OC for an OC (10); i.e., unlabeled MPP should compete with labeled MPP at the extracellular binding surface of hOCT2 to 1) prevent unidirectional influx of [3H]MPP while 2) supporting exchange for the intracellular [3H]MPP. The rapid decline in the rate of loss of label from the cells reflected the anticipated inhibitory impact of increasing competition between labeled MPP and the increasing concentration of unlabeled substrate at the cytoplasmic face of OCT2. It also underscores the advantage of using a labeled substrate with a comparatively high affinity for the transporter, such as MPP for hOCT2, because it delays the inhibitory influence of the accumulated test compound on probe efflux. Figure 6C shows the effect of increasing the concentration of extracellular unlabeled MPP on the rate of [3H]MPP efflux from preloaded cells. Neither 1 or 3 μM MPP, concentrations well below the Kt for MPP uptake (Fig. 1), induced a significant loss of [3H]MPP from the cells (Fig. 6). However, the presence of 10 μM MPP, equal to the Kt for OCT2-mediated MPP uptake (Fig. 1), and all concentrations greater than that, did stimulate a net loss of labeled MPP (Fig. 6). This trans-effect was consistent with the fact that MPP is a substrate for hOCT2. Efflux increased up to 100 μM, but concentrations >100 μM had no further stimulatory effect. In other words, increasing extracellular MPP beyond ∼100 μM was incapable of stimulating more efflux, due to the effective saturation of available OCT2 transporters by extracellular MPP.
Fig. 6.
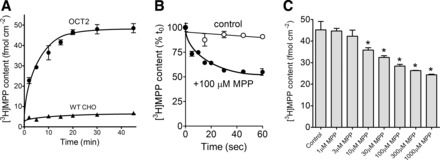
Competitive counterflow (CCF) profiles. A: time course of 12 nM [3H]MPP uptake in OCT2 and wild-type (WT) CHO cells. Each point is the mean of triplicate determinations of cell content of [3H]MPP into WT CHO cells or CHO cells that stably expressed hOCT2 (lines fit by eye). Data shown are from a single representative experiment. B: time course of [3H]MPP efflux from hOCT2-expressing CHO cells. Before the measurement of efflux, cells were exposed to 12 nM [3H]MPP for 30 min. At time 0, the medium was rinsed and replaced with medium containing either 12 nM [3H]MPP (control) or 12 nM [3H]MPP plus 100 μM MPP. Each point is the mean (±SE) of cell [3H]MPP content (normalized to content at time 0) measured in 3 wells from a representative experiment. C: effect of increasing extracellular concentrations of unlabeled MPP on efflux of [3H]MPP from hOCT2-expressing CHO cells. Before the measurement of efflux, cells were exposed to 12 nM [3H]MPP for 30 min. At time 0, the medium was rinsed and replaced with medium containing either 12 nM [3H]MPP (control) or 12 nM [3H]MPP plus the indicated concentration of unlabeled MPP. The height of each bar represents triplicate determinations of the [3H]MPP remaining in cells after a 60-s efflux period. Data shown are from a single representative experiment. Bars marked with * were different from the control (P < 0.05).
Figure 7 shows the effect of several additional known OCT2 substrates, TEA, metformin, and cimetidine, on the efflux of [3H]MPP from OCT2-expressing CHO cells. The cells were exposed to a concentration of each agent equal to ∼10 times the IC50 value for that compound as an inhibitor of OCT2-mediated MPP uptake (Table 1), concentrations that, for each compound, may be expected to occupy ∼90% of available binding sites at the extracellular face of the transporter. As expected, each compound supported CCF because each has been shown to be a transported substrate of OCT2, in addition to being an inhibitory ligand (39). Figure 7 also shows that, as expected, compounds that do not interact with OCT2 (glucose, mannitol, sucrose, and succinic acid) do not support CCF.
Fig. 7.
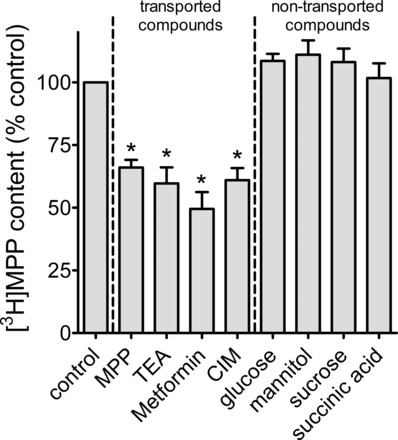
CCF as elicited by known OCT2 substrates and by compounds that have no interaction with OCT2. Before the measurement of CCF, cells were exposed to 12 nM [3H]MPP for 30 min. At time 0, the medium was rinsed and replaced with medium containing either 12 nM [3H]MPP (control) or 12 nM [3H]MPP plus the indicated compound. For the compounds known to be substrates of OCT2, the concentration of unlabeled compound was ∼10 times the IC50 measured in the present study (MPP, 100 μM; TEA, 1 mM; metformin, 10 mM; cimetidine, 2 mM). Each of the nontransported compounds was present at a concentration 10 mM. The height of each bar represents the mean (±SE) of [3H]MPP remaining in cells after a 60-s efflux period, normalized to control content. Data shown are from 2–7 separate experiments (each run in triplicate). Bars marked with * were different from the control (P < 0.05).
