

Abstract
Anti-ADAMTS13 autoantibodies are the main cause of acquired thrombotic thrombocytopenic purpura. Binding of these antibodies to ADAMTS13 eventually results in the formation of antigen-antibody immune complexes. Circulating ADAMTS13-specific immune complexes have been described in patients with acquired thrombotic thrombocytopenic purpura, although the prevalence and persistence of these immune complexes over time have hitherto remained elusive. Here, we analyzed a large cohort of patients with acquired thrombotic thrombocytopenic purpura for the presence of free and complexed anti-ADAMTS13 antibodies. In the acute phase (n=68), 100% of patients had free IgG antibodies and 97% had ADAMTS13-specific immune complexes. In remission (n=28), 75% of patients had free antibodies (mainly IgG) and 93% had ADAMTS13-specific immune complexes. Free antibodies were mainly of subclasses IgG1 and IgG4, whereas IgG4 was by far the most prevalent in ADAMTS13-specific immune complexes. Comparison of ADAMTS13 inhibitor and anti-ADAMTS13 IgG (total and subclasses) antibody titers in acute phase and in remission samples showed a statistically significant decrease in all parameters in remission. Although non-significant, a trend towards reduced or undetectable titers in remission was also observed for ADAMTS13-specific immune complexes of subclasses IgG1, IgG2 and IgG3. No such trend was discernible for IgG4; IgG4 immune complexes persisted over years, even in patients who had been treated with rituximab and who showed no features suggesting relapse.
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a life-threatening disease characterized by hemolytic anemia, severe thrombocytopenia and fluctuating organ dysfunction (mainly renal and cerebral) due to the deposition of platelet-rich thrombi in the microvasculature.1 A severe deficiency of the plasma enzyme ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type-1 repeats), due to genetic mutations (congenital)2 or anti-ADAMTS13 autoantibodies (acquired),3,4 is the main mechanism in the pathogenesis of TTP. Autoantibodies against ADAMTS13 are predominantly of the IgG class,5–7 particularly subclasses IgG4 and IgG1,8–10 but autoantibodies belonging to classes IgM and IgA have also been described.5,9–11
Elevated levels of soluble circulating autoantibody-antigen immune complexes are the hallmark of many autoimmune diseases.12–14 Deposition of circulating immune complexes in tissues, mainly in capillary beds, promoting inflammation and tissue damage, is the most relevant pathological mechanism underlying immune complex-mediated diseases. Early reports attributed the benefit of plasma exchange (PEX) therapy in patients with TTP in part to the removal of circulating immune complexes,15,16 however, their presence remained hypothetical for years as the underlying mechanism leading to TTP had not yet been identified.17,18 With the isolation of inhibiting IgG antibodies against ADAMTS13 from plasma of TTP patients, the presence of ADAMTS13-specific immune complexes became plausible,19 with the following observations supporting their existence: (i) removal of anti-ADAMTS13 IgG antibodies also removed measurable residual ADAMTS13 antigen;20 (ii) residual ADAMTS13 activity was no longer measurable after plasma IgG depletion;21 and (iii) human IgG bound to ADAMTS13 were identified by adaptation of a commercial ADAMTS13 antigen enzyme-linked immunosorbent assay (ELISA).22
Recently, we demonstrated the presence of ADAMTS13-specific immune complexes in a patient with refractory acquired TTP using a co-immunoprecipitation technique.23 This observation prompted us to investigate the prevalence of ADAMTS13-specific immune complexes in a large cohort of patients with acquired TTP during the acute phase and in remission. Results are discussed in terms of immune complexes as a novel biomarker that may contribute to a better understanding of the pathogenesis of acquired TTP and its responsiveness to treatment.
Methods
Patients’ plasma samples
The study included 78 patients diagnosed with idiopathic acquired TTP. Sixty-eight patients were tested during the acute phase, with 48 patients experiencing their first acute episode and 20 a relapse. For 18 of these patients, corresponding samples in clinical remission were also available. Ten patients were analyzed only during remission, giving a total of 28 patients tested in remission. The patients’ demographics and clinical features are summarized in Table 1.
Table 1.
Demographic and clinical features of patients with acquired TTP.
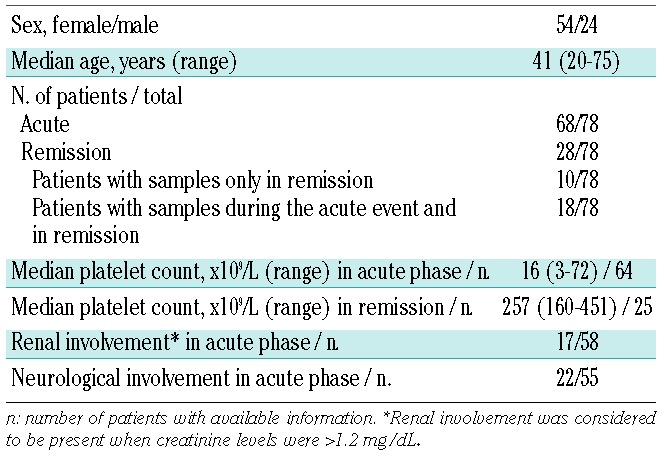
The inclusion criteria for patients with acute acquired TTP were: presence of severe ADAMTS13 deficiency (<10%), thrombocytopenia (platelet count <150×109/L), microangiopathic hemolytic anemia (hemoglobin <12 g/dL) with presence of schistocytes on the peripheral blood smear, and elevated lactate dehydrogenase levels (>450 IU/L). Fever, neurological symptoms or renal failure were not mandatory. Remission was defined as a normal platelet count (>150×109/L) and no plasma exchange treatment for ≥30 consecutive days. Relapse was defined as the reappearance of clinical manifestation and/or laboratory data compatible with TTP after remission.
