

Abstract
The clinical heterogeneity among first relapses of childhood ETV6/RUNX1-positive acute lymphoblastic leukemia indicates that further genetic alterations in leukemic cells might affect the course of salvage therapy and be of prognostic relevance. To assess the incidence and prognostic relevance of additional copy number alterations at relapse of the disease, we performed whole genome array comparative genomic hybridization of leukemic cell DNA from 51 patients with first ETV6/RUNX1-positive relapse enrolled in and treated according to the relapse trials ALL-REZ of the Berlin-Frankfurt-Münster Study Group. Within this cohort of patients with relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia, the largest analyzed for genome wide DNA copy number alterations to date, alterations were present in every ETV6/RUNX1-positive relapse and a high proportion of them occurred in recurrent overlapping chromosomal regions. Recurrent losses affected chromosomal regions 12p13, 6q21, 15q15.1, 9p21, 3p21, 5q and 3p14.2, whereas gains occurred in regions 21q22 and 12p. Loss of 12p13 including CDKN1B was associated with a shorter remission duration (P=0.009) and a lower probability of event-free survival (P=0.001). Distribution of X-chromosomal copy number alterations was gender-specific: whole X-chromosome loss occurred exclusively in females, gain of Xq only in males. Loss of the glucocorticoid receptor gene NR3C1 (5q31.3) was associated with a poor response to induction treatment (P=0.003), possibly accounting for the adverse prognosis of some of the ETV6/RUNX1-positive relapses.
Introduction
Relapsed childhood acute lymphoblastic leukemia (ALL) is a clinically and biologically heterogeneous disease. Among patients with relapsed ALL, treatment response and outcome are dependent on the immunophenotype and on specific genetic aberrations in ALL cells, such as structural or numerical chromosome changes which characterize particular subtypes.1 As in initial ALL, the chromosomal translocation t(12;21)(p13;q22) generating the fusion gene ETV6/RUNX1 is the most common structural genetic aberration in pediatric B-cell precursor (BCP) ALL at first relapse.2–7 Although generally associated with favorable risk features and advantageous long-term survival rates,6,8 the high frequency at relapse of disease opposes this positive prognostic characteristic.9,10
In the current relapse trial of the Berlin-Frankfurt-Münster (BFM) study group, ALL-REZ BFM 2002, risk stratification and, accordingly, therapy intensity are based on well-established prognostic factors: time of relapse with respect to the cessation of frontline treatment, site of relapse (isolated or combined bone marrow or isolated extramedullary involvement), and immunophenotype (B- or T-lineage). Risk-adapted treatment of patients with first ALL relapse is based on these key prognostic determinants which are used to assign patients to three main risk groups with significantly different event-free survival (EFS) rates.11 Within the largest group, the intermediate-risk group, minimal residual disease assessment at the end of induction therapy is of strong prognostic relevance and is applied for stratification of post-remission therapy including further treatment intensification by stem cell transplantation.12 Given the clinical characteristics of ETV6/RUNX1-positive ALL, such as late recurrence of the disease and BCP immunophenotype, most patients with this form of ALL are stratified into the intermediate risk group (Online Supplementary Table S1). Only 10% of ETV6/RUNX1-positive relapses occur early, and very early relapses are rare. A second continuous complete remission can be achieved in the majority of patients with ETV6/RUNX1-positive relapses. However, a considerable proportion exhibits a poor molecular response to treatment and eventually experiences a subsequent relapse. The underlying causes of the differences in response to treatment and subsequently outcome in the clinically heterogeneous group of ETV6/RUNX1-positive ALL relapses are not known. The current view is that despite the inability of the fusion oncoprotein ETV6/RUNX1 to induce leukemic transformation on its own, the ETV6/RUNX1 fusion constitutes the initiating mutation in ETV6/RUNX1-positive leukemogenesis.13–16 The identification of leukemia-initiating and -propagating cells in ETV6/RUNX1-positive ALL has recently corroborated this insight.17 Secondary genetic alterations are most likely required for ETV6/RUNX1-positive leukemogenesis and might be important for the differences in clinical outcome.
More than 80% of initial ETV6/RUNX1-positive ALL display additional genetic alterations of the ETV6 and RUNX1 gene loci in fluorescence in situ hybridization (FISH) analyses. These include deletions of the untranslocated ETV6 gene (70%), an extra copy of RUNX1 (23%) and duplication of the derivative chromosome der(21) t(12;21) (10%).18 At relapse, additional alterations of chromosomes 12 and 21 are often detected in leukemic cells as well (85%).19 Current genome wide high resolution analyses of DNA copy number alterations (CNA) have identified numerous genetic alterations in childhood ALL.20–23 In initial ETV6/RUNX1-positive ALL, genes related to B-lymphocyte development and differentiation, cell cycle regulation, tumor suppression and apoptosis are recurrently affected.20,21,24–27 Recent DNA copy number analyses on matched initial diagnosis and relapse ALL samples revealed that CNA acquired at relapse primarily affect genes implicated in cell cycle regulation, B-cell development, drug metabolism and drug response.25,28,29 In the present study, leukemic cell DNA from 51 patients with a first relapse of ETV6/RUNX1-positive ALL enrolled in and treated according to the ALL-REZ BFM relapse trials were examined by whole genome array comparative genomic hybridization (CGH) for cooperating genetic lesions. This cohort represents the largest number of patients analyzed so far, enabling the investigation of an association between identified CNA and relapse-specific clinical and prognostic parameters for the first time.