CCF of Inhibitory Ligands
CCF was used to determine whether ligands selected from the list of representatives for each OCT2 inhibitory cluster identified by Kido et al. (24) are also transported by OCT2. Carvedilol was the cluster I representative (long, flexible cations), and adrenosterone was the cluster III representative (noncharged heterotypic ring structures). Known transported substrates of OCT2 have the structural characteristics of the cluster II compounds (smaller, globular, cations), so we tested two representatives of this group of inhibitory ligands: diphenidol and phenyltoloxamine. The concentration of the unlabeled test compound was generally 10 times its IC50 as an inhibitor of OCT2-mediated MPP uptake (because of solubility issues, the carvedilol concentration was limited to 4 times its IC50 value). Figure 8 shows that none of these compounds supported a net loss of [3H]MPP through CCF from OCT2-expressing cells. These data support the contention that these inhibitors of OCT2, including diphenidol and phenyltoloxamine, both members of the cluster II series of compounds described by Kido et al. (24), are not transported substrates of OCT2.
Fig. 8.
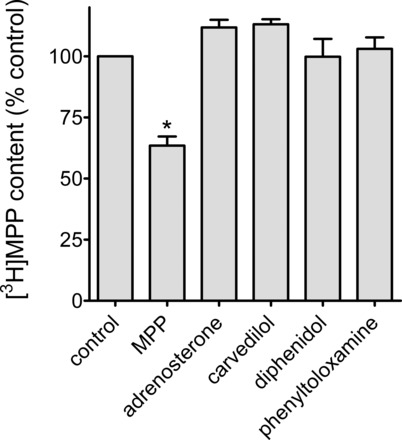
CCF as elicited by known OCT2 inhibitory ligands. Before measurement of CCF, cells were exposed to 12 nM [3H]MPP for 30 min. At time 0, the medium was rinsed and replaced with medium containing either 12 nM [3H]MPP (control) or 12 nM [3H]MPP plus the indicated compound (MPP, 100 μM; adrenosterone, 100 μM ; carvedilol, 225 μM; diphenidol, 70 μM ; phenyltoloxamine, 60 μM). The height of each bar represents the mean (±SE) of [3H]MPP remaining in cells after a 60-s efflux period, normalized to control content. Data shown are from 3–5 separate experiments (each run in triplicate). Bar marked with * (MPP) refers to the only compound that supported significant CCF (P < 0.05).
Chemical Assessment of OCT2-Mediated Metformin and Diphenidol Transport
The failure of phenyltoloxamine and diphenidol to support CCF was unexpected; both have structural characteristics commonly found in compounds shown to be substrates for OCT2 (small, mildly hydrophobic, and cationic at physiological pH). Indeed, as noted earlier, both were included in this study because they were explicitly listed as “cluster II-inhibitory ligands,” i.e., the class in which most OCT substrates are found (24). The failure to support CCF is, of course, a “negative result.” Therefore, it was desirable to get independent confirmation whether a “CCF-negative” ligand can, nevertheless, be a transported substrate. Unfortunately, neither compound is available in radiolabeled form, nor are they fluorescent. Consequently, we used direct chemical detection (using liquid chromatography with tandem mass spectroscopy) to determine the extent to which hOCT2 supports accumulation of diphenidol. To provide a “positive control,” accumulation of metformin, a known substrate of OCT2 that does support CCF (Fig. 7), was determined in parallel with the measurement of diphenidol accumulation. OCT2-expressing and wild-type CHO cells were exposed for 5 min to either metformin or diphenidol, in the presence or absence of 1 mM MPP. The concentration of each test substrate was set at its IC50 value, which should be sufficient to drive OCT2-mediated transport at ∼50% of its maximal value. As shown in Fig. 9, OCT2 supported a robust accumulation of metformin that was blocked by 1 mM MPP to a level equal to that measured in wild-type CHO cells. In contrast, there was no inhibitable, OCT2-specific accumulation of diphenidol.
Fig. 9.