Frozen citrated plasma samples were obtained from four international centers. The study was approved by the ethic committees of the University Hospital of Berne, Switzerland; Medical University of Vienna, Austria; Lille University Hospital, France; and Icahn School of Medicine, New York, USA.
ADAMTS13 assays
ADAMTS13 activity (ADAMTS13:Ac) and ADAMTS13 functional inhibitor titers were measured using fluorometric FRETS-VWF73 assay as described elsewhere.24,25 The limit of quantification of ADAMTS13:Ac was 0.05 U/mL (5%); values <0.10 U/mL (<10%) were considered severely reduced; levels of 0.10–0.50 U/mL as reduced and levels >0.5 U/mL as normal. An inhibitor titer <0.7 BU/mL was considered negative. Plasma ADAMTS13 antigen (ADAMTS13:Ag) levels were determined by ELISA as described previously.20 The reference range of the assay was 403 – 907 ng/mL; the limit of quantification was 10 ng/mL. Levels of 100 – 403 ng/mL were considered reduced, levels <100 ng/mL as severely reduced, and values <10 ng/mL as undetectable.
Detection of free anti-ADAMTS13 antibodies by enzyme-linked immunosorbent assay
Free antibodies were detected as described elsewhere25 with modifications (Online Supplementary Methods).
Detection of ADAMTS13-specific immune complexes
Microtiter plates were coated with a polyclonal rabbit antihuman ADAMTS13 antibody. After blocking the non-specific sites, diluted patient’s plasma samples and controls were added and incubated overnight at 4°C. The next day, the immunoglobulin fraction of the bound immune complexes was detected with horseradish peroxidase-conjugated class-specific secondary antibodies followed by detection with an appropriate chromogenic substrate. For details, see Online Supplementary Methods and Online Supplementary Figure S1.
An ELISA to detect total IgG-immune complexes was not established, however, the total amount of IgG-immune complexes was investigated in selected patients by co-immunoprecipitation, as described previously23 (Online Supplementary Methods).
Statistical calculations
Levels of free and complexed anti-ADAMTS13 antibodies were expressed as titers (Online Supplementary Methods).
Data are presented as the median with the range in brackets. The statistical significance of the differences between values for the acute phase and remission was assessed by the Mann-Whitney rank sum test. The strength of the relation between different variables was analyzed using Spearman rank correlation. A P-value of <0.05 was considered statistically significant. All statistical analyses were performed with SigmaPlot (Systat Software, San Jose, CA, USA).
Results
Detection of free and complexed anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura during the acute phase
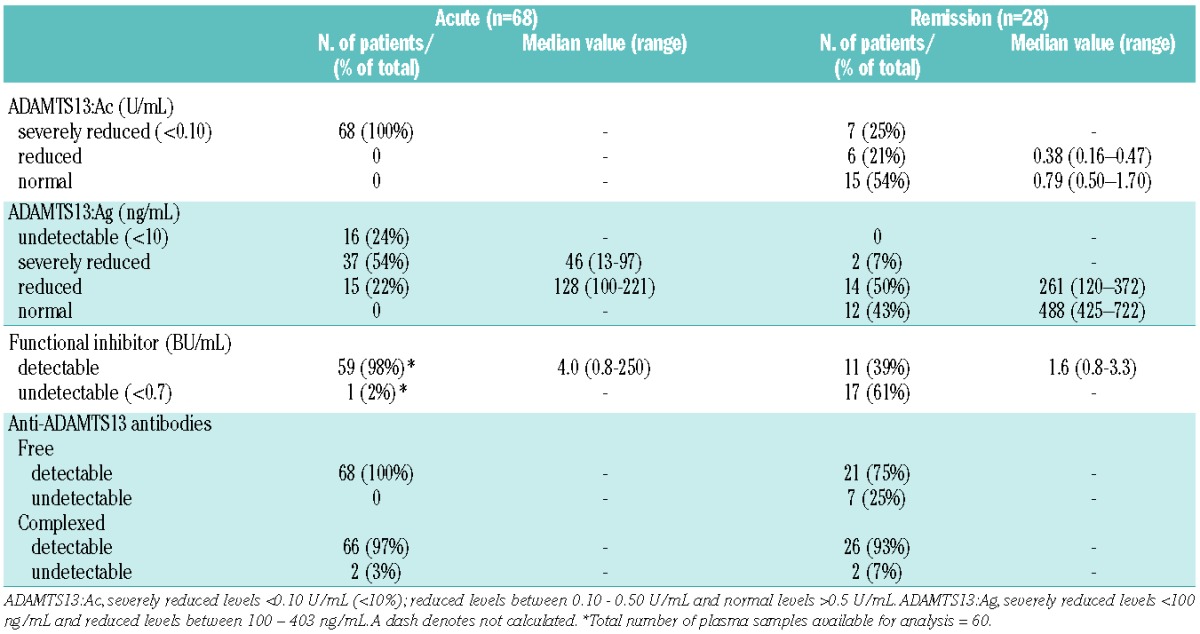
Samples withdrawn from 68 patients with acquired TTP during an acute event and before treatment were analyzed. ADAMTS13:Ac was <0.1 U/mL (<10%) in all patients; ADAMTS13:Ag levels were undetectable in 16/68 (24%) patients, severely reduced (median 46 ng/mL; range, 13–97) in 37/68 (54%) patients and reduced (median 128 ng/mL; range, 100–221) in 15/68 (22%) patients. ADAMTS13 functional inhibitors were present in 59 of 60 (98%) patients tested, with a median inhibitor titer of 4.0 BU/mL (range, 0.8–250) (Table 2).
Table 2.
Overview of the most relevant ADAMTS13-related parameters in patients with acquired TTP during the acute event and in remission.
Analysis of the anti-ADAMTS13 antibody profile showed free antibodies present in all the patients during the acute phase, with free IgG, IgM and IgA anti-ADAMTS13 antibodies detected in 68/68 (100%), 3/68 (4%), and 17/68 (25%) patients, respectively (Figure 1A). Median antibody titers for IgG were 200 (range, 25–12800), 100 (range, 25–6400) for IgA and 100 (range, 25–200) for IgM. A comparison of titers between the functional inhibitor and free IgG revealed a statistically significant correlation (r =0.434, P<0.001; data not shown).
Figure 1.

Prevalence of free and complexed anti-ADAMTS13 antibodies in patients with acquired TTP during the acute and remission phases. Plasma samples were analyzed by ELISA for the presence of free and complexed IgG (total and subclasses), IgA and IgM anti-ADAMTS13 antibodies during the acute event (n=68) and in remission (n=28). In the acute phase, 68/68 and 66/68 patients had detectable free and ADAMTS13-specific immune complexes; in remission, 21/28 and 26/28 patients had detectable free and ADAMTS13-specific immune complexes. The histograms represent frequencies of patients positive for each class of antibodies: (A, C) free anti-ADAMTS13 antibodies in acute phase and in remission and (B, D) ADAMTS13-specific immune complexes in acute phase and in remission. Percentages of patients for IgG, IgA and IgM (free and complexed) were calculated using the total number of patients having positive antibodies. Percentages of patients positive for IgG1-4 subclasses (free and complexed) were calculated using the number of IgG positive patients as 100%. Ab: antibodies; IC: immune complexes.
IgG4 was detected in 63/68 (93%) of IgG-positive patients, followed by IgG1 (52/68, 76%), IgG2 (26/68, 38%), and IgG3 (15/68, 22%) (Figure 1A). IgG4 was detected either alone (14/68, 21%) or in association with other IgG subclasses (Online Supplementary Table S1). Overall, the prevalence of free anti-ADAMTS13 antibodies during the acute phase was in agreement with that previously reported.8,9
ADAMTS13-specific immune complexes were detected in 66/68 (97%) patients during the acute event; among the positively tested samples, ADAMTS13 immune complexes of classes IgG, IgM and IgA were found in 63/66 (95%), 2/66 (3%) and 15/66 (23%) patients, respectively (Figure 1B). Fifty-eight of 63 (92%) IgG-complex-positive patients had IgG4-complexes, 23/63 (37%) patients had IgG1-complexes, 18/63 (29%) had IgG2-complexes and only 4/63 (6%) patients had IgG3-complexes (Figure 1B). For most patients, a match in the distribution of IgG subclasses for free and complexed antibodies was noted. In only five patients who tested positive for free IgG antibodies, were IgG-immune complexes not detected by ELISA; two of these samples gave positive immune complex results using co-immunoprecipitation.
Comparison of titers between free antibodies and ADAMTS13-specific immune complexes revealed a statistically significant correlation for IgG1 (r =0.465, P<0.001) and IgG4 (r =0.573, P<0.001) antibodies (data not shown).
Detection of free and complexed anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura during remission
Our cohort included 28 patients in remission, ten of whom were studied only in remission, the other 18 patients were also analyzed at presentation with an acute event. Samples were taken at a median remission of 12 months (range, 2–36). ADAMTS13:Ac was normal (median 0.79 U/mL; range, 0.50–1.70) in 15/28 (54%) patients, reduced (median 0.38 U/mL; range, 0.16–0.47) in 6/28 (21%) patients and <0.1 U/mL (<10%) in the remaining seven (25%) patients. ADAMTS13:Ag levels were normal (median 488 ng/mL; range, 425–722) in 12/28 (43%) patients, reduced (median 261 ng/mL; range, 120–372) in 14/28 (50%) patients and severely reduced (23 and 95 ng/mL, respectively) in 2/28 (7%) patients. ADAMTS13 functional inhibitors were present in 11/28 (39%) patients, with a median titer of 1.6 BU/mL (range, 0.8–3.3) and negative in 17/28 (61%) patients (Table 2).
Free anti-ADAMTS13 antibodies were detected in 21/28 (75%) patients in remission, with IgG, IgM and IgA antibodies being detected in 20/21 (95%), 1/21 (5%) and 6/21 (29%) patients, respectively (Figure 1C). The IgG subclass distribution of anti-ADAMTS13 antibodies in remission did not differ substantially from that during the acute event, with IgG4 again being most prevalent (13/20, 65%), followed by IgG1 (12/20, 60%), IgG2 (1/20, 5%), and IgG3 (1/20, 5%) (Figure 1C). However, median antibody titers were significantly lower in remission: 50 (range, 25–200) for IgG and 50 (range, 25–100) for IgA; IgM was detectable in only one patient at a very low titer (25). Among the 21 patients who presented with anti-ADAMTS13 antibodies in remission (75%), only 11 had functional inhibitors, six of whom with ADAMTS13 activity <0.1 U/mL (<10%).
ADAMTS13-specific immune complexes were present in 26/28 (93%) patients, with IgG-complexes, IgM-complexes and IgA-complexes detected in 26/26 (100%), 3/26 (12%) and 6/26 (23%) patients, respectively (Figure 1D). As observed for the acute event, IgG4-complexes (24/26 patients, 92%) were the most prevalent, followed by IgG1-complexes (6/26; 23%), IgG2-complexes (5/26; 19%) and IgG3-complexes (2/26; 8%) (Figure 1D).
Comparison of titers between free antibodies and ADAMTS13-specific immune complexes revealed a statistically significant correlation in remission only for IgG4 antibodies (r =0.6481, P<0.001) (data not shown).
In 14/16 patients with detectable immune complexes, ADAMTS13-specific immune complexes were present even with normal ADAMTS13:Ac and ADAMTS13:Ag levels. Moreover, ADAMTS13-specific immune complexes (mainly IgG4) were detected in the absence of free IgG anti-ADAMTS13 antibodies in seven out of eight patients in remission. Only one patient tested negative for both free and complexed antibodies. On the other hand, patients with undetectable or very low ADAMTS13:Ac during remission exhibited higher titers of free and complexed antibodies (mainly IgG4) as well as functional inhibitor titers than those with a normalized ADAMTS13:Ac.
Comparison of free and complexed anti-ADAMTS13 antibodies during the acute event and in remission
Next, we compared ADAMTS13-related parameters of the acute event with those upon remission in all 18 patients with available samples from both phases. During the acute event, ADAMTS13:Ac was <0.10 U/mL (<10%) in all the patients. In remission, ADAMTS13:Ac was normal (median 0.75 U/mL; range, 0.50–1.70) in 10/18 (56%) patients, decreased in 5/18 (28%) patients (median 0.34 U/mL; range, 0.16–0.44), and <0.10 U/mL (<10%) in 3/18 (17%) patients (Figure 2A). ADAMTS13:Ag levels were reduced in 16 patients (median 39 ng/mL; range, 15–206) in the acute phase and undetectable in two patients. In remission, ADAMTS13:Ag levels returned to normal levels (median 481 ng/mL; range, 425–722) in 9/18 (50%) patients and were low but detectable (median 241 ng/mL; range, 23–353) in 9/18 (50%) patients (Figure 2A). A statistically significant increase (P<0.001) in ADAMTS13:Ac (median 0.51 U/mL) and ADAMTS13:Ag levels (median 32 ng/mL versus 389 ng/mL) was observed in remission.
Figure 2.

Comparison of ADAMTS13-related parameters and free and complexed anti-ADAMTS13 antibodies in individual patients between the acute phase and in remission. Plasma samples from 18 patients with acquired TTP taken at presentation (Acute) and in clinical remission (Remission) were analyzed for ADAMTS13 activity, ADAMTS13 antigen, ADAMTS13 functional inhibitor, anti-ADAMTS13 antibodies and ADAMTS13-specific immune complexes. (A) Comparison of ADAMTS13:Ac, ADAMTS13:Ag, ADAMTS13 functional inhibitor and IgG anti-ADAMTS13 antibody titers. The lower values of the normal range (solid line) for ADAMTS13:Ac and ADAMTS13:Ag are 0.5 U/mL and 403 ng/mL, respectively. The limits of quantification (dashed line) for ADAMTS13:Ac, ADAMTS13:Ag and ADAMTS13 functional inhibitor is 0.05 U/mL, 100 ng/mL and 0.7 BU/mL, respectively. (B) Comparison of free and complexed IgG1-4 anti-ADAMTS13 antibody titers between the acute and remission phase. (*) and (**) indicate statistically significant differences at P<0.05 and P<0.001, respectively. ns: non-significant difference. IC: immune complexes.
ADAMTS13 functional inhibitors were present in the acute phase in all 18 patients, with a median titer of 7.7 BU/mL (range, 1–18) (Figure 2A). In remission, only 7/18 (39%) patients had detectable inhibitors (median titer 2.2 BU/mL; range, 0.8–3.3), with a statistically significant decrease in titers (P<0.001) from the acute phase. Free IgG anti-ADAMTS13 antibody titers also showed a statistically significant decrease in remission (median IgG titer 600 versus 25; P<0.001) (Figure 2A). Highly significant decreases were noted for the subclasses IgG1 (250 versus 25; P<0.001) and IgG4 (1400 versus 25; P<0.001); for IgG2 and IgG3 the differences in median titers were statistically significant (P=0.002 and P=0.016, respectively) (Figure 2B). By contrast, there was no statistically significant difference in ADAMTS13-specific immune complex titers between the acute event and in remission. While for IgG1-, IgG2- and IgG3-immune complexes, a trend towards reduced or undetectable titers in remission was perceivable, no such tendency was observed for IgG4-immune complexes (Figure 2B).
The presence of ADAMTS13-specific IgG-complexes in the acute phase and in remission was also investigated by co-immunoprecipitation in 17/18 patients. As expected, variations in IgG subclass composition, ELISA titers and band intensities between patients were noted. For individual patients however, band intensities and ELISA titers roughly correlated, indicating that both methods were capable of adequately detecting ADAMTS13-specific immune complexes (Online Supplementary Figure S2).
A similar comparison for ADAMTS13-specific IgA and IgM antibody titers revealed that most patients who tested positive for these two classes of antibodies in the acute event still had detectable levels of free and complexed IgA and IgM antibodies in remission, and that there was no statistically significant difference in titers between the two phases (Online Supplementary Figure S3).
Course of free and complexed anti-ADAMTS13 antibodies in three patients with acquired thrombotic thrombocytopenic purpura during treatment
To understand the impact of different treatment modalities on the levels of anti-ADAMTS13 antibodies, longitudinal samples from three patients with acquired TTP covering at least one acute event and subsequent remission were analyzed.
At admission with a first acute episode, patient A had undetectable ADAMTS13:Ac and very low ADAMTS13:Ag levels, high functional inhibitor and free IgG anti-ADAMTS13 antibody titers, and detectable ADAMTS13-specific IgG2- and IgG4-complexes (Figure 3A). The patient was treated with daily PEX and corticosteroids but achieved clinical remission only after splenectomy and rituximab administration. ADAMTS13:Ac and ADAMTS13:Ag levels remained undetectable or very low during the acute phase, reaching normal levels 1 year after remission. Functional inhibitor and free IgG anti-ADAMTS13 antibody titers remained high during the acute phase but gradually decreased in remission until they were no longer detectable. ADAMTS13-specific immune complexes (mainly IgG4-complexes) were detectable throughout the observation period by both ELISA and co-immunoprecipitation. Thus, despite absence of functional free antibodies and normal ADAMTS13:Ac and ADAMTS13:Ag levels, immune complexes were still detectable 2 years into remission.
Figure 3.
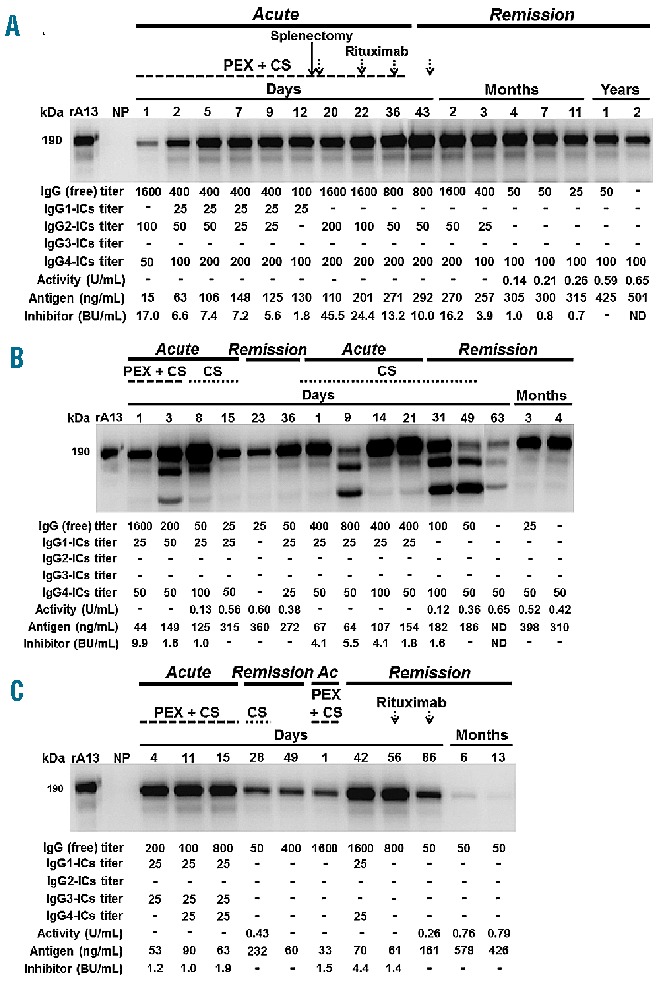
Dynamic course of ADAMTS13-related parameters in three patients with acquired TTP. The dynamic course of ADAMTS13:Ac, ADAMTS13:Ag, ADAMTS13 functional inhibitor, free IgG anti-ADAMTS13 antibody titers and ADAMTS13-specific immune complexes (ICs) was analyzed in three patients who experienced one (A) or two (B–C) acute events with subsequent clinical remission within the observation period. For each patient, treatment with plasma exchange (PEX), corticosteroids (CS), splenectomy or rituximab is indicated. The immunoblots show the ADAMTS13-specific ICs of the IgG class as determined by co-immunoprecipitation. Pooled normal human plasma (NP) served as a negative control. The bands of ~160 and ~140 kDa seen in (B) for some time points most likely represent cleavage fragments of ADAMTS13, as fragments of equal molecular weight were obtained after digestion of rADAMTS13 with plasmin (data not shown). Similar findings were obtained by Feys et al.44 in samples of plasma from a patient with acquired TTP. The position of ADAMTS13 in the gel (~190 kDa) was determined by loading 5 ng of rADAMTS13 (rA13). Ac: acute event. The lower panel of each blot shows the corresponding free and complexed anti-ADAMTS13 antibody titers as detected by ELISA, as well as ADAMTS13:Ac, ADAMTS13:Ag and inhibitor titers. ADAMTS13:Ac and ADAMTS13:Ag levels over 0.5 U/mL and 403 ng/mL are considered normal. For ADAMTS13:Ac, a dash denotes levels <0.10 U/mL (<10%). A dash denotes an undetectable level. ND: not determined.
Patients B and C relapsed during follow-up. Patient B presented with undetectable ADAMTS13:Ac, very low ADAMTS13:Ag levels, high functional inhibitor and free IgG antibody titers and detectable IgG1- and IgG4-complexes (Figure 3B). After treatment with PEX and corticosteroids all ADAMTS13 parameters normalized by day 15, whereas free and complexed anti-ADAMTS13 antibodies were detectable by ELISA and co-immunoprecipitation until remission. The patient experienced a relapse 76 days after the first acute event (day 1 of second acute event) exhibiting a drop in ADAMTS13:Ac and ADAMTS13:Ag levels, an increase in inhibitor as well as a moderate increase in free and complexed anti-ADAMTS13 antibody titers. The patient was treated with corticosteroids and achieved clinical remission after 30 days. Over the next 3 months, ADAMTS13:Ac and ADAMTS13:Ag levels returned to normal and inhibitor and free anti-ADAMTS13 antibodies disappeared. In contrast, IgG4-complexes were still detectable by ELISA and co-immunoprecipitation 4 months after remission.
Patient C was treated with daily PEX and cortico steroids during the first acute event (Figure 3C). During the acute phase and early remission, ADAMTS13:Ac was undetectable or very low accompanied by low ADAMTS13:Ag levels and moderate functional inhibitor and free IgG anti-ADAMTS13 antibody titers. When the patient relapsed (78 days after admission), ADAMTS13:Ac was undetectable and free IgG anti-ADAMTS13 antibody and functional inhibitor titers increased. The patient was treated with PEX and cortico steroids until remission. Two doses of rituximab were given during the early remission, which resulted in an increase of ADAMTS13:Ac and ADAMTS13:Ag and reduced functional inhibitors. IgG titers decreased but were still detectable up to 13 months after remission. ADAMTS13-specific immune complexes were detectable over time by co-immunoprecipitation but hardly by ELISA (Figure 3C). After a 13-month follow-up, ADAMTS13-specific immune complexes were no longer detectable by either co-immunoprecipitation or ELISA.
Influence of rituximab treatment on free and complexed anti-ADAMTS13 antibody levels
Clinical remission samples were available from ten patients who had received rituximab during the acute event. Despite this small population, a possible effect of rituximab on anti-ADAMTS13 antibody levels was assessed. The median remission for these patients was 2 years (range, 0.5–4 years). Five patients still had detectable levels of free IgG anti-ADAMTS13 antibodies (median titer 50; range 25–100) of subclasses IgG1 and IgG4 (Figure 4). ADAMTS13-specific immune complexes were detected in nine of the ten patients, and were mainly subclass IgG4 (8/9 patients), with a median titer of 100 (range, 50–200), although IgG1-complexes and IgG2-complexes were also detected in a few patients (2/9 patients in both instances). There were no statistically significant differences in ADAMTS13:Ac, ADAMTS13 functional inhibitor titers or free and complexed anti-ADAMTS13 antibody titers between rituximab-treated and non-treated patients in remission (data not shown). Among the patients treated with rituximab, only one (10%) relapsed, whereas 5/18 (28%) patients (median remission of 6 months; range, 6 months–1.5 year) not treated with rituximab relapsed.
Figure 4.
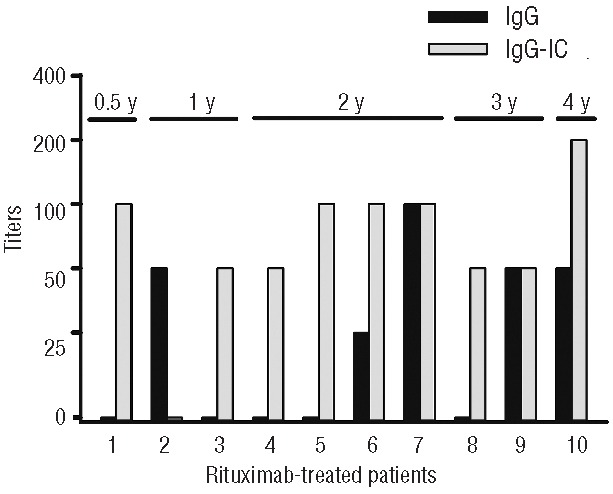
Detection of free and complexed antibodies in patients with acquired TTP treated with rituximab and in remission for at least 1 year. Plasma samples from patients (1–10) treated with rituximab during the acute event were analyzed by ELISA for the presence of free and complexed IgG anti-ADAMTS13 antibodies during clinical remission. The duration of remission (y=years) is shown for each patient. ADAMTS13-specific immune complexes were mainly of subclass IgG4. IC: immune complexes.
Discussion
The pathogenesis of acquired TTP is characterized by severe ADAMTS13 deficiency due to anti-ADAMTS13 autoantibodies, inhibiting its activity or enhancing its clearance.3,5,7,26 Binding of these antibodies to ADAMTS13 eventually results in the formation of antigen-antibody immune complexes that might have a potentially pathogenic role. In this study, we determined the prevalence of free and complexed anti-ADAMTS13 antibodies in 78 patients with acquired TTP. ADAMTS13-specific immune complexes were detected in most patients during the acute event (97%; n=68) and in remission (93%; n=28), with IgG4-complexes being the predominant subclass.
ADAMTS13-specific immune complexes were detected with a newly developed ELISA-based assay and, as a complementary method, a co-immunoprecipitation assay.23 In the ELISA, a polyclonal anti-ADAMTS13 antibody was used to capture ADAMTS13-specific immune complexes present in plasma; the composition of the immunoglobulin fraction of the immune complexes was revealed by antibodies directed against human IgG1-4, IgM and IgA. A limitation of the ELISA set-up is that it only detects immune complexes bearing an ADAMTS13 molecule with accessible epitope(s) for the capturing antibody. As a consequence, large complexes harboring many bound antibody molecules on ADAMTS13 are likely to be underestimated. Co-immunoprecipitation circumvents this problem as the immune complexes are isolated from plasma through the Fc portion of the IgG fraction using Protein G, and co-isolated ADAMTS13 is detected by subsequent western blot analysis using an anti-human ADAMTS13 antibody. However, this method fails to reveal IgG subclass composition of the immune complexes, and quantification relies on densitometric evaluation of immunoblot signals, which is prone to inaccuracy. Thus, both methods have their strengths and weaknesses. Nonetheless, we have proven that, for most samples, the assays are comparable in sensitivity and specificity, making them suitable for immune complex detection in plasma. By contrast, previously employed methods detected immune complexes only indirectly by measuring ADAMTS13:Ag or ADAMTS13:Ac in IgG-depleted plasma20,21 or by a poorly reported direct detection of the IgG antibody portion of the ADAMTS13-immune complexes using a commercial kit to quantify ADAMTS13:Ag levels in a limited number of patients.22
The most relevant finding of our study was the detection of IgG-immune complexes in 93% of patients in remission, independently of the duration of the remission. This strongly indicates that ADAMTS13-specific immune complexes circulate in patients with acquired TTP patients throughout the entire course of the disease and many years after clinical remission. That immune complexes were detectable in remission despite normalization of other ADAMTS13-related parameters might be explained by a low-level, steady-state production of autoantibodies that is triggered by follicular dendritic cells. These accessory immune cells are frequently found in autoimmune diseases to retain immune complexes on their surface for years thereby providing a constant antigen depot for memory B-cell stimulation.27 Since only a limited number of patients (6/28; 21%) tested in remission relapsed, the clinical relevance of circulating ADAMTS13-specific immune complexes in remission remains to be established in prospective studies.
Free anti-ADAMTS13 IgG were mainly of subclasses IgG1 and IgG4, in accordance with previous studies,8,9 and no IgG subclass switch was observed between the acute phase and in remission. For most patients, the distribution of subclasses for free and complexed IgG anti-ADAMTS13 antibodies correlated, suggesting a similar avidity for ADAMTS13 among IgG subclasses. Interestingly though, ADAMTS13-specific immune complexes were predominantly of subclass IgG4 (92% of patients during the acute phase and also 92% in remission), whereas all other subclasses were detected at a substantially lower rate (Figure 1).
IgG4 is usually the least effective IgG subclass in terms of final effector functions because of its inability to activate complement via the classical pathway and its low affinity to cellular Fcγ receptors (FcγR).28 The extent to which IgG4 elicits effector functions depends on the immunoglobulin’s environment. As a free monomer, IgG4 only interacts with high-affinity FcγRIA, while when part of an immune complex, binding to the low-affinity FcγRIIA, FcγRIIB and FcγRIIIA is also observed.29,30 Interestingly, the size of the immune complexes also affects the engagement with FcγR. Compared to small IgG4-complexes, large IgG4-complexes show enhanced binding to the low-affinity FcγR29,30 suggesting that only the latter species has the potential to trigger activation signals and effector functions.
However, IgG4 tends to form small immune complexes, since in vivo, IgG4 antibodies are involved in a half-molecule exchange process rendering IgG4 antibodies functionally monovalent for a specific antigen.31,32 Furthermore, epitope mapping studies revealed that in most cases, anti-ADAMTS13 antibodies from TTP patients target a hot-spot inside the ADAMTS13 spacer domain.9,33,34 It is, therefore, conceivable that a substantial portion of the immune complexes in TTP patients is composed of ADAMTS13 bound to a single antibody molecule. Thus, one would expect that ADAMTS13-specific IgG4-complexes are indeed small and therefore exhibit limited effector functions. Notably, small immune complexes are reported to escape clearance by the reticuloendothelial system.35 The resulting longer plasma half-life of IgG4-complexes might contribute to their prefential detection in the TTP patients’ samples.
Although the effector functions are limited, a pathogenic role has been described for IgG4 in certain autoimmune diseases, such as recruitment and activation of neutrophils, induction of leukocyte-dependent dermal-epidermal separation and leukocyte-dependent tissue damage. At least in some cases, these effects appear to be independent of complement activation and FcγR binding;36–38 however, the role of IgG4 antibodies in the pathogenesis of acquired TTP needs further investigation.
Analysis of three patients during treatment and remission confirmed the presence of ADAMTS13-specific immune complexes throughout the observation period (Figure 3). This is consistent with results for a previously described patient with refractory acquired TTP.23 Furthermore, the inverse correlation between free and complexed anti-ADAMTS13 antibodies seen in this patient during treatment was also discernible for the three patients during the early acute event while on PEX. However, no clear such trend was found in remission. These findings may reflect the continuous variation in levels of ADAMTS13, auto-antibodies and immune complexes throughout PEX therapy which also determines the rate of immune complex formation and clearance. It appears that the courses of free and complexed antibodies during treatment are complex and depend on treatment modalities as well as the responses of individual patients.
The current standard treatment for acute idiopathic TTP is PEX and immunosuppressive therapy with corticosteroids. Over the last years, rituximab, a humanized antibody that targets CD20 expressed on mature and memory B cells but not on plasma cells,39 has successfully been administered as an adjuvant therapy for refractory and relapsing acquired TTP, and treated patients seem to benefit through long-lasting remission.40–43 In our study, we found that the ten patients treated with rituximab during the acute event still had detectable free and/or complexed anti-ADAMTS13 antibodies in remission; in some of the patients with long-term remission, ADAMTS13-specific immune complexes were detectable even after years. Moreover, there was no significant difference in free and complexed anti-ADAMTS13 antibody titers in these patients compared to those in patients not treated with rituximab.
In summary, we have reported the findings of the first comprehensive study on the presence of ADAMTS13-specific immune complexes in a large cohort of patients with acquired TTP. ADAMTS13-specific immune complexes persisted in a vast majority of patients during remission, even in the presence of normal ADAMTS13:Ac and ADAMTS13:Ag levels. Whether determination of circulating ADAMTS13-specific immune complexes in remission is of value in predicting potential relapse needs to be further investigated. Most immune complexes contained IgG4, a subclass with limited effector functions, likely only neutralizing ADAMTS13:Ac; nonetheless, future work needs to address the role of ADAMTS13-bound IgG4 antibodies in the pathogenesis of acquired TTP.
Acknowledgments
The authors would like to thank Dr. François Provot (Department of Nephrology, Lille University Hospital, France), Dr. Jean-Louis Bacri and Dr. Raynald Binaut (Department of Nephrology, Valenciennes Hospital, France) for providing patients’ plasma samples and clinical information. We are indebted to Karima Benamara for the expert editing of the manuscript.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589–600 [DOI] [PubMed] [Google Scholar]
- 2.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–94 [DOI] [PubMed] [Google Scholar]
- 3.Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578–84 [DOI] [PubMed] [Google Scholar]
- 4.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rieger M, Mannucci PM, Kremer Hovinga JA, Herzog A, Gerstenbauer G, Konetschny C, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood. 2005;106(4):1262–7 [DOI] [PubMed] [Google Scholar]
- 6.Tsai HM, Raoufi M, Zhou W, Guinto E, Grafos N, Ranzurmal S, et al. ADAMTS13-binding IgG are present in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2006;95(5):886–92 [PMC free article] [PubMed] [Google Scholar]
- 7.Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4(8):1707–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7(10):1703–10 [DOI] [PubMed] [Google Scholar]
- 9.Pos W, Sorvillo N, Fijnheer R, Feys HB, Kaijen PH, Vidarsson G, et al. Residues Arg568 and Phe592 contribute to an antigenic surface for anti-ADAMTS13 antibodies in the spacer domain. Haematologica. 2011;96(11):1670–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettoni G, Palla R, Valsecchi C, Consonni D, Lotta LA, Trisolini SM, et al. ADAMTS-13 activity and autoantibodies classes and subclasses as prognostic predictors in acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2012;10(8):1556–65 [DOI] [PubMed] [Google Scholar]
- 11.Ferrari S, Scheiflinger F, Rieger M, Mudde G, Wolf M, Coppo P, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig iso-type, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood. 2007;109(7):2815–22 [DOI] [PubMed] [Google Scholar]
- 12.Novak J, Julian BA, Tomana M, Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol. 2008;28(1):78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weissmann G. Rheumatoid arthritis and systemic lupus erythematosus as immune complex diseases. Bull NYU Hosp Jt Dis. 2009;67(3):251–3 [PubMed] [Google Scholar]
- 14.Toong C, Adelstein S, Phan TG. Clearing the complexity: immune complexes and their treatment in lupus nephritis. Int J Nephrol Renovasc Dis. 2011;4:17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayer AS, Theofilopoulos AN, Eisenberg R, Friedman SG, Guze LB. Thrombotic thrombocytopenic purpura-like syndrome associated with infective endocarditis. A possible immune complex disorder. JAMA. 1977;238(5):408–10 [PubMed] [Google Scholar]
- 16.Bukowski RM, King JW, Hewlett JS. Plasmapheresis in the treatment of thrombotic thrombocytopenic purpura. Blood. 1977;50(3):413–7 [PubMed] [Google Scholar]
- 17.Celada A, Perrin LH. Circulating immune complexes in thrombotic thrombocytopenic purpura (TTP). Blood. 1978;52(4):855. [PubMed] [Google Scholar]
- 18.Neame PB, Hirsh J. Circulating immune complexes in thrombotic thrombocytopenic purpura (TTP). Blood. 1978;51(3):559–60 [PubMed] [Google Scholar]
- 19.Furlan M, Robles R, Solenthaler M, Lämmle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91(8):2839–46 [PubMed] [Google Scholar]
- 20.Rieger M, Ferrari S, Kremer Hovinga JA, Konetschny C, Herzog A, Koller L, et al. Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA). Thromb Haemost. 2006;95(2):212–20 [DOI] [PubMed] [Google Scholar]
- 21.Froehlich-Zahnd R, George JN, Vesely SK, Terrell DR, Aboulfatova K, Dong JF, et al. Evidence for a role of anti-ADAMTS13 autoantibodies despite normal ADAMTS13 activity in recurrent thrombotic thrombocytopenic purpura. Haematologica. 2012;97(2):297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S, Jin M, Lin S, Cataland S, Wu H. ADAMTS13 activity and antigen during therapy and follow-up of patients with idiopathic thrombotic thrombocytopenic purpura: correlation with clinical outcome. Haematologica. 2011;96(10):1521–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrari S, Knoebl P, Kolovratova V, Plaimauer B, Turecek PL, Varadi K, et al. Inverse correlation of free and immune complex-sequestered anti-ADAMTS13 antibodies in a patient with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2012;10(1):156–8 [DOI] [PubMed] [Google Scholar]