Methods
Patients and samples
Leukemic bone marrow samples from 51 patients with first relapse of an ETV6/RUNX1-positive BCP-ALL were collected at relapse diagnosis after written informed consent was obtained from the patients, their parents or guardians in accordance with the ethical committee of the Charité and the declaration of Helsinki. All patients were enrolled in and treated according to ALL-REZ relapse trials 90, 95/96, and 2002 of the BFM study group, which were approved by the Institutional Review Boards of the Charité and the FU-Berlin, Germany. Diagnostic bone marrow samples were selected to contain >60% leukemic cells prior to further enrichment by Ficoll-density gradient separation of mononuclear cells. The presence of the ETV6/RUNX1 fusion was detected by reverse transcriptase polymerase chain reaction analysis as described previously.9
DNA isolation
Leukemic cell samples were prepared and DNA isolated from bone marrow or peripheral blood mononuclear cells of all patients and of 20 healthy controls, serving as gender-specific control DNA, as described previously.30 All DNA samples were amplified using the GenomePlex Whole Genome Amplification Kit (Sigma-Aldrich Chemie GmbH, Munich, Germany) following the manufacturer’s directions.
Array comparative genomic hybridization
Array CGH was performed on a submegabase resolution whole genome tiling-path bacterial artificial chromosome (BAC) DNA array consisting of approximately 36,000 BAC clones obtained from several sources as described previously.31 The array CGH data were visualized and analyzed using CGHPRO software (for details see the Online Supplementary Information).32
Fluorescence in situ hybridization
Array CGH findings of chromosome 5q31.3 (NR3C1), 6q21, and 12p13 (CDKN1B) were validated by interphase FISH on bone marrow smears obtained at relapse diagnosis. BAC clones RP11-D16614 (5q31.3; genomic position Mb 142.7–142.9) and RP11-M18773 (5p15.33; Mb 0.5–0.7; BACPAC Resources Center) were used as chromosome 5 probes and fluorescence-labeled with Vysis fluorophore-labeled dUTP (RP11-D16614: Spectrum Or-ange; RP11-M18773: FITC) by nick translation (Abbott, Wiesbaden, Germany). The cut-off value for deletion of NR3C1 (7.78%) (5q31.3; RP11-D16614) was determined by interphase FISH analysis of blood smears from 20 healthy donors. For chromosome 6q21 and 12p13 findings, the 6q21 (SEC63)/SE6 probe (6q21: Spectrum Orange; SE6: Spectrum Green; Kreatech Diagnostics, Amsterdam, the Netherlands) and the BAC clone RP11-180M5 (12p13, Spectrum Orange, BlueGenome, Cambridge, UK) were applied, respectively. CEP 12 probe (D12Z1, Spectrum Green, Abbott) served as the control.
Statistical analysis
Statistical analysis was performed using STATA software (version 9.0, StataCorp, Texas, USA) and SPSS for Windows software (version 18.0.1, SPSS Inc., Chicago, USA). Associations between highly recurrent CNA (≥10% of relapses) and established prognostic determinants at ALL relapse (ALL-REZ BFM trials) were assessed by the Fisher exact and Mann-Whitney-U tests. Probabilities of event-free survival (pEFS) from the date of relapse diagnosis until the last recall or subsequent event (second relapse and death) were estimated by Kaplan-Meier analyses. Differences were considered by the log-rank test. To test the independence of predictive factors for pEFS, Cox regression analysis was performed by forward testing. The likelihood-ratio test was used for comparison of models. Due to the investigational nature of this analysis, we did not apply a stringent multiple comparisons ad-justment. However, all tests were conducted at the 1% significance level.
Results
Patients and representativeness
DNA from 51 leukemic bone marrow samples with a median blast count of 92% prior to enrichment (minimum 71%; maximum 98%) was hybridized to the tiling-path BAC array. Detailed clinical and genetic data are provided in Online Supplementary Tables S1 and S2. The median follow-up time was 10.1 years (minimum 4.9; maximum 20.6). Comparison of the study cohort (n=51) with the total cohort of patients diagnosed with a first relapse of ETV6/RUNX1-positive BCP-ALL with bone marrow involvement enrolled in the ALL-REZ BFM relapse trials over the same period (n=113) confirmed the representativeness of the patients included (Online Supplementary Table S1).
Copy number alterations are a common feature of relapsed ETV6/RUNX1-positive acute lymphoblastic leukemia
Every relapse sample exhibited at least one additional chromosomal aberration. Chromosomal losses were approximately 2.5-fold more frequent than chromosomal gains and generally affected smaller genomic regions (Figure 1).
Figure 1.

Summation of the total of identified CNA in 51 ETV6/RUNX1-positive ALL relapses. The figure shows the relative frequency of CNA identified in all the investigated relapses. Copy number losses are indicated in red, copy number gains in green. The numbering 0-50-100 indicates the number of relapses showing the CNA. The diagram is based on an automatic readout of the array CGH data without a correction of the data regarding copy number variants.
Whole chromosome gains were observed in 12 relapses, nine of which displayed a whole chromosome gain of a single chromosome only. One relapse exhibited a hyperdiploid karyotype (+10, +16, +18, +21). Overall +10 was the most frequent whole chromosome gain (6/51). Whole chromosome losses were detected in 12 relapses: the most common whole chromosome loss was that of chromosome X (9/51) (Online Supplementary Table S3).
Recurrently altered regions at relapse and their clinical association
A substantial number of CNA in relapsed ETV6/RUNX1-positive ALL occurred in recurrently affected regions. About 65% of detected CNA were identified in regions that were affected in at least two of the analyzed relapses, and 48% in more than five relapses which were, therefore, considered as highly recurrent (Table 1).
Table 1.
Recurrent CNA in relapsed childhood ETV6/RUNX1-positive ALL.