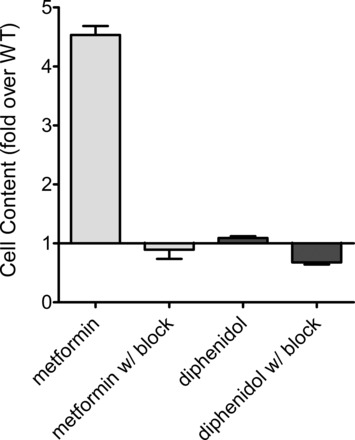
Uptake of metformin and diphenidol by OCT2 measured by liquid chromatography with tandem mass spectrometry. WT and hOCT2-expressing CHO cells were exposed for 5 min to buffers containing either metformin or diphenidol. The concentration of each approximated its IC50 value for inhibition of OCT2-mediated MPP transport (500 μM for metformin; 35 μM for diphenidol). In parallel, cells were exposed to these same substrate concentrations plus 1 mM MPP (100 times its Kt value). The height of each bar reflects the mean (±SE) accumulation of test substrate in OCT2-expressing cells, expressed relative to content measured in WT cells. The data presented are from 1 of 2 separate experiments, each measured in triplicate. Absolute WT accumulations in the 2 experiments were WT metformin, 35.2 ± 14.4 pmol/cm; WT metformin w/block, 27.0 ± 14.3 pmol/cm; WT diphenidol, 79.1 ± 14.5 pmol/cm; WT diphenidol w/block, 102 ± 15 pmol/cm.
DISCUSSION
Between 1964 and 1999, ∼8% of the drugs approved by the US Food and Drug Administration were subsequently withdrawn from the US market (38), underscoring the value of predicting unwanted DDIs as early as possible in the drug discovery process. Indeed, the influence of drug interactions at the level of membrane transport led regulatory agencies to issue guidance on conducting in vitro and clinical studies during drug development (30), which, in turn, has led to development of decision trees to determine whether a drug candidate may be a substrate or inhibitor of key transporters commonly reported to be involved in DDIs (14). The accuracy of such tools depends, ultimately, on understanding the underlying mechanism(s) of ligand interaction with the processes involved in drug transport.
Here, we assessed the mechanism of inhibitory interaction of selected ligands with the human ortholog of OCT2, the first step in renal secretion of many cationic drugs. In the absence of contrary evidence, it is frequently assumed, either explicitly (e.g., Refs. 2 and 50) or tacitly (e.g., Ref. 37), that the inhibition of OCT-mediated transport produced by small cations is competitive. However, our observations indicate that such assumptions are unwarranted; we found examples of three distinct kinetic mechanisms of ligand interaction with OCT2: competitive, noncompetitive, and mixed-type.
Metformin and cimetidine are cluster II substrates of hOCT2 (e.g., Refs. 50 and 56) and were, in fact, competitive inhibitors of hOCT2-mediated MPP transport (Fig. 4; Table 2). Indeed, one of the predictions of the subcluster analysis is that cluster II ligands should display “competitive binding” with hOCT2 (24). Despite the substantial attention given to studies of OCT selectivity in recent years, there are comparatively few other examples of kinetically competitive interactions of ligands with OCT2 as assessed by determining the effect of the test agent on Kt and Jmax of the transported probe. This criterion is important to establish because the other commonly applied test for competition between a transported substrate and an inhibitory ligand, the Dixon plot (1/J vs. [I]) (e.g., Ref. 29 and 55), while capable of discriminating between competitive and noncompetitive interactions, does not discriminate between competitive and certain types of mixed-type inhibitory interactions (47) (we expand on this in an upcoming section). With this caveat in mind, cimetidine and the antimalarial drug pyrimethamine have been shown to be competitive inhibitors of hOCT2-mediated metformin transport (21, 27); cimetidine has been shown to competitively inhibit hOCT2-mediated TEA transport (5); and NMN has been shown to competitively inhibit rOCT2-mediated TEA transport (3). Each of these individual interactions indicate that the ligand pairs in question share a common binding site or, alternatively, a set of mutually exclusive binding sites on a larger binding surface within the translocation pathway of OCT2.
The noncompetitive interactions seen here between adrenosterone and carvedilol and hOCT2-mediated MPP transport (Fig. 5; Table 2) were, arguably, consistent with their presence within structural subclusters identified by Kido et al. (24) as being effectively free of known transported substrates of OCT2. Adrenosterone, an endogenous steroid hormone that has been promoted as a dietary supplement capable of reducing body fat and increasing muscle mass (8), is a representative member of cluster III, which consists primarily of neutral sterols. It is relevant to note, therefore, that previous studies of corticosterone's inhibition of OCT2-mediated transport described behavior inconsistent with a competitive mechanism, including evidence of a noncompetitive mode of interaction (5, 52).
Carvedilol was also a noncompetitive inhibitor of hOCT2-mediated MPP transport. Carvedilol, a nonselective β blocker, is a cluster I-inhibitory ligand (24). These compounds are distinguished from the cluster II and cluster III ligands by their size (MW generally >400) and flexibility (as characterized by their asphericity and comparatively large geometrical radii of gyration and second-order principal static moments). Of 18 known transported substrates of OCT2, none falls into the cluster I category, leading to speculation that cluster I ligands may act by occluding the substrate binding site (24). That description, however, implies that cluster I compounds and OCT2 substrates may occupy mutually exclusive binding sites, a profile inconsistent with the noncompetitive profile observed for carvedilol.
The molecular basis of the noncompetitive inhibitory profiles observed here for adrenosterone and carvedilol is not clear. Adrenosterone is not charged, and carvedilol's pKa of ∼8 would result in a substantial fraction (∼25%) being uncharged at physiological pH. Consequently, it is conceivable these compounds could diffuse across the membrane and exert an inhibitory interaction at the cytoplasmic (trans) face of OCT2 that could not readily be displaced by simply increasing the extracellular (cis) concentration of MPP. Just such a scenario proved to be the basis of the apparent noncompetitive profile of quinine's inhibition of rOCT2-mediated TEA transport (3).
TEA is a prototypic cluster II substrate of hOCT2 (11, 16), so it was somewhat unexpected to find that, along with diphenidol and phenyltoloxamine, TEA's inhibition of hOCT2-mediated MPP transport displayed a mixed-type, rather than a competitive profile (Fig. 5; Table 2). As small, globular charged ligands, all three of these compounds would fall into cluster II, along with the other known substrates of OCT2 (24). As noted earlier, the cluster II ligands cimetidine and NMN interact competitively with OCT2-mediated TEA transport, consistent with the view that these three compounds interact at a common binding site or, as noted earlier, a set of mutually exclusive binding sites within a larger binding surface. Based on the observation that a site-directed mutation in rOCT1 (D475E) changed the kinetics of transport of some (e.g., TEA and choline), but not all (MPP) substrates, Gorboulev et al. (17) suggested “that rOCT1 contains a large cation-binding pocket with several interaction domains.” We subsequently found that a single site-directed replacement (E447L) altered the interaction of TEA and cimetidine with rbOCT2 but had no effect on the interaction MPP had with the transporter (54). These observations, along with others (see Ref. 26), support the view that (some) ligands can bind simultaneously to OCT2 at sites that are spatially distinct. When these sites overlap one another, competitive (mutually exclusive) interactions are expected, consistent with the MPP-metformin and MPP-cimetidine interactions noted here, and with the metformin-cimetidine, metformin-pyrimethamine, TEA-cimetidine, and TEA-NMN interactions noted previously (3, 5, 21, 27). However, ligand binding sites that are spatially distinct, but which exert an allosteric influence on one another, may display mixed-type inhibitor profiles of the type observed here for inhibition of hOCT2-mediated MPP transport by TEA, diphenidol, and phenyltoloxamine. Indeed, several studies have presented evidence supporting the presence of multiple binding sites for selected OCT ligands (18, 34).
Mixed-type inhibition of the type seen here can reflect several types of ligand interaction, but they all involve simultaneous binding of substrate and inhibitor (or second substrate). The simplest interaction is one in which 1) binding of the inhibitor results in a complex that has reduced (although finite) affinity for substrate, but 2) the resulting transporter/substrate/inhibitor complex does not support transport.1 The equilibria describing this type of interaction (linear mixed-type inhibition or LMT) (47) are shown below
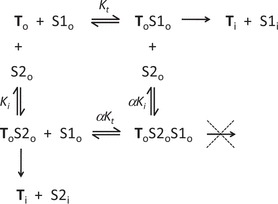 |
To and Ti indicate the transporter in its outwardly vs. inwardly facing conformations; S1 and S2 are two transported substrates (e.g., MPP and TEA); Kt and Ki are the Michaelis constants for interaction of S1 and S2, respectively, with the transporter; and α is the factor by which Kt (or Ki) changes (via allostery) when the other substrate binds to the transporter. In linear mixed-type inhibition, α > 1 < ∞ (if α = 1, inhibition is “noncompetitive”; if α = ∞, inhibition is “competitive”). Examination of this scheme reveals that during exposure to S1, if S2 is present, some of the transporter will be in the nontransporting ToS2oS1o form, even at infinitely high concentrations of S1, so Jmax-i will be less than Jmax. Also, any concentration of S2 will result in a fraction of the transporters available for interaction with S1 being in the lower affinity ToS2o form, resulting in an increase in apparent Kt. A key prediction of LMT inhibition is that increasing concentrations of S2 will result in complete inhibition of S1 transport; other schemes of mixed-type inhibition (e.g., hyperbolic mixed-type) or partial competitive inhibition are characterized by the inability of the inhibition ligand to completely block transport of S1 (47). Thus it is relevant to emphasize that concentrations of TEA, diphenidol, and phenyltoloxamine >20-times larger than their respective IC50 values reduced hOCT2-mediated [3H]MPP transport to levels not different from that produced by similar concentrations of unlabeled MPP. In other words, hOCT2 does not appear to support “cotransport” of two bound substrates; when two substrates bind, translocation of one requires dissociation of the other.
Our evidence suggests that ligand interaction with hOCT2 can involve multiple, nonoverlapping sites, which has important implications with respect to the development of predictive models of drug-drug interaction with OCT2 and its role as the entry step in renal OC secretion. Structure-activity relationships are typically based on the degree to which a structurally diverse array of test compounds inhibits biological activity (e.g., transport). However, if ligands can interact with multiple sites it introduces the potential for “substrate-dependent” inhibitory profiles. Therefore, the profile of inhibitory interactions with hOCT2 reported here using MPP as a substrate might be expected to differ markedly had another substrate (e.g., TEA, or ASP) been used. Consequently, models of the spatial arrangements of structural features required for ligand interaction with multidrug transporters, i.e., pharmacophores, are likely to vary depending on the substrate used. This has been shown to be the case for P-gp (e.g., Refs. 13 and 41), and we recently presented evidence suggesting this is true for MATE1 (4). The present study argues for a more concerted effort to evaluate the kinetic mechanism of ligand interaction with OCT2, and other multidrug transporters, in efforts to develop robust, predictive models of drug binding to this critical element in renal OC secretion.
The present results showed that possession of cluster II structural characteristics is insufficient to ensure that a ligand will have a competitive interaction with other cluster II compounds. Importantly, they also showed that possession of such characteristics is insufficient to support predictions concerning the extent to which the ligand is itself a substrate for transport. As noted by Kido et al. (24), known hOCT2 substrates are congregated in cluster II. Diphenidol and phenyltoloxamine were included in our study because of their designation as cluster II-inhibitory ligands of hOCT2 (24), and we were surprised to find that neither supported CCF. Adrenosterone and corticosterone also appear to be nontransported inhibitors of OCT2, but their cluster III characteristics are consistent with this observation. The antiretroviral drug nelfinavir is also a non-transported, high-affinity (IC50 of 13 μM) inhibitor of hOCT2-mediated MPP transport (22), but its large size (MW 567) and weak charge (pKa of <7) make it a poor fit for a cluster II designation. Diphenidol and phenyltoloxamine, in contrast, are models of the structural characteristics of cluster II (24). Their failure to support CCF was sufficiently unexpected that we used liquid chromatography with tandem mass spectroscopy to measure directly the effect of expression of hOCT2 on accumulation of diphenidol (Fig. 9), with the results supporting the same conclusion: diphenidol (and, by extension, phenyltoloxamine) is not a transported substrate of hOCT2, despite general structural features that would seem to predict that it be.
CCF offers an important advantage over the “trans-stimulation” strategy more commonly used to test indirectly whether a compound can serve as a transported substrate. The trans-assay as commonly applied involves preloading the cells with a suspected substrate and then determining whether the presence of this outwardly directed gradient stimulates uptake of a labeled substrate (the alternative approach is to preload the cells with the labeled substrate and then measure efflux of the label following exposure to the test agent in the extracellular solution). In either case, a trans-stimulation of flux, although not “proof” that the test compound is a substrate (it could be an “allosteric stimulator”), is generally considered strong supportive evidence that it is (e.g., Ref. 12). However, failure to observe a trans-effect is not sufficient to conclude that the test compound is a not a substrate. A uniporter like OCT2 can undergo the conformational changes associated with translocation either when occupied by substrate or when “empty” (thereby accounting for the documented electrogenicity of OCT-mediated OC transport) (9). Trans-stimulation only occurs if the “occupied” transporter undergoes these conformational changes more readily than the empty transporter. Although that may be “probable,” such behavior is not assured. CCF makes no such assumptions. If an extracellular test agent can be transported, it will induce a net loss of substrate if the substrate is distributed at steady state.
There is a limitation to the use of CCF as an indirect measure of transport: it cannot provide a precise measure of the rate of test agent transport (because the rapid increase in intracellular concentration of the inhibitory agent can be expected to begin to reduce the rate of labeled substrate efflux with kinetics that are ill defined). With this caveat, we suggest that CCF can provide a means to establish a high-throughput assay capable of collecting data required to develop a predictive model of structural features that permit the ligand/transporter complex to undergo the conformational changes that result in successful substrate translocation.
In summary, ligand interaction with hOCT2 involves a complex array of kinetic mechanisms, including competitive, noncompetitive, and mixed-type mechanisms that are not apparent in selectivity assays based on simple inhibition of transport activity. In addition, using CCF, several compounds predicted to be transported substrates of hOCT2 based on their inhibition of hOCT2 were shown to be nontransported inhibitors. CCF is a comparatively simple assay capable of determining whether an inhibitory ligand is also capable of serving as a transported substrate, providing a potential means for developing models of the structural determinants required to support effective transporter turnover.
GRANTS
This work was supported by National Institutes of Health (NIH) award 5R01DK58251 and NIH Grants 5P30E006694 and 5T32HL07249.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.N.H. performed experiments; J.N.H. and S.H.W. analyzed data; J.N.H. and S.H.W. interpreted results of experiments; J.N.H. and S.H.W. prepared figures; J.N.H. drafted manuscript; S.H.W. provided conception and design of research; S.H.W. edited and revised manuscript; S.H.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the technical contributions of Mark Morales and Alexandra Cooke, and useful discussions with Dr. William Dantzler.
Footnotes
The stipulation that the simultaneous binding of two ligands results in a nontransporting complex runs counter to the observation of Koepsell and colleagues (18, 34) that OCTs have high- and low-affinity binding sites for selected ligands and that transport may occur when both are occupied. We suggest, however, that the high-affinity site(s) characterized in these studies, which display KD values in the picomolar range, place them in a different kinetic category from the multiple sites in the present model. As noted by these authors (34), dissociation of ligands from these high-affinity sites can be expected to have half-times of many hours and, consequently, these sites are more likely to play a modulatory role to the transport process. The model outlined here suggests an interaction of ligands with multiple sites from which transport may occur.
REFERENCES
- 1. Ahlin G, Karlsson J, Pedersen JM, Gustavsson L, Larsson R, Matsson P, Norinder U, Bergstrom CA, Artursson P. Structural requirements for drug inhibition of the liver specific human organic cation transport protein. J Med Chem 51: 5932–5942, 2008 [DOI] [PubMed] [Google Scholar]
- 2. Amphoux A, Vialou V, Drescher E, Bruss M, La Cour CM, Rochat C, Millan MJ, Giros B, Bonisch H, Gautron S. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 50: 941–952, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Arndt P, Volk C, Gorboulev V, Budiman T, Popp C, Ulzheimer-Teuber I, Akhoundova A, Koppatz S, Bamberg E, Nagel G, Koepsell H. Interaction of cations, anions, and weak base quinine with rat renal cation transporter rOCT2 compared with rOCT1. Am J Physiol Renal Physiol 281: F454–F468, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Astorga B, Ekins S, Morales M, Wright SH. Molecular determinants of ligand selectivity for the human multidrug and toxin extrusion proteins, MATE1 and MATE-2K. J Pharmacol Exp Ther 341: 743–755, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barendt WM, Wright SH. The human organic cation transporter (hOCT2) recognizes the degree of substrate ionization. J Biol Chem 277: 22491–22496, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Bednarczyk D, Ekins S, Wikel JH, Wright SH. Influence of molecular structure on substrate binding to the human organic cation transporter, hOCT1. Mol Pharm 63: 489–498, 2003 [DOI] [PubMed] [Google Scholar]
- 7. Bednarczyk D, Mash EA, Aavula BR, Wright SH. NBD-TMA: a novel fluorescent substrate of the peritubular organic cation transporter of renal proximal tubules. Pflügers Arch 440: 184–192, 2000 [DOI] [PubMed] [Google Scholar]
- 8. Brooker L, Parr MK, Cawley A, Flenker U, Howe C, Kazlauskas R, Schanzer W, George A. Development of criteria for the detection of adrenosterone administration by gas chromatography-mass spectrometry and gas chromatography-combustion-isotope ratio mass spectrometry for doping control. Drug Test Anal 1: 587–595, 2009 [DOI] [PubMed] [Google Scholar]
- 9. Budiman T, Bamberg E, Koepsell H, Nagel G. Mechanism of electrogenic cation transport by the cloned organic cation transporter 2 from rat. J Biol Chem 275: 29413–29420, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Busch AE, Quester S, Ulzheimer JC, Waldegger S, Gorboulev V, Arndt P, Lang F, Koepsell H. Electrogenic properties and substrate specificity of the polyspecific rat cation transporter rOCT1. J Biol Chem 271: 32599–32604, 1996 [DOI] [PubMed] [Google Scholar]
- 11. Dresser MJ, Gray AT, Giacomini KM. Kinetic and selectivity differences between rodent, rabbit, and human organic cation transporters (OCT1). J Pharmacol Exp Ther 292: 1146–1152, 2000 [PubMed] [Google Scholar]
- 12. Dresser MJ, Xiao G, Leabman MK, Gray AT, Giacomini KM. Interactions of n-tetraalkylammonium compounds and biguanides with a human renal organic cation transporter (hOCT2). Pharm Res 19: 1244–1247, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Garrigues A, Loiseau N, Delaforge M, Ferte J, Garrigos M, Andre F, Orlowski S. Characterization of two pharmacophores on the multidrug transporter P-glycoprotein. Mol Pharmacol 62: 1288–1298, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Wah YS, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov 9: 215–236, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gorboulev V, Shatskaya N, Volk C, Koepsell H. Subtype-specific affinity for corticosterone of rat organic cation transporters rOCT1 and rOCT2 depends on three amino acids within the substrate binding region. Mol Pharmacol 67: 1612–1619, 2005 [DOI] [PubMed] [Google Scholar]
- 16. Gorboulev V, Ulzheimer JC, Akhoundova A, Ulzheimer-Teuber I, Karbach U, Quester S, Baumann C, Lang F, Busch AE, Koepsell H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol 16: 871–881, 1997 [DOI] [PubMed] [Google Scholar]
- 17. Gorboulev V, Volk C, Arndt P, Akhoundova A, Koepsell H. Selectivity of the polyspecific cation transporter rOCT1 is changed by mutation of aspartate 475 to glutamate. Mol Pharmacol 56: 1254–1261, 1999 [DOI] [PubMed] [Google Scholar]
- 18. Gorbunov D, Gorboulev V, Shatskaya N, Mueller T, Bamberg E, Friedrich T, Koepsell H. High-affinity cation binding to transporter OCT1 induces movement of helix 11 and blocks transport after mutations in a modelled interaction domain between two helices. Mol Pharmacol 73: 50–61, 2008 [DOI] [PubMed] [Google Scholar]
- 19. Gründemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature 372: 549–552, 1994 [DOI] [PubMed] [Google Scholar]
- 20. Hayer-Zillgen M, Bruss M, Bonisch H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br J Pharmacol 136: 829–836, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ito S, Kusuhara H, Yokochi M, Toyoshima J, Inoue K, Yuasa H, Sugiyama Y. Competitive inhibition of the luminal efflux by MATEs, but not basolateral uptake by OCT2, is the likely mechanism underlying the pharmacokinetic drug-drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther 340: 393–403, 2012 [DOI] [PubMed] [Google Scholar]
- 22. Jung N, Lehmann C, Rubbert A, Knispel M, Hartmann P, van Lunzen J, Stellbrink HJ, Faetkenheuer G, Taubert D. Relevance of the organic cation transporters 1 and 2 for antiretroviral therapy in human immunodeficiency virus infection. Drug Metab Dispos 36: 1616–1623, 2008 [DOI] [PubMed] [Google Scholar]
- 23. Kekuda R, Prasad PD, Wu X, Wang H, Fei YJ, Leibach FH, Ganapathy V. Cloning and functional characterization of a potential-sensitive, polyspecific organic cation transporter (OCT3) most abundantly expressed in placenta. J Biol Chem 273: 15971–15979, 1998 [DOI] [PubMed] [Google Scholar]
- 24. Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem 54: 4548–4558, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet 20: 379–386, 2005 [DOI] [PubMed] [Google Scholar]
- 26. Koepsell H. Substrate recognition and translocation by polyspecific organic cation transporters. Biol Chem 392: 95–101, 2011 [DOI] [PubMed] [Google Scholar]
- 27. Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, Inoue K, Yuasa H, Sugiyama Y. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther 89: 837–844, 2011 [DOI] [PubMed] [Google Scholar]
- 28. Lacko L, Burger M. Common carrier system for sugar transport in human red cells. Nature 191: 881–882, 1961 [DOI] [PubMed] [Google Scholar]
- 29. Lee WK, Reichold M, Edemir B, Ciarimboli G, Warth R, Koepsell H, Thevenod F. Organic cation transporters OCT1, 2, and 3 mediate high-affinity transport of the mutagenic vital dye ethidium in the kidney proximal tubule. Am J Physiol Renal Physiol 296: F1504–F1513, 2009 [DOI] [PubMed] [Google Scholar]
- 30. Lepist EI, Ray AS. Renal drug-drug interactions: what we have learned and where we are going. Expert Opin Drug Metab Toxicol 8: 433–448, 2012 [DOI] [PubMed] [Google Scholar]
- 31. Malo C, Berteloot A. Analysis of kinetic data in transport studies: new insights from kinetic studies of Na+-d-glucose cotransport in human intestinal brush-border membrane vesicles using a fast sampling, rapid filtration apparatus. J Membr Biol 122: 127–141, 1991 [DOI] [PubMed] [Google Scholar]
- 32. Marcelin-Jimenez G, Morales-Martinez M, Angeles-Moreno AP, Mendoza-Morales L. Ultra-fast chromatographic micro-assay for quantification of diphenidol in plasma: application in an oral multi-dose switchability trial. Biomed Chromatogr 22: 1143–1148, 2008 [DOI] [PubMed] [Google Scholar]
- 33. Ming X, Ju W, Wu H, Tidwell RR, Hall JE, Thakker DR. Transport of dicationic drugs pentamidine and furamidine by human organic cation transporters. Drug Metab Dispos 37: 424–430, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Minuesa G, Volk C, Molina-Arcas M, Gorboulev V, Erkizia I, Arndt P, Clotet B, Pastor-Anglada M, Koepsell H, Martinez-Picado J. Transport of lamivudine [(−)-beta-L-2′,3′-dideoxy-3′-thiacytidine] and high-affinity interaction of nucleoside reverse transcriptase inhibitors with human organic cation transporters 1, 2, and 3. J Pharmacol Exp Ther 329: 252–261, 2009 [DOI] [PubMed] [Google Scholar]
- 35. Moaddel R, Ravichandran S, Bighi F, Yamaguchi R, Wainer IW. Pharmacophore modelling of stereoselective binding to the human organic cation transporter (hOCT1). Br J Pharmacol 151: 1305–1314, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Motohashi h Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, Inui K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol 13: 866–874, 2002 [DOI] [PubMed] [Google Scholar]
- 37. Muller J, Lips KS, Metzner L, Neubert RH, Koepsell H, Brandsch M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem Pharmacol 70: 1851–1860, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Nassar AE, Talaat RE, Tokuno H. Drug interactions: concerns and current approaches. IDrugs 10: 47–52, 2007 [PubMed] [Google Scholar]
- 39. Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol 201: 105–167, 2011 [DOI] [PubMed] [Google Scholar]
- 40. Okuda M, Saito H, Urakami Y, Takano M, Inui KI. cDNA cloning and functional expression of a novel rat kidney organic cation transporter, OCT2. Biochem Biophys Res Commun 224: 500–507, 1996 [DOI] [PubMed] [Google Scholar]
- 41. Pascaud C, Garrigos M, Orlowski S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J 333: 351–358, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pelis RM, Dangprapai Y, Wunz TM, Wright SH. Inorganic mercury interacts with cysteine residues (C451 and C474) of hOCT2 to reduce its transport activity. Am J Physiol Renal Physiol 292: F1583–F1591, 2007 [DOI] [PubMed] [Google Scholar]
- 43. Pelis RM, Wright SH. Renal transport of organic anions and cations. Compr Physiol 1: 1795–1835, 2011 [DOI] [PubMed] [Google Scholar]
- 44. Rosenberg T, Wilbrandt W. Uphill transport induced by counterflow. J Gen Physiol 41: 289–296, 1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ross CR, Holohan PD. Transport of organic anions and cations in isolated renal plasma membranes. Annu Rev Pharmacol Toxicol 23: 65–85, 1983 [DOI] [PubMed] [Google Scholar]
- 46. Schomig E, Lazar A, Grundemann D. Extraneuronal monoamine transporter and organic cation transporters 1 and 2: a review of transport efficiency. Handb Exp Pharmacol 175: 151–180, 2006 [DOI] [PubMed] [Google Scholar]
- 47. Segel IH. Enzyme Kinetics. New York: Wiley, 1975, p. 161–192 [Google Scholar]
- 48. Stein WD. The Movement of Molecules Across Cell Membranes. New York: Academic, 1967, p. 143–148 [Google Scholar]
- 49. Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. Molecular determinants of substrate/inhibitor binding to the human and rabbit renal organic cation transporters, hOCT2 and rbOCT2. Mol Pharmacol 67: 1067–1077, 2005 [DOI] [PubMed] [Google Scholar]
- 50. Tahara H, Kusuhara H, Endou H, Koepsell H, Imaoka T, Fuse E, Sugiyama Y. A species difference in the transport activities of h2 receptor antagonists by rat and human renal organic anion and cation transporters. J Pharmacol Exp Ther 315: 337–345, 2005 [DOI] [PubMed] [Google Scholar]
- 51. Varma MV, Feng B, Obach RS, Troutman MD, Chupka J, Miller HR, El-Kattan A. Physicochemical determinants of human renal clearance. J Med Chem 52: 4844–4852, 2009 [DOI] [PubMed] [Google Scholar]
- 52. Volk C, Gorboulev V, Budiman T, Nagel G, Koepsell H. Different affinities of inhibitors to the outwardly and inwardly directed substrate binding site of organic cation transporter 2. Mol Pharmacol 64: 1037–1047, 2003 [DOI] [PubMed] [Google Scholar]
- 53. Wright SH, Wunz TM, Wunz TP. Structure and interaction of inhibitors with the TEA/H+ exchanger of rabbit renal brush border membranes. Pflügers Arch 429: 313–324, 1995 [DOI] [PubMed] [Google Scholar]
- 54. Zhang X, Shirahatti NV, Mahadevan D, Wright SH. A conserved glutamate residue in transmembrane helix 10 influences substrate specificity of rabbit OCT2 (SLC22A2). J Biol Chem 280: 34813–34822, 2005 [DOI] [PubMed] [Google Scholar]
- 55. Zolk O, Solbach TF, Konig J, Fromm MF. Functional characterization of the human organic cation transporter 2 variant p.270Ala>Ser. Drug Metab Dispos 37: 1312–1318, 2009 [DOI] [PubMed] [Google Scholar]
- 56. Zolk O, Solbach TF, Konig J, Fromm MF. Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379: 337–348, 2009 [DOI] [PubMed] [Google Scholar]