

Abstract
Myeloproliferative neoplasms are a varied group of disorders that can have prolonged chronic phases, but eventually accelerate and can transform into a secondary acute myeloid leukemia that is ultimately fatal. Triapine is a novel inhibitor of the M2 subunit of ribonucleotide reductase. Sequential inhibition of ribonucleotide reductase with triapine and an M1 ribonucleotide reductase inhibitor (fludarabine) was noted to be safe, and led to a 29% complete plus partial response rate in myeloproliferative neoplasms. This article reports the findings of a phase II trial of triapine (105 mg/m2/day) followed by fludarabine (30 mg/m2/day) daily for 5 consecutive days in 37 patients with accelerated myeloproliferative neoplasms and secondary acute myeloid leukemia. The overall response rate was 49% (18/37), with a complete remission rate of 24% (9/37). Overall response rates and complete remissions were seen in all disease subsets, including secondary acute myeloid leukemia, in which the overall response rate and complete remission rate were 48% and 33%, respectively. All patients with known JAK2 V617F mutations (6/6) responded. The median overall survival of the entire cohort was 6.9 months, with a median overall survival of both overall responders and complete responders of 10.6 months. These data further demonstrate the promise of sequential inhibition of ribonucleotide reductase in patients with accelerated myeloproliferative neoplasms and secondary acute myeloid leukemia. This study was registered with clinicaltrials.gov (NCT00381550).
Introduction
Myeloproliferative neoplasms (MPN) are a diverse group of disorders that include polycythemia vera, essential thrombocythemia, chronic myeloid leukemia (CML), and primary myelofibrosis, while chronic myelomonocytic leukemia (CMML) and atypical (Philadelphia-negative) CML are classified as overlap syndromes of MPN and myelodysplastic syndrome. There are approximately 23,000 new cases of MPN (including CMML) per year in the United States; these diseases lead to significant amounts of morbidity and mortality.1,2 While MPN may have protracted chronic phases, they often accelerate and many eventually transform into an ultimately fatal acute leukemia. Patients with MPN in accelerated phase, CML in blast crisis (CML-BC), and secondary acute myeloid leukemia (AML) in transformation have a dismal prognosis. In a large retrospective analysis, patients with secondary AML transformed from myelofibrosis were shown to have a median overall survival of <3 months.3 Allogeneic stem cell transplantation (SCT) benefits selected patients; however, the use of allogeneic SCT is limited by the older age of MPN patients at presentation, increased rates of transplant-related mortality, and unknown optimal timing.4–6 Thus, development of novel, safe and effective strategies in aggressive MPN continues to be a pressing need.
Ribonucleotide reductase is an enzyme that converts ribonucleotides into deoxyribonucleotides and is, therefore, a pivotal enzyme in the processes of DNA synthesis, DNA damage repair, and cellular growth and survival. Inhibition of ribonucleotide reductase depletes the intracellular pools of deoxyribonucleotides and their triphosphorylated forms (dNTP). Ribonucleotide reductase is composed of two subunits: a regulatory (M1) subunit, which is inhibited by nucleoside analogs, and a catalytic (M2) subunit, which requires iron and generation of a tyrosyl free radical for activity.7 Hydroxyurea effectively inhibits the M2 subunit, and has been used clinically for decades in patients with AML, CML, CMML and other MPN.8–12 Triapine, a novel thiosemicarbazone derivative, is an extremely potent inhibitor of the M2 subunit of ribonucleotide reductase (100–1000 fold more potent than hydroxyurea), and has been studied as a single agent in hematologic malignancies with demonstrable safety and clinical activity.13–17
Multiple studies have shown that inhibition of either the M1 subunit (via fludarabine, gemcitabine, or clofarabine) or M2 subunit (via hydroxyurea) of ribonucleotide reductase can enhance the anti-tumor activity of subsequently administered nucleoside analogs.16,18–22 Fludarabine, a nucleoside analog, has demonstrated potent activity in lymphoid malignancies.23 As a single agent, fludarabine exhibits activity in AML at very high doses, but is limited by significant neurotoxicity.24,25 Nonetheless, fludarabine can interact synergistically with multiple cytotoxic agents, partly due to its role in inhibiting the ribonucleotide reductase M1 subunit.26 Clinical trials in refractory AML combining fludarabine and cytarabine in a sequential fashion demonstrated clinical responses in adults and children, and revealed the potentiation of cytarabine cytotoxicity ex vivo.27–30
Preclinical studies demonstrated that triapine administration was associated with decreases in intracellular circulating leukemic blast dNTP pools with subsequent inhibition of DNA synthesis.16 On the basis of these data, we conducted a phase I clinical trial of triapine followed by escalating doses of fludarabine in adults with relapsed and refractory acute leukemias, high-risk myelodysplastic syndrome, or transformed MPN including CMML.31 We tested two schedules: schedule A, consisting of daily doses of triapine at 105 mg/m2 over 4 h for 5 days followed by a 30-min infusion of fludarabine within 1 h of completion of triapine, and schedule B, consisting of triapine at 200 mg/m2 over 24 h followed by fludarabine for 5 days. There were no dose-limiting toxicities in this study; however, the dose of fludarabine (30 mg/m2 daily) was not further escalated due to concern about neurotoxicity. Schedule A (triapine 105 mg/m2 daily) led to a 21% complete remission + partial remission rate, with the majority of responses being in patients with underlying MPN (29% response rate in MPN). In contrast, schedule B (triapine 200 mg/m2 over 24 h) did not lead to any clinical responses.
Given the promising results of schedule A, we designed and conducted a phase II study of triapine 105 mg/m2 daily followed by fludarabine 30 mg/m2 daily for 5 days in 37 patients with aggressive MPN and secondary, transformed AML. We confirm our previous findings that this combination of agents is clinically active in accelerated MPN and secondary AML derived from MPNs, and further identify the JAK2 V617F mutation as a potential predictor of response to sequential ribonucleotide reductase inhibition.
Methods
Patients
Between August 2006 and November 2010, 37 adults with pathological confirmation of accelerated MPN (>5% blasts in bone marrow, new onset/worsening myelofibrosis, new onset or >25% increase in hepatomegaly/splenomegaly, or new onset/worsening constitutional symptoms), CML-BC, CMML (>5% blasts), or secondary AML, arising from a preexisting MPN or CMML, defined by standard criteria,32 were entered on study at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center. This study was approved by the Johns Hopkins Medical Institutional Review Board.
Treatment plan
Triapine was administered as a 4-h intravenous infusion daily for 5 consecutive days at a dose of 105 mg/m2/day. Fludarabine 30 mg/m2/day was administered as a 30-min intravenous infusion daily, beginning within 1 h of completion of the triapine infusion, on each of the 5 consecutive days. Each cycle was 21 days in duration. Patients were eligible to receive additional cycles of treatment until disease progression or toxicity.
Measurement of toxicities
Non-hematologic toxicities were graded according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 3.0. Treatment of triapine was interrupted for grade 3 non-hematologic toxicities for up to 48 h. All subsequent doses were withheld until toxicity resolved to grade ≤1 (or baseline).
Measurement of response
Bone marrow aspiration and biopsies were performed prior to treatment, during week 3 of the first cycle, at the time of hematologic recovery from all cycles of therapy (defined as neutrophil count >500/mm3 and platelets >20,000/mm3 independently of transfusion), or at any time that leukemia regrowth was suspected. The overall response rate was defined as complete remission, partial remission, or hematologic improvement, lasting for ≥30 days. Given the different subsets of diseases, standardized response criteria were used for CMML (the Myelodysplastic Syndrome International Working Group criteria),33 CMML transforming to acute myeloid leukemia (standard AML response criteria)34, accelerated MPN (Giles et al.)35, and transformation of MPN to secondary AML (Mascarenhas et al.).36
Statistical considerations
The primary objectives of this study were to determine the efficacy and toxicity of triapine followed by fludarabine in patients with aggressive MPN. The study was designed to detect an improvement in overall response rate from 10–30% with a two-sided type 1 error rate of 5% and 90% power using a two-stage approach. A stopping requirement was placed such that the study would terminate if fewer than three out of 18 patients had a response. An additional 24 patients were planned but the study was completed after a total of 37 patients had been enrolled because of lack of drug availability.
Overall survival was calculated from the first day of treatment to death or last known follow-up and estimated using the Kaplan-Meier method. A landmark analysis37 was performed to compare response groups using day 42 of treatment as the landmark time. Overall survival was compared between groups of patients using Cox proportional hazard models, adjusting for age and gender. The database was locked on June 1, 2013. Analyses were completed using statistical freeware R version 3.0.1.38
Results
Patients’ characteristics
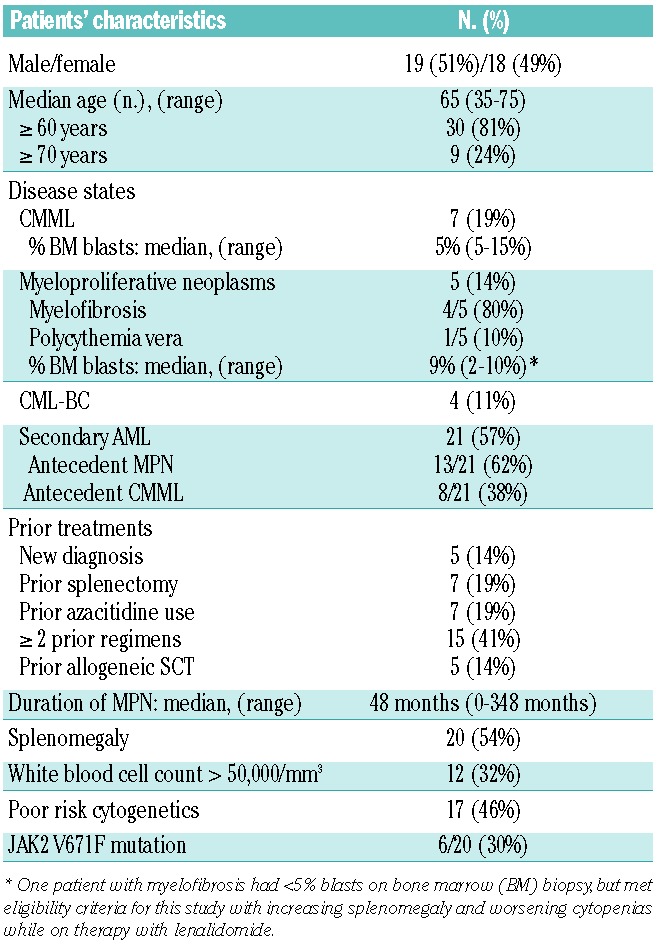
The pretreatment clinical demographics of the 37 patients enrolled in this trial are outlined in Table 1. The median age of the patients enrolled was 65 years, with the vast majority being ≥60 years (81%). Seven (19%) patients had CMML, five (14%) patients had MPN (myelofibrosis: n=4, all of whom were high-risk according to the Dynamic International Prognostic Scoring Scale-plus;39 polycythemia vera: n=1, resistant to hydroxyurea with 10% bone marrow blasts), four (11%) patients had CML-BC, and 21 (57%) patients had secondary AML, either derived from an antecedent MPN (13/21), or from an antecedent CMML (8/21). The median duration of MPN prior to study enrollment was 48 months. The majority (86%) of the patients had received prior treatment courses before entering the study, and 15 patients (41%) had been treated with two or more prior regimens. Seven (19%) patients had received azacitidine prior to entering the study, with six of these seven patients having secondary AML. Twenty (54%) patients had splenomegaly (as measured by physical examination and/or radiological imaging), while 17 (46%) patients had poor-risk cytogenetics. JAK2 V617F mutational analysis was performed on 20/37 (54%) patients, with 6/20 (30%) patients having a detectable JAK2 V617F mutation.
Table 1.
Patients’ demographics.
Toxicity
A total of 100 cycles of triapine and fludarabine were administered to 37 patients, with a median of three cycles per patient (range, 1–6). Nine out of 37 (24%) patients received four or more cycles of therapy. Early death (prior to day 30) occurred in 4/37 (11%) patients (secondary AML: n=2, CML: n=2) due to sepsis (n=2), multi-organ failure (n=1), or cardiogenic shock (n=1).
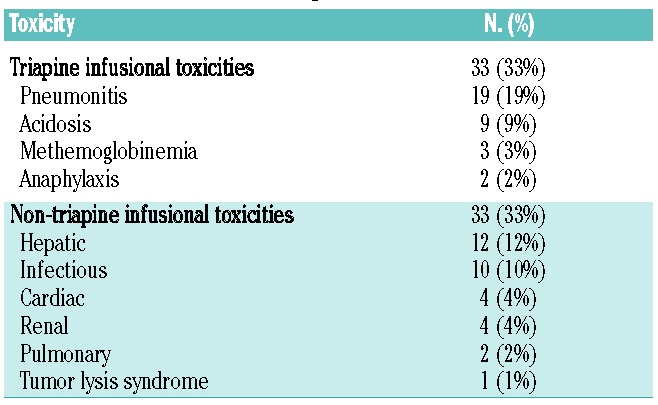
As presented in Table 2, there were six cases of ≥ grade 4 toxicities: two cases of grade 4 acidosis, one case of grade 4 acute kidney injury, one case of grade 5 acute respiratory distress syndrome and two cases of grade 5 sepsis. Overall, in 100 cycles of treatment, 66 grade ≥3 toxicities were seen, of which 33 (50%) were transiently related to triapine infusion. These toxicities manifested as pneumonitis (19%: defined as hypoxia and/or dyspnea directly related to the triapine infusion), acidosis (9%), methemoglobinemia ≥15% (3%), and anaphylaxis (2%), and were reversed rapidly with supportive care without recurrence. There were three cases of clinical methemoglobinemia ≥15% (range, 15–16.2%) while on this study, two patients required methylene blue for severe symptoms. The most common non-triapine infusional toxicities were hepatic (12%) and infectious (10%). Ten (27%) patients required dose reductions of triapine, predominantly due to infusion-related toxicities of triapine. No patient required more than one dose reduction of triapine. An additional nine (24%) patients required dose reductions of fludarabine because of transient decreases in creatinine clearance to <60 mL/min.
Table 2.
Grade ≥3 non-hematologic toxicities.
Clinical outcomes
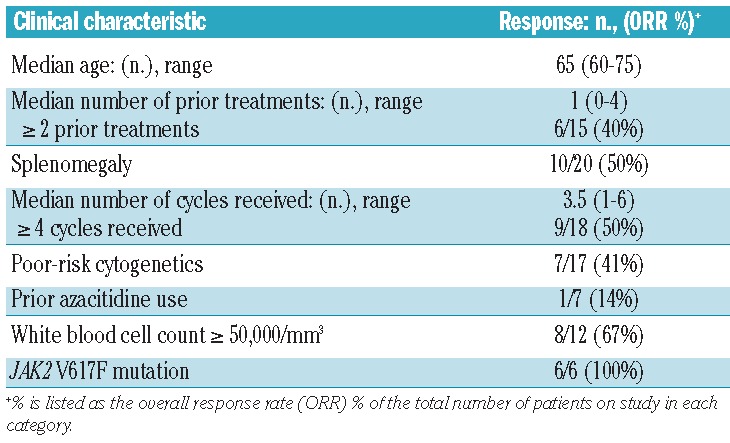
The overall response rate to triapine and fludarabine in this population was 49% (18/37). In the first stage, nine of 18 patients achieved a response, with nine of the remaining 19 patients achieving a response in the second stage. Overall, nine patients achieved a complete remission (24%) (including one patient with complete remission with incomplete platelet recovery), four patients achieved a partial remission (11%), and five patients achieved hematologic improvement (14%). An additional five patients had stable disease for at least one cycle. The overall response rate for each disease subset was: CMML 57% (4/7), MPN 60% (3/5), CML-BC 25% (1/4), and secondary AML 48% (10/21) (Table 3). Of the nine patients achieving a complete remission, seven had secondary AML (antecedent MPN: n=5, antecedent CMML: n=2), one had CML-BC (and achieved a complete cytogenetic response) and one had CMML. The patients with secondary AML derived from MPN who achieved a complete remission (n=5/13) were further sub-classified based on recent response criteria defined by Mascarenhas et al.36 Four out of these five patients who had achieved a complete remission had reverted back to a chronic MPN state (response defined as acute leukemia response-complete) with continued fibrosis seen on subsequent bone marrow biopsies, while one had a complete remission with complete molecular response, and no evidence of a chronic MPN state by morphology. The overall response rates were further broken down according to specific clinical risk factors (Table 4). The median age of responders was 65 years. Of note, the two oldest patients in the study (both with secondary AML) were 75 years old and both achieved complete remission. Fifty percent of patients with splenomegaly had some type of response to therapy, with eight of the ten responding patients also having improvements in both symptoms and size of splenomegaly (as measured by physical examination and/or radiological findings). Additionally, 41% (7/17) of patients with poor-risk cytogenetics (all of whom had either complex and/or monosomal karyotype) responded, with two of the seven patients having complete cytogenetic clearance, and one obtaining partial cytogenetic clearance of a complex karyotype. Only one of seven patients who received prior azacitidine therapy had a response (a patient with CMML who had a hematologic improvement).
Table 3.
Response rates by disease state.
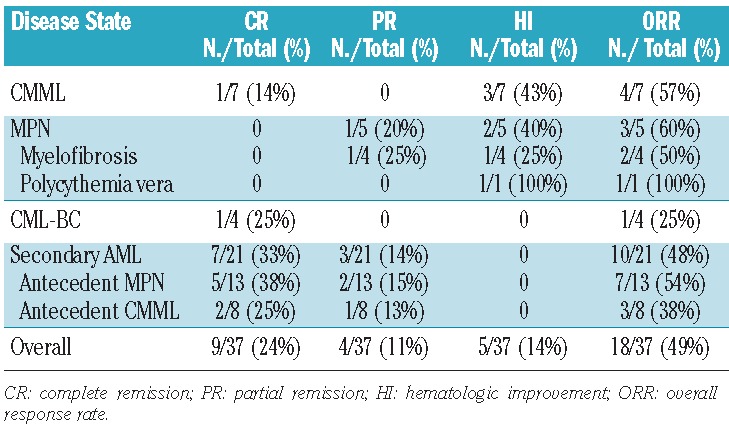
Table 4.
Characteristics of the 18/37 responders.
All six patients with a JAK2 V617F mutation had a response, including three (50%) who achieved a complete remission. In comparison, the overall response rate in patients with wild-type JAK2 was 29% (4/14 patients, P=0.01), with an overall complete remission rate of 14% (2/14 patients, P=0.13). Each of the three patients with JAK2 V617F mutations achieving a complete remission had secondary AML arising from MPN (myelofibrosis: n=2, essential thrombocythemia: n=1). The two patients who achieved a complete remission with a known wild-type JAK2 had secondary AML (from antecedent CMML: n=1) and CML-BC (n=1).
The median overall survival of the study population was 6.9 months (95% CI: 4.3–10.7 months), with the 6-month and 12-month overall survival rates being 54% and 24%, respectively. The median overall survival of responders (patients achieving complete or partial remission or having a hematologic improvement) and complete responders was 10.6 months (95% CI: 8.7–15.8 2 months) and 10.6 months (95% CI: 8.7–21.5 months), respectively. Patients with a known JAK2 V617F mutation had a median overall survival of 10.5 months (95% CI: 5.0–41.5 months) compared with 5.6 months (95% CI: 2.7–30.4 months) in patients without a JAK2 V617F mutation (adjusted P=0.17). The median duration of first response to relapse or death was 121 days (range, 52–422 days). Figure 1 illustrates the overall survival curves for the entire cohort (Figure 1A), and landmark overall survival comparisons between responders and non-responders (Figure 1B,C).
Figure 1.
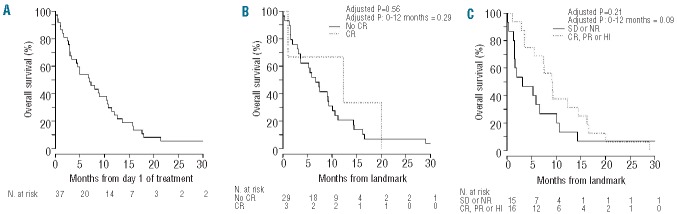
Kaplan-Meier overall survival curves for (A) the entire study population, (B) patients who achieved complete remission (CR) versus those who did not, and (C) responders versus non-responders. Comparisons between response groups (B–C) are based on a landmark analysis, where overall survival was measured from day 42 of treatment until death or last known follow-up, and response group was based on patients’ status on day 42. Patients who died before day 42 (n=5) were excluded from these analyses. P values are from Cox proportional hazards models that adjusted for patients’ age and gender. CR: complete remission; PR: partial remission; HI: hematologic improvement; SD: stable disease; NR: no response.
Five patients received an allogeneic SCT after therapy with triapine and fludarabine (CMML: n=2, CML: n=2, secondary AML: n=1). The median age of patients receiving an allogeneic SCT was 60 years (range, 43–68). Three patients (CML: n=2, CMML: n=1) had myeloablative conditioning and two patients (secondary AML: n=1, CML: n=1) had non-myeloablative conditioning. Two of the five patients (CMML: n=1, CML: n=1) who underwent an allogeneic SCT had a complete response to triapine and fludarabine, two patients (CMML: n=1, CML-BC: n=1) had refractory disease on study and then received additional treatment prior to allogeneic SCT, and one patient with secondary AML had stable disease prior to allogeneic SCT. Three patients (CML: n=2, CMML: n=1) died of complications of the allogeneic SCT (infection: n=2, hepatic sinusoidal obstructive syndrome: n=1), including the two patients who achieved a complete remission, and two patients relapsed after the transplant. One patient with secondary AML (arising from CMML) achieved stable disease on study and then had a non-myeloablative haploidentical allogeneic SCT (5 months after being enrolling into the study), with a subsequent relapse 17 months after the transplant, and is alive in complete remission for >15 months after re-induction cytotoxic therapy and donor lymphocyte infusion.
Discussion
MPN continue to be a major therapeutic challenge, especially once they undergo aggressive transformation. This phase II study involving triapine, a novel ribonucleotide reductase M2 inhibitor, combined with fludarabine (an M1 ribonucleotide reductase inhibitor), extends our initial observations that this regimen has clinical activity in aggressive/accelerated phase MPN including transformations into AML. This study met its primary end-point of an overall response rate >30% (49%). The complete remission rate of 24% in this heavily pretreated population of patients is promising. Patients with secondary AML had an overall response rate and complete remission rate of 48% and 33%, respectively, which is particularly encouraging in these patients who have a dismal prognosis. There did not seem to be an upper age limit of responders, as patients responded to therapy up to the age of 75 years.
The encouraging response rates (n=6/6) in patients with JAK2 V617F mutations are of particular interest, and reached statistical significance (P=0.01) when compared to those in JAK2 wild-type patients (overall response rate: 100% versus 29%). The three patients who achieved a complete remission with JAK2 V617F mutations had secondary AML derived from a JAK2 V617F MPN clone. The results of triapine and fludarabine in MPN (including transformations to AML) with JAK2 V617F mutations are intriguing; however, given the small number of patients in these analyses, larger scale trials will need to be performed to confirm these suggestions.
Landmark analyses comparing responders to non-responders by day 42 of treatment did not reveal significant differences in overall survival. This landmark analysis does, however, have its limitations. The time point of 42 days was chosen prior to study accrual as the time at which an initial response rate should be noted for each patient in the study. However, seven of nine complete responders achieved a complete remission after day 42 of treatment (range, 21–96 days) and these seven patients were thus not listed among those with a complete remission for statistical comparisons using the landmark analysis. Additionally, if a patient who had a partial remission or hematologic improvement before day 42 went on to achieve a complete remission after day 42, this response was not included in the specific comparison of complete remission versus no complete remission for the landmark statistical comparisons.
Nonetheless, the clinical responses were short-lived, with the median duration of response being 4 months, emphasizing the high degree of chemoresistance seen in accelerated MPN. Moreover, the median overall survival of 6.9 months underscores the extremely poor prognosis of the patients in this study. Because of the short duration of response, this combination of agents may work best as a bridge to further therapy, such as allogeneic SCT. However, only five patients were able to receive an allogeneic SCT after treatment with these ribonucleotide reductase inhibitors (including 4/5 who died), which may reflect the older age of patients in the study (median age: 64 years). Three of the five patients who were able to undergo allogeneic SCT were given myeloablative preparative regimens. As allogeneic SCT techniques become more refined, it may be possible to select patients who could benefit from a non-myeloablative preparative regimen compared to a myeloablative one.
The combination of triapine and fludarabine led to unique toxicities, most notably acute pulmonary toxicities, such as methemoglobinemia and metabolic acidosis. These toxicities are distinctively related to the triapine infusion. Although these toxicities were not inconsequential, they often resolved promptly without intervention and without subsequent organ dysfunction. Upon dose reduction of triapine in ten patients, there were no additional grade ≥3 toxicities.
Contemporaneous inhibition of the M1 and M2 subunits of ribonucleotide reductase is a concept worthy of further exploration in patients with accelerated MPN. Although triapine is 100–1000 fold more potent than hydroxyurea, the therapeutic efficacy of triapine and hydroxyurea has never been compared. It would be rational to further explore hydroxyurea in combination with other cytotoxic agents and M1 ribonucleotide reductase inhibitors (such as fludarabine, gemcitabine and clofarabine) for synergistic ribonucleotide reductase inhibition. This approach may be better tolerated than the combination of triapine and fludarabine. Given the epigenetic dysregulation seen in patients with accelerated MPN (with or without transformation to AML), additional treatment approaches for these patients include hypomethylating agents, such as azacitidine.40 Thepot et al. evaluated the use of azacitidine in 54 patients with AML or myelodysplastic syndrome secondary to MPN, finding an overall response rate and complete remission rate of 52% and 24%, respectively, with a median overall survival of 11 months.41 However, in AML patients, the complete remission rate (including complete remission with incomplete count recovery) was only 11%. As in our study, the duration of response was short (9 months); nonetheless azacitidine still represents another promising treatment strategy for patients in this high-risk group, either as a bridge to an allogeneic SCT or for symptomatic improvement. Unfortunately, the response was poor in the subset of patients in this study who received prior azacitidine therapy (1/7 overall response rate). The majority of these patients (6/7) had secondary AML, and it is possible that prior azacitidine use changes the biology of the disease to a more aggressive, treatment-resistant disease, although the small numbers of patients in this study preclude any significant generalizations or conclusions.
Since this study was conceived and executed, novel JAK2 inhibitors, such as ruxolitinib, have emerged as integral parts of the therapeutic armamentarium for MPN.42 Given the encouraging results of this study, simultaneous inhibition of the M1 and M2 subunits of ribonucleotide reductase should be investigated with JAK2 inhibitors, either in combination or in sequence. Hydroxyurea was shown in one study to increase STAT-5 phosphorylation in a dose-dependent manner, thus leading to activation of the STAT-JAK pathway.43 Combining lower doses of hydroxyurea with a JAK2 inhibitor would theoretically overcome this potentially deleterious activation. There are, however, other reports suggesting that hydroxyurea has a preferential effect in patients with JAK2 V617F mutations with a high allele burden.44 Given the high response rates seen in the study in patients with JAK2 V617F mutations, it would be worth exploring whether M2 ribonucleotide reductase inhibitors or combination ribonucleotide reductase inhibition has any significant impact on JAK2 allele burden. JAK2 inhibitors, such as ruxolitinib, seem to be clinically effective in myelofibrosis patients with or without JAK2 V617F mutations and it is unclear whether these agents work by JAK1 or JAK2 inhibition, with or without modulation of inflammatory cytokines.45 Ruxolitinib also has demonstrated activity as a single agent in patients with secondary AML (transformed from antecedent MPN), and is being studied in combination with other active agents in MPN.46 Future studies should, therefore, investigate combination therapy with ribonucleotide reductase inhibitors and JAK2 inhibitors in patients with accelerated MPN states and secondary AML.
In summary, sequential ribonucleotide reductase inhibition with an M2 inhibitor (triapine) followed by an M1 inhibitor (fludarabine) led to an overall response rate of 49% in patients with accelerated MPN, CML, CMML and secondary AML. Responses were seen in all disease states, including secondary AML, and were particularly notable in patients with JAK2 V617F mutations. The responses were, however, temporary, which underscores the need for further investigative trials in this population of patients. Future studies combining ribonucleotide reductase inhibitors, with or without the addition of JAK2 inhibitors, should be explored in patients with secondary AML and high-risk accelerated MPN.
Acknowledgments
The authors would like to thank the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center nursing and research staff for the superb medical care, and the patients and their families, without whose partnership we could never have conducted the trial and from whom we have learned critical information that will help us to improve the treatment of these diseases. The authors would also like to acknowledge the instrumental role that Dr. Igor Espinoza-Delgado had in developing this study.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This trial was supported in part by NCI Cooperative Agreement U01 CA70095 and NCI Cancer Center Support Grant 2P30 CA06973-46.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.[cited; Available from: http://www.lls.org/diseaseinformation/
- 2.Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22(7):1308–19 [DOI] [PubMed] [Google Scholar]
- 3.Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–7 [DOI] [PubMed] [Google Scholar]
- 4.Merup M, Lazarevic V, Nahi H, Andreasson B, Malm C, Nilsson L, et al. Different outcome of allogeneic transplantation in myelofibrosis using conventional or reduced-intensity conditioning regimens. Br J Haematol. 2006;135(3):367–73 [DOI] [PubMed] [Google Scholar]
- 5.Guardiola P, Esperou H, Cazals-Hatem D, Ifrah N, Jouet JP, Buzyn A, et al. Allogeneic bone marrow transplantation for agnogenic myeloid metaplasia. French Society of Bone Marrow Transplantation. Br J Haematol. 1997;98(4):1004–9 [DOI] [PubMed] [Google Scholar]
- 6.Przepiorka D, Giralt S, Khouri I, Champlin R, Bueso-Ramos C. Allogeneic marrow transplantation for myeloproliferative disorders other than chronic myelogenous leukemia: review of forty cases. Am J Hematol. 1998;57(1):24–8 [DOI] [PubMed] [Google Scholar]
- 7.Eklund H, Uhlin U, Farnegardh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol. 2001;77(3):177–268 [DOI] [PubMed] [Google Scholar]
- 8.Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332(17):1132–6 [DOI] [PubMed] [Google Scholar]
- 9.Hehlmann R, Heimpel H, Hasford J, Kolb HJ, Pralle H, Hossfeld DK, et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. The German CML Study Group. Blood. 1993;82(2):398–407 [PubMed] [Google Scholar]
- 10.Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100(13):4272–90 [DOI] [PubMed] [Google Scholar]
- 11.Wattel E, Guerci A, Hecquet B, Economopoulos T, Copplestone A, Mahe B, et al. A randomized trial of hydroxyurea versus VP16 in adult chronic myelomonocytic leukemia. Groupe Francais des Myelodysplasies and European CMML Group. Blood. 1996;88(7):2480–7 [PubMed] [Google Scholar]
- 12.Wright JA, Chan AK, Choy BK, Hurta RA, McClarty GA, Tagger AY. Regulation and drug resistance mechanisms of mammalian ribonucleotide reductase, and the significance to DNA synthesis. Biochem Cell Biol. 1990;68(12):1364–71 [DOI] [PubMed] [Google Scholar]
- 13.Cory JG, Cory AH, Rappa G, Lorico A, Liu MC, Lin TS, et al. Inhibitors of ribonucleotide reductase. Comparative effects of amino- and hydroxy-substituted pyridine-2-carboxaldehyde thiosemicarbazones. Biochem Pharmacol. 1994;48(2):335–44 [DOI] [PubMed] [Google Scholar]
- 14.Liu MC, Lin TS, Cory JG, Cory AH, Sartorelli AC. Synthesis and biological activity of 3- and 5-amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem. 1996;39(13):2586–93 [DOI] [PubMed] [Google Scholar]
- 15.Moore EC, Booth BA, Sartorelli AC. Inhibition of deoxyribonucleotide synthesis by pyridine carboxaldehyde thiosemicarbazones. Cancer Res. 1971;31(3):235–8 [PubMed] [Google Scholar]
- 16.Giles FJ, Fracasso PM, Kantarjian HM, Cortes JE, Brown RA, Verstovsek S, et al. Phase I and pharmacodynamic study of triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk Res. 2003;27(12):1077–83 [DOI] [PubMed] [Google Scholar]
- 17.Gojo I, Tidwell ML, Greer J, Takebe N, Seiter K, Pochron MF, et al. Phase I and pharmacokinetic study of triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk Res. 2007;31(9):1165–73 [DOI] [PubMed] [Google Scholar]
- 18.Bhalla K, Swerdlow P, Grant S. Effects of thymidine and hydroxyurea on the metabolism and cytotoxicity of 1-B-D arabinofuranosylcytosine in highly resistant human leukemia cells. Blood. 1991;78(11):2937–44 [PubMed] [Google Scholar]
- 19.Iwasaki H, Huang P, Keating MJ, Plunkett W. Differential incorporation of ara-C, gemcitabine, and fludarabine into replicating and repairing DNA in proliferating human leukemia cells. Blood. 1997;90(1):270–8 [PubMed] [Google Scholar]
- 20.Rauscher F, 3rd, Cadman E. Biochemical and cytokinetic modulation of L1210 and HL-60 cells by hydroxyurea and effect on 1-beta-D-arabinofuranosylcytosine metabolism and cytotoxicity. Cancer Res. 1983;43(6):2688–93 [PubMed] [Google Scholar]
- 21.Tanaka M, Kimura K, Yoshida S. Mechanism of synergistic cell killing by hydroxyurea and cytosine arabinoside. Jpn J Cancer Res. 1985;76(8):729–35 [PubMed] [Google Scholar]
- 22.Zhou B, Mi S, Mo X, Shih J, Tsai J, Hu E, et al. Time and sequence dependence of hydroxyurea in combination with gemcitabine in human KB cells. Anticancer Res. 2002;22(3):1369–77 [PubMed] [Google Scholar]
- 23.Chun HG, Leyland-Jones B, Cheson BD. Fludarabine phosphate: a synthetic purine antimetabolite with significant activity against lymphoid malignancies. J Clin Oncol. 1991;9(1):175–88 [DOI] [PubMed] [Google Scholar]
- 24.Chun HG, Leyland-Jones BR, Caryk SM, Hoth DF. Central nervous system toxicity of fludarabine phosphate. Cancer Treat Rep. 1986;70(10):1225–8 [PubMed] [Google Scholar]
- 25.Warrell RP, Jr, Berman E. Phase I and II study of fludarabine phosphate in leukemia: therapeutic efficacy with delayed central nervous system toxicity. J Clin Oncol. 1986;4(1):74–9 [DOI] [PubMed] [Google Scholar]
- 26.Kano Y, Akutsu M, Tsunoda S, Suzuki K, Ichikawa A, Furukawa Y, et al. In vitro cytotoxic effects of fludarabine (2-F-ara-A) in combination with commonly used antileukemic agents by isobologram analysis. Leukemia. 2000;14(3):379–88 [DOI] [PubMed] [Google Scholar]
- 27.Gandhi V, Estey E, Keating MJ, Plunkett W. Fludarabine potentiates metabolism of cytarabine in patients with acute myelogenous leukemia during therapy. J Clin Oncol. 1993;11(1):116–24 [DOI] [PubMed] [Google Scholar]
- 28.Estey E, Thall P, Andreeff M, Beran M, Kantarjian H, O’Brien S, et al. Use of granulocyte colony-stimulating factor before, during, and after fludarabine plus cytarabine induction therapy of newly diagnosed acute myelogenous leukemia or myelodysplastic syndromes: comparison with fludarabine plus cytarabine without granulocyte colony-stimulating factor. J Clin Oncol. 1994;12(4):671–8 [DOI] [PubMed] [Google Scholar]
- 29.Dinndorf PA, Avramis VI, Wiersma S, Krailo MD, Liu-Mares W, Seibel NL, et al. Phase I/II study of idarubicin given with continuous infusion fludarabine followed by continuous infusion cytarabine in children with acute leukemia: a report from the Children’s Cancer Group. J Clin Oncol. 1997;15(8):2780–5 [DOI] [PubMed] [Google Scholar]
- 30.Fleischhack G, Hasan C, Graf N, Mann G, Bode U. IDA-FLAG (idarubicin, fludarabine, cytarabine, G-CSF), an effective remission-induction therapy for poor-prognosis AML of childhood prior to allogeneic or autologous bone marrow transplantation: experiences of a phase II trial. Br J Haematol. 1998;102(3):647–55 [DOI] [PubMed] [Google Scholar]
- 31.Karp JE, Giles FJ, Gojo I, Morris L, Greer J, Johnson B, et al. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk Res. 2008;32(1):71–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–302 [DOI] [PubMed] [Google Scholar]
- 33.Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419–25 [DOI] [PubMed] [Google Scholar]
- 34.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21(24):4642–9 [DOI] [PubMed] [Google Scholar]
- 35.Giles FJ, Cooper MA, Silverman L, Karp JE, Lancet JE, Zangari M, et al. Phase II study of SU5416–a small-molecule, vascular endothelial growth factor tyrosine-kinase receptor inhibitor–in patients with refractory myeloproliferative diseases. Cancer. 2003;97(8):1920–8 [DOI] [PubMed] [Google Scholar]
- 36.Mascarenhas J, Heaney ML, Hexner E, Abdel-Wahab O, Rampal R, Ravandi F, et al. Proposed criteria for response assessment in patients treated in clinical trials for myeloproliferative neoplasms in blast phase (MPN-BP): formal recommendations from the post-myeloproliferative neoplasm acute myeloid leukemia consortium. Leuk Res. 2012;36:1500–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol. 1983;1(11):710–9 [DOI] [PubMed] [Google Scholar]
- 38.R: A Language and Environment for Statistical Computing, R Core Team, R Foundation for Statistical Computing; 2013 [Google Scholar]
- 39.Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS Plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–7 [DOI] [PubMed] [Google Scholar]
- 40.Wang JC, Chen W, Nallusamy S, Chen C, Novetsky AD. Hypermethylation of the P15INK4b and P16INK4a in agnogenic myeloid metaplasia (AMM) and AMM in leukemic transformation. Br J Haematol. 2002;116(3):582–86 [DOI] [PubMed] [Google Scholar]
- 41.Thepot S, Itzykson R, Seegers V, Raffoux E, Quesnel B, Chait Y, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010;116(19):3735–42 [DOI] [PubMed] [Google Scholar]
- 42.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubovcakova L, Lundberg P, Grisouard J, Hao-Shen H, Romanet V, Andraos R, et al. Differential effects of hydroxyurea and INC424 on mutant allele burden and myeloproliferative phenotype in a JAK2-V617F polycythemia vera mouse model. Blood. 2013;121(7):1188–99 [DOI] [PubMed] [Google Scholar]
- 44.Sirhan S, Lasho TL, Hanson CA, Mesa RA, Pardanani A, Tefferi A. The presence of JAK2V617F in primary myelofibrosis or its allele burden in polycythemia vera predicts chemosensitivity to hydroxyurea. Am J Hematol. 2008;83(5):363–5 [DOI] [PubMed] [Google Scholar]
- 45.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including post-myeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119(20):4614–8 [DOI] [PMC free article] [PubMed] [Google Scholar]