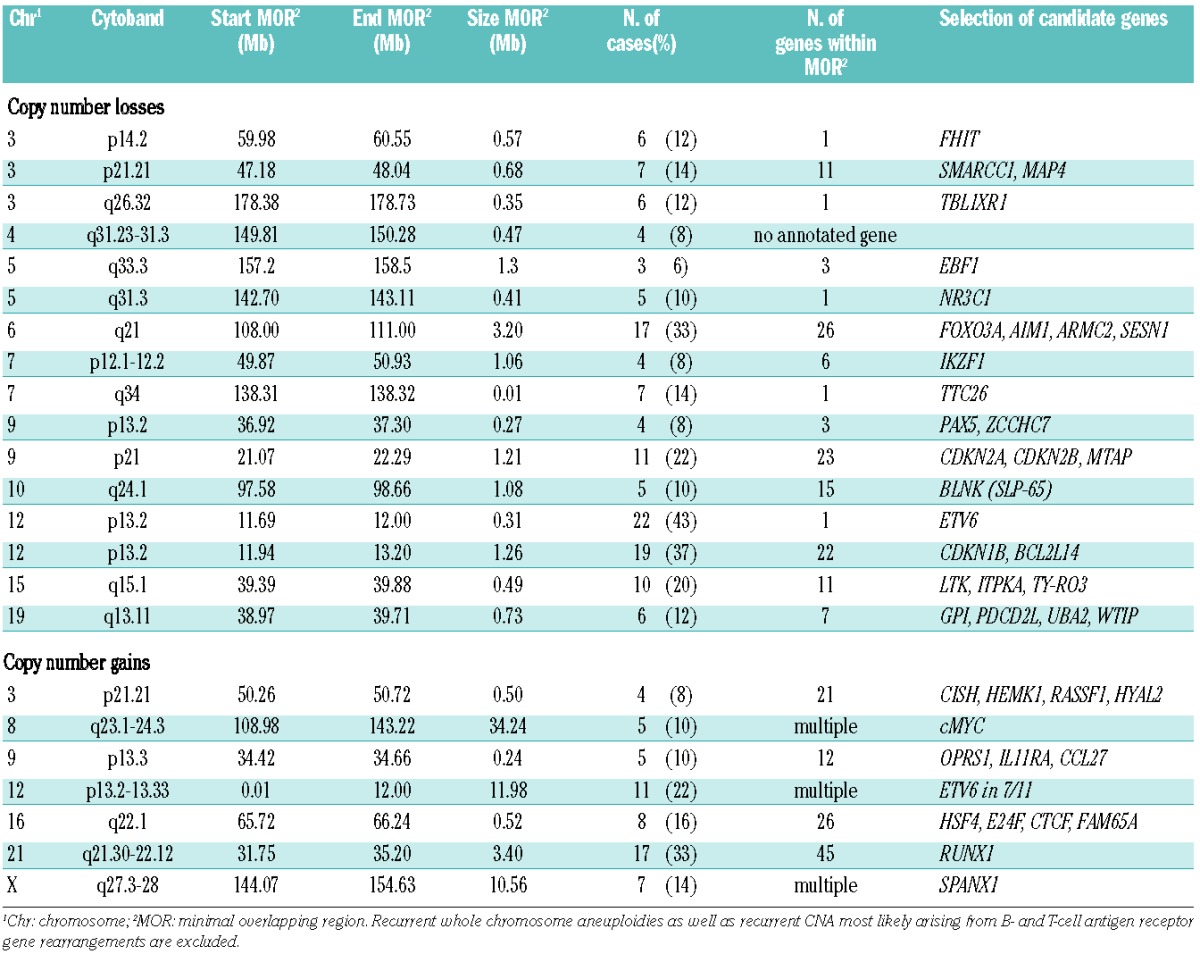
Deletions of chromosomal region 12p13 (49% of relapses) varied in size ranging from loss of large parts of chromosome 12p to microdeletions of only 0.31 Mb, limited to ETV6 (22/51, 43%) (Table 1). BCL2L14 and CDKN1B, located adjacent and approximately 0.8 Mb centromeric to ETV6, respectively, were deleted in 23/51 (45%) and 19/51 (37%) relapses. The minimal overlapping region of loss including CDKN1B and BCL2L14 (Table 1) contained several genes except for ETV6. A subset of 16 relapses showed losses including CDKN1B, BCL2L14 and ETV6. FISH validation of the 12p13 findings (Online Supplementary Figure S1) confirmed the deletion in all of the investigated samples. Bone marrow smears were available from 12/19 (63%) of the relapsed patients with loss of the minimal overlapping region on chromosome 12p13 including CDKN1B among other genes. All of the deletions were heterozygous and detectable in 64–99% of screened nuclei (Online Supplementary Table S6). Analysis of the clinical data revealed that copy number losses including CDKN1B (37%), were significantly associated with a shorter first remission (P=0.009) and with inferior pEFS (P=0.001) and probability of overall survival (P<0.001) (Table 2). Patients with loss of this region had a pEFS (± SE) at 10 years of 42% ± 11% compared to 81% ± 7% for those without loss (Figure 2; Table 2). Independence of the association of losses at 12p13 including CDKN1B and BCL2L14 with the pEFS was tested by multivariate Cox regression analysis testing all relevant parameters. The final model included time of relapse as the strongest prognostic variable, the immunophenotype and loss of 12p13 including CDKN1B and BCL2L14 showing a weaker prognostic impact (hazard ratio 2.28, 95% CI: 0.95–19.47, P=0.059) than in the univariate analysis (hazard ratio 4.60, 95% CI: 1.69–12.50, P=0.003) (Online Supplementary Table S4).
Table 2.
Detailed clinical and prognostic characteristics of the patients with loss of chromosome 12p13.2, including CDKN1B and BCL2L14, of chromosome 6q21, and combined loss of 6q21 and ETV6.
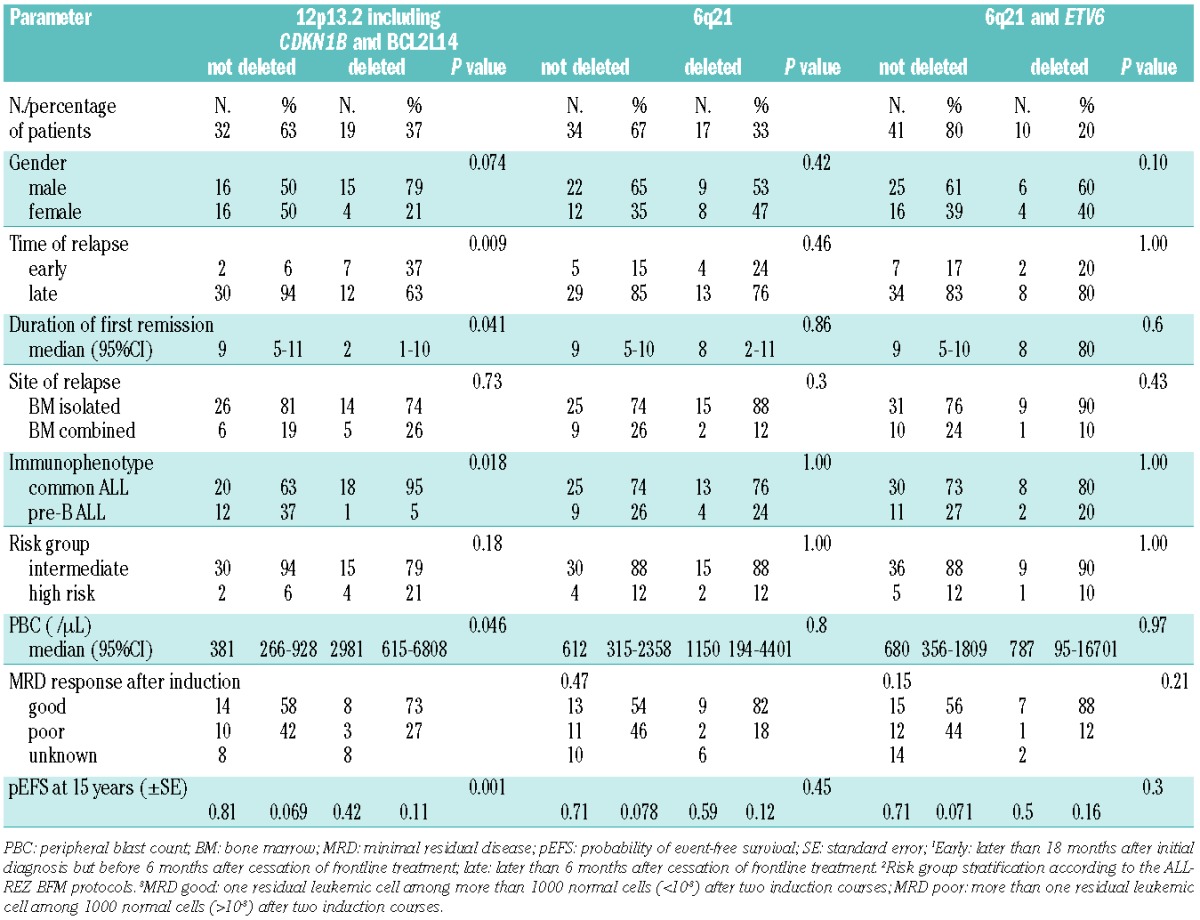
Figure 2.

Probability of event-free survival (pEFS) and overall survival (pOS) (± standard error) as a function of CNA of chromosome region 12p13 including CDKN1B. pEFS (A) and pOS (B) differ significantly between the two groups defined by CDKN1B status (P=0.001, and P<0.001, respectively). The pEFS is 42% (±11%) and the pOS is 47 (±16%) in the group of patients with loss of this region compared to 81%±7% (pEFS) and 90%±5% in the group without loss of the region.
The second most frequent alteration was loss of 6q21 (17/51, 33%). Confirmation of the deletion by interphase FISH (Online Supplementary Figure S1) verified heterozygous deletions in all of the investigated samples (bone marrow smears obtainable in 8/17 cases, 47%) (Online Supplementary Table S7). The commonly deleted region (Table 1) contained multiple genes, including FOXO3A, AIM1, ARMC2 and SESN1. The protein tyrosine kinase FYN was involved in 31% of 6q21 losses. Concomitant losses of 6q21 and 12p13.2 were noticeably frequent in the analyzed relapse samples (12/51, 24%) and included CDKN1B (12p13.2) in 16% (8/51) of cases. This subgroup was characterized by a high incidence of subsequent relapses (5/8, 63%). Statistical analysis showed a tendency towards both an inferior outcome (P=0.047) and an inferior pEFS (P=0.045) which was not attributable to the combined loss of 6q21 with ETV6 (10/51, 20%) (Table 2; Online Supplementary Figure S2). Since loss of 6q21 and CDKN1B were among the most frequent CNA, statistical analysis was done to assess whether the occurrence of both was associated. The comparison by cross-classified tables (Fisher exact test) showed no statistically significant association.
The commonly deleted region of 9p21.3 (11/51, 22%; Table 1) comprises 19 genes including CDKN2A (encoding p16INK4A and p14ARF), CDKN2B (p15INK4B) and MTAP (methylthioadenosine phosphorylase). Patients displaying loss of 9p21.3 in their leukemic cells had higher peripheral blast cell counts (P=0.002), higher leukocyte counts (P=0.001) and were older (P=0.0005) at relapse diagnosis.
Chromosomal gains mainly involved chromosomes 12 and 21. The most frequently gained region was 21q22 (17/51, 33%) followed by gain of 12p (22%). Gains of 12p were always accompanied by gain of 21q, a finding compatible with the existence of an extra der(21)t(12;21).
Copy number alterations affecting genes associated with B-lymphocyte differentiation
Several of the identified copy number losses affected genes associated with B-lymphocyte development and differentiation. These included IKZF1 and PAX5 (4/51, 8%), the early B-cell factor gene EBF1 (3/51, 6%) and the B-cell differentiation regulator SLP-65 (BLNK) (5/51, 10%) (Table 1).
X-chromosomal copy number alterations show a gender-specific distribution
As mentioned above, the most frequently detected aberration affecting an entire chromosome was a whole chromosome loss of the X-chromosome (9/51, 18%). This loss was identified exclusively in female patients (P<0.0001) and was associated with older age at relapse diagnosis (P=0.006).
In contrast to whole chromosome loss of the X chromosome, a gain of Xq (7/51, 14%) was observed exclusively in male patients. As regards the clinical features of patients displaying gain of Xq, all were found to be in continuous complete remission.
Loss of 5q31.3 is associated with poor response to treatment
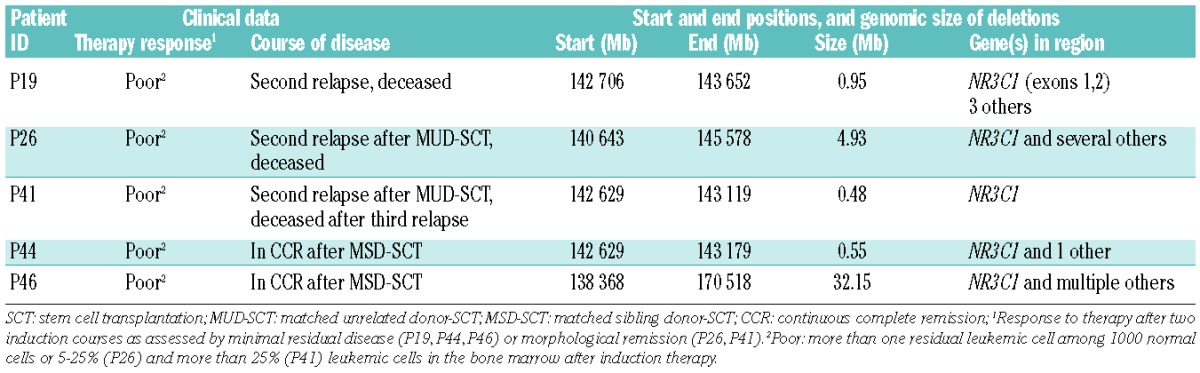
The extent of loss of 5q31.3 (5/51, 10%) varied from a large deletion to submicroscopic deletions in most samples (Table 3). The minimal overlapping region of 0.4 Mb (Table 1) contained exclusively the glucocorticoid receptor gene NR3C1. The 5q31.3 deletion was confirmed by FISH analyses on bone marrow smears from relapse diagnosis (Online Supplementary Figure S1). The proportion of cells exhibiting a deletion of NR3C1 varied between 38% and 74%. Although heterozygous deletions were more frequent than homozygous deletions, the latter occurred in up to 17% of screened nuclei (Online Supplementary Table S5). Statistical evaluation of the clinical data of the five patients with loss of NR3C1 in their leukemic cells (Table 3) revealed both a significantly worse molecular response to induction treatment (P=0.004) and a delayed morphological response (P=0.009) compared to ETV6/RUNX1-positive relapses without loss of NR3C1. Although molecular response status (minimal residual disease) was not known for two of the five patients, morphological evaluation revealed that these patients had not achieved remission after induction treatment (≥5% leukemic blasts). Both patients were, therefore, classified as molecular poor responders for the statistical analysis. Regarding the clinical course of disease in the five patients with loss of NR3C1 (Table 3), three experienced a subsequent relapse and eventually died. Two patients underwent matched sibling donor hematopoietic stem cell transplantation in second remission and are in continuous complete remission for 8 and 9 years.
Table 3.
Clinical and genetic characteristics of ALL relapses with loss of 5q31.3.
Discussion
This genome-wide study on ETV6/RUNX1-positive leukemic cell samples at first relapse was aimed at identifying CNA that possibly not only contribute to leukemic transformation of this leukemic subtype but also influence response to relapse therapy and subsequent recurrence of disease. A representative, large cohort of patients was analyzed. The results indicate that ETV6/RUNX1-positive ALL relapses are hallmarked by further numerical genomic alterations and that some of the identified CNA show an association with clinical characteristics.
The finding that chromosomal losses clearly outnumber gains and generally involve smaller genomic regions is consistent with the results on ETV6/RUNX1-positive ALL at first presentation.20,21,25 In addition, recent studies showed an increase in the number of CNA at relapse.25,28,29 The two most frequent CNA were losses of the chromosome bands 12p13 (49%) and 6q21 (33%). Loss of 12p13, including ETV6, is a known frequent alteration in initial ALL.5 In initial ETV6/RUNX1-positive ALL, loss of ETV6 has been described in 42 – 70% of cases.21,25,26 At relapse, a recent study found ETV6 losses in 57% of ETV6/RUNX1-positive ALL,25 a proportion which coincides with a previous interphase FISH-based analysis by our group in which ETV6 losses were detected in 52% of relapses19 and with the present study. Further candidate genes affected by losses at 12p13 were BCL2L14, encoding a pro-apoptotic member of the Bcl-2 family,33 and CDKN1B, coding for a cyclin-dependent kinase inhibitor, a negative regulator of cell cycle progression from G1 to S phase.34 Thirty-seven percent of relapses showed losses of the minimal overlapping region including these genes. Losses involving CDKN1B and BCL2L14, among other genes, were associated with significantly shorter remissions and inferior pEFS and probability of OS. However, multivariate analysis did not detect an independent prognostic influence and revealed that the association with pEFS was confounded by the time of relapse diagnosis and immunophenotype. Heterozygous deletions of CDKN1B have been recurrently described in initial ALL and have frequently been detected in ETV6/RUNX1-positive samples.35 Though an association with CDKN1B gene deletion and a dismal prognosis has not been described in ALL, lower expression of CDKN1B has been shown to correlate with a poor prognosis in solid tumors.36,37
Numerical and structural aberrations involving chromosome 6q are common in childhood ALL.38 In our relapsed ETV6/RUNX1-positive cohort, the rate of 6q21 losses (33%) was clearly higher than the reported incidence in initial disease (7–18%).20,21,24,25 This frequency exceeds the occurrence of reported 6q losses in relapsed ALL in general (17%)39 and of 6q21 losses in ETV6/RUNX1-positive relapses in particular (7%).25
In line with previous studies assessing the association between loss of 6q and clinical features in initial ALL,38,40 no prognostic impact was found in the present study on ETV6/RUNX1-positive relapses. However, the combined loss of 12p13 (including CDKN1B and BCL2L14, among other genes) and 6q21 was frequent in the analyzed relapses (8/51, 16%). Subsequent relapses were disproportionately more frequent among patients with this combination of losses than in those without (subsequent relapses in 63% versus 24%), which was also reflected by an inferior pEFS. Since both of the identified regions of overlap were relatively large (3.2 Mb, 6q21; 1.26 Mb, 12p13) and included several genes, the observed association with a dismal prognosis is difficult to interpret. Nonetheless, it can be presumed that genes affected by the deletion are essential for normal cellular homeostasis. FOXO3A, a transcription factor involved in the control of proliferation and apoptosis as well as in the induction of CDKN1B gene transcription,41 was included in the deleted region at 6q21. Thus co-inactivation of both FOXO3A (6q21) and CDKN1B (12p13) caused by copy number losses might have an adverse prognostic impact.
The incidence of deletions at 9p21.3 (22%), including CDKN2A, MTAP and CDKN2B, in initial ETV6/RUNX1-positive ALL varies between 12% and 29%.20,21,24 In our relapsed cohort (22%), a significant association was seen between loss of 9p21.3 and higher peripheral blast cell and leukocyte counts as well as older age at relapse diagnosis. Consistent with other studies which analyzed childhood BCP-ALL, no correlation was observed between loss of this region and the outcome of patients.42,43
Involvement of B-cell differentiation genes
Recently, several studies reported recurrent CNA of genes associated with B-lymphocyte differentiation in ALL at initial presentation.21,22,29 These genes included the B-lineage transcription factors PAX5, EBF1 and IKZF1, as well as genes with other established functions in B-cell development such as FYN, RAG1 and RAG2. Several of these genes were affected by CNA in ETV6/RUNX1-positive relapsed ALL as well, but mostly at lower frequencies than described before. Loss of PAX5 was only detected in 8% of the analyzed samples, a proportion clearly lower than those found by Tzusuki et al.,24 Mullighan et al.,21 and Lilljebjörn et al.26 at first presentation (6/24, 25%; 13/47, 27%; 6/24, 25%, respectively) and at relapse of ETV6/RUNX1-positive ALL (3/14, 21%).25
Two recent studies compared the incidence of CNA in paired initial and relapsed BCP-ALL29 and ETV6/RUNX1-positive ALL25 samples. PAX5 losses were observed at both stages of disease in 35%29 and 21%,25 respectively. In the comparative analysis of 14 paired ETV6/RUNX1-positive ALL samples,25 acquired losses of EBF1 (2/14) and IKZF1 (1/14) were detected at relapse. In our larger cohort of 51 relapsed ETV6/RUNX1-positive ALL, EBF1 and IKZF1 deletions occurred in 6% and 8% of cases, respectively. Deletions of IKZF1 have also been reported at initial presentation of ETV6/RUNX1-positive ALL, albeit at a lower frequency.21,26,44 However, IKZF1 is frequently deleted in BCR/ABL-positive45 and other high-risk BCP-ALL subtypes.46
X-chromosome copy number alterations
CNA of the X-chromosome showed a gender-specific distribution at relapse. Loss of the whole X-chromosome was the most frequent whole chromosome aneuploidy (18% in the entire cohort) and was confined to female patients (9/20 females, 45%). Previous cytogenetic studies found a lower incidence of whole chromosome loss of X (3–8%) at initial presentation of ALL,47,48 though an association with female gender and ETV6/RUNX1-positivity was observed as well. In contrast, gain of Xq (14% of relapses) was restricted to male patients (13/31, 42%). Gain of Xq has been described as one of the most frequent CNA in initial ETV6/RUNX1-positive ALL (6/17, 35%), occurring exclusively in ALL samples from male patients.20 Consistent with our results, gain of Xq constantly included the telomere of Xq while the centromeric breakpoints affected variable regions of the long arm. Borst et al.27 detected gain of Xq in 11% (six males, one female) and Kawamata et al.23 in 2% of ETV6/RUNX1-positive ALL at first presentation.
Loss of glucocorticoid receptor gene NR3C1
Most significantly, loss of 5q31.3 (10% of ETV6/RUNX1-positive ALL relapses) was associated with an adverse impact on response to treatment. The NR3C1 gene encoding the human glucocorticoid receptor was the single gene identified within the minimal overlapping region of loss. Confirmation by BAC-based FISH analyses detected homozygous as well as heterozygous deletions in the five relapses. Since glucocorticoids represent an essential component in frontline and relapse treatment protocols for ALL, we hypothesize that the poor treatment response of the five patients with loss of 5q31.3 might have originated from their NR3C1 gene deletions.
Resistance to glucocorticoids is frequently observed clinically at relapse, and in vitro sensitivity assays show that leukemic cells from patients with relapsed ALL are much more resistant to glucocorticoids than at first presentation.49 However, although cell line models implicate mutations and loss of heterozygosity of the glucocorticoid receptor gene as mechanisms of in vitro glucocorticoid resistance,50–52 results from mutational screening studies suggest that the contribution of somatic mutations to glucocorticoid resistance is rather negligible in initial and relapsed ALL patients.51,53,54 In initial ALL, the frequency of NR3C1 loss was approximately 10% in ETV6/RUNX1-positive ALL but considerably lower in ETV6/RUNX-negative cases.21,26 The analysis of paired initial and relapse bone marrow samples showed NR3C1 deletions were acquired at relapse in 9% of BCP-ALL relapses28 and in 21% of ETV6/RUNX1-positive relapses.25
In our study, patients with NR3C1 loss displayed clinical features distinct from those of the majority of patients with ETV6/RUNX1-positive relapses. None of the patients with loss of NR3C1 had achieved molecular remission at the end of induction therapy at relapse and, therefore, received further treatment intensification with hematopoietic stem cell transplantation. Consideration of the course of disease demonstrates the benefit of risk stratification by minimal residual disease as well as the need for further biological risk markers that enable a more detailed understanding of poor treatment response and possibly help to rationalize specific treatment intensification before stem cell transplantation in poor responding ETV6/RUNX1-positive relapses.
In summary, array CGH analysis of relapsed childhood ETV6/RUNX1-positive leukemic cells allowed the identification of various highly recurrent CNA. The correlation with clinical characteristics revealed a prognostic relevance for several of the detected alterations. Most significantly, array CGH enabled the molecular characterization of subgroups with inferior outcomes and poor cytological/molecular response to induction treatment. The results of this study underscore the clinical and prognostic relevance of additional CNA in ETV6/RUNX1-positive ALL and constitute a basis for further analyses of the underlying molecular mechanisms that might contribute to the clinical heterogeneity within the large subgroup of ETV6/RUNX1-positive relapses. Prospectively, high resolution genomic profiling will yield valuable information for further improvement of risk stratification to provide more refined and individualized therapy in relapsed childhood ALL.
Acknowledgments
The authors would like to thank all participating patients, parents, and physicians who enabled this research.
Footnotes
The online version of this article has a Supplementary Appendix
Funding
This study was supported by a grant from the José Carreras Leukämie-Stiftung e.V., Munich, Germany.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol. 2005;23(26):6306–15 [DOI] [PubMed] [Google Scholar]
- 2.Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, et al. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92(11):4917–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Romana SP, Poirel H, Leconiat M, Flexor MA, Mauchauffe M, Jonveaux P, et al. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood. 1995;86(11):4263–9 [PubMed] [Google Scholar]
- 4.Shurtleff SA, Buijs A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia. 1995;9(12):1985–9 [PubMed] [Google Scholar]
- 5.Raynaud S, Cave H, Baens M, Bastard C, Cacheux V, Grosgeorge J, et al. The 12;21 translocation involving TEL and deletion of the other TEL allele: two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood. 1996;87(7):2891–9 [PubMed] [Google Scholar]
- 6.Rubnitz JE, Wichlan D, Devidas M, Shuster J, Linda SB, Kurtzberg J, et al. Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children’s Oncology Group study. J Clin Oncol. 2008;26(13):2186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhojwani D, Kang H, Moskowitz NP, Min DJ, Lee H, Potter JW, et al. Biologic pathways associated with relapse in childhood acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2006;108(2):711–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loh ML, Goldwasser MA, Silverman LB, Poon WM, Vattikuti S, Cardoso A, et al. Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood. 2006;107(11):4508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger K, Adams HP, Buchwald D, Beyermann B, Kremens B, Niemeyer C, et al. TEL-AML1 fusion transcript in relapsed childhood acute lymphoblastic leukemia. The Berlin-Frankfurt-Munster Study Group. Blood. 1998;91(5):1716–22 [PubMed] [Google Scholar]
- 10.Seeger K, Buchwald D, Peter A, Taube T, von Stackelberg A, Schmitt G, et al. TEL-AML1 fusion in relapsed childhood acute lymphoblastic leukemia. Blood. 1999;94(1):374–6 [PubMed] [Google Scholar]
- 11.Henze G, Von Stackelberg A. Treatment of Relapsed Acute Lymphoblastic Leukemia. Memphis, TN: Humana Press, 2002 [Google Scholar]
- 12.Eckert C, Biondi A, Seeger K, Cazzaniga G, Hartmann R, Beyermann B, et al. Prognostic value of minimal residual disease in relapsed childhood acute lymphoblastic leukaemia. Lancet. 2001;358(9289):1239–41 [DOI] [PubMed] [Google Scholar]
- 13.Morrow M, Horton S, Kioussis D, Brady HJ, Williams O. TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity. Blood. 2004;103(10):3890–6 [DOI] [PubMed] [Google Scholar]
- 14.Nimer SD, Moore MA. Effects of the leukemia-associated AML1-ETO protein on hematopoietic stem and progenitor cells. Oncogene. 2004;23(24):4249–54 [DOI] [PubMed] [Google Scholar]
- 15.Ford AM, Bennett CA, Price CM, Bruin MC, Van Wering ER, Greaves M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc Natl Acad Sci USA. 1998;95(8):4584–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354(9189):1499–503 [DOI] [PubMed] [Google Scholar]
- 17.Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319(5861):336–9 [DOI] [PubMed] [Google Scholar]
- 18.Stams WA, Beverloo HB, den Boer ML, de Menezes RX, Stigter RL, van Drunen E, et al. Incidence of additional genetic changes in the TEL and AML1 genes in DCOG and COALL-treated t(12;21)-positive pediatric ALL, and their relation with drug sensitivity and clinical outcome. Leukemia. 2006;20(3):410–6 [DOI] [PubMed] [Google Scholar]
- 19.Peter A, Heiden T, Taube T, Korner G, Seeger K. Interphase FISH on TEL/AML1 positive acute lymphoblastic leukemia relapses–analysis of clinical relevance of additional TEL and AML1 copy number changes. Eur J Haematol. 2009;83(5):420–32 [DOI] [PubMed] [Google Scholar]
- 20.Lilljebjorn H, Heidenblad M, Nilsson B, Lassen C, Horvat A, Heldrup J, et al. Combined high-resolution array-based comparative genomic hybridization and expression profiling of ETV6/RUNX1-positive acute lymphoblastic leukemias reveal a high incidence of cryptic Xq duplications and identify several putative target genes within the commonly gained region. Leukemia. 2007;21(10):2137–44 [DOI] [PubMed] [Google Scholar]
- 21.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64 [DOI] [PubMed] [Google Scholar]
- 22.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258–66 [DOI] [PubMed] [Google Scholar]
- 23.Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111(2):776–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuzuki S, Karnan S, Horibe K, Matsumoto K, Kato K, Inukai T, et al. Genetic abnormalities involved in t(12;21) TEL-AML1 acute lymphoblastic leukemia: analysis by means of array-based comparative genomic hybridization. Cancer Sci. 2007;98(5):698–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuster L, Grausenburger R, Fuka G, Kaindl U, Krapf G, Inthal A, et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011;117(9):2658–67 [DOI] [PubMed] [Google Scholar]
- 26.Lilljebjorn H, Soneson C, Andersson A, Heldrup J, Behrendtz M, Kawamata N, et al. The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet. 2010;19(16):3150–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borst L, Wesolowska A, Joshi T, Borup R, Nielsen FC, Andersen MK, et al. Genome-wide analysis of cytogenetic aberrations in ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia. Br J Haematol. 2012;157(4):476–82 [DOI] [PubMed] [Google Scholar]
- 28.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322(5906):1377–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112(10):4178–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graf Einsiedel H, Taube T, Hartmann R, Wellmann S, Seifert G, Henze G, et al. Deletion analysis of p16(INKa) and p15(INKb) in relapsed childhood acute lymphoblastic leukemia. Blood. 2002;99(12):4629–31 [DOI] [PubMed] [Google Scholar]
- 31.Erdogan F, Chen W, Kirchhoff M, Kalscheuer V, Hultschig C, Muller I, et al. Impact of low copy repeats on the generation of balanced and unbalanced chromosomal aberrations in mental retardation. Cytogenet Genome Res. 2006;115(3–4): 247–53 [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Erdogan F, Ropers HH, Lenzner S, Ullmann R. CGHPRO – a comprehensive data analysis tool for array CGH. BMC Bioinformatics. 2005;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo B, Godzik A, Reed JC. Bcl-G, a novel pro-apoptotic member of the Bcl-2 family. J Biol Chem. 2001;276(4):2780–5 [DOI] [PubMed] [Google Scholar]
- 34.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78(1):59–66 [DOI] [PubMed] [Google Scholar]
- 35.Komuro H, Valentine MB, Rubnitz JE, Saito M, Raimondi SC, Carroll AJ, et al. p27KIP1 deletions in childhood acute lymphoblastic leukemia. Neoplasia. 1999;1(3):253–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, et al. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3(2):231–4 [DOI] [PubMed] [Google Scholar]
- 37.Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997;3(2):227–30 [DOI] [PubMed] [Google Scholar]
- 38.Hayashi Y, Raimondi SC, Look AT, Behm FG, Kitchingman GR, Pui CH, et al. Abnormalities of the long arm of chromosome 6 in childhood acute lymphoblastic leukemia. Blood. 1990;76(8):1626–30 [PubMed] [Google Scholar]
- 39.Takeuchi S, Koike M, Seriu T, Bartram CR, Schrappe M, Reiter A, et al. Frequent loss of heterozygosity on the long arm of chromosome 6: identification of two distinct regions of deletion in childhood acute lymphoblastic leukemia. Cancer Res. 1998;58(12):2618–23 [PubMed] [Google Scholar]
- 40.Heerema NA, Sather HN, Sensel MG, Lee MK, Hutchinson R, Lange BJ, et al. Clinical significance of deletions of chromosome arm 6q in childhood acute lymphoblastic leukemia: a report from the Children’s Cancer Group. Leuk Lymphoma. 2000;36(5–6):467–78 [DOI] [PubMed] [Google Scholar]
- 41.Chandramohan V, Jeay S, Pianetti S, Sonenshein GE. Reciprocal control of Forkhead box O 3a and c-Myc via the phosphatidylinositol 3-kinase pathway coordinately regulates p27Kip1 levels. J Immunol. 2004;172(9):5522–7 [DOI] [PubMed] [Google Scholar]
- 42.Mirebeau D, Acquaviva C, Suciu S, Bertin R, Dastugue N, Robert A, et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica. 2006;91(7):881–5 [PubMed] [Google Scholar]
- 43.van Zutven LJ, van Drunen E, de Bont JM, Wattel MM, Den Boer ML, Pieters R, et al. CDKN2 deletions have no prognostic value in childhood precursor-B acute lymphoblastic leukaemia. Leukemia. 2005;19(7):1281–4 [DOI] [PubMed] [Google Scholar]
- 44.Dorge P, Meissner B, Zimmermann M, Moricke A, Schrauder A, Bouquin JP, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2013;98(3):428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4 [DOI] [PubMed] [Google Scholar]
- 46.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riesch M, Niggli FK, Leibundgut K, Caflisch U, Betts DR. Loss of X chromosome in childhood acute lymphoblastic leukemia. Cancer Genet Cytogenet. 2001;125(1):27–9 [DOI] [PubMed] [Google Scholar]
- 48.Forestier E, Andersen MK, Autio K, Blennow E, Borgstrom G, Golovleva I, et al. Cytogenetic patterns in ETV6/RUNX1-positive pediatric B-cell precursor acute lymphoblastic leukemia: a Nordic series of 245 cases and review of the literature. Genes Chromosomes Cancer. 2007;46(5):440–50 [DOI] [PubMed] [Google Scholar]
- 49.Klumper E, Pieters R, Veerman AJ, Huismans DR, Loonen AH, Hahlen K, et al. In vitro cellular drug resistance in children with relapsed/refractory acute lymphoblastic leukemia. Blood. 1995;86(10):3861–8 [PubMed] [Google Scholar]
- 50.Powers JH, Hillmann AG, Tang DC, Harmon JM. Cloning and expression of mutant glucocorticoid receptors from glucocorticoid-sensitive and -resistant human leukemic cells. Cancer Res. 1993;53(17):4059–65 [PubMed] [Google Scholar]
- 51.Hillmann AG, Ramdas J, Multanen K, Norman MR, Harmon JM. Glucocorticoid receptor gene mutations in leukemic cells acquired in vitro and in vivo. Cancer Res. 2000;60(7):2056–62 [PubMed] [Google Scholar]
- 52.Hala M, Hartmann BL, Bock G, Geley S, Kofler R. Glucocorticoid-receptor-gene defects and resistance to glucocorticoid-induced apoptosis in human leukemic cell lines. Int J Cancer. 1996;68(5):663–8 [DOI] [PubMed] [Google Scholar]
- 53.Tissing WJ, Meijerink JP, den Boer ML, Brinkhof B, van Rossum EF, van Wering ER, et al. Genetic variations in the glucocorticoid receptor gene are not related to glucocorticoid resistance in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2005;11(16):6050–6 [DOI] [PubMed] [Google Scholar]
- 54.Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65(21):9712–8 [DOI] [PubMed] [Google Scholar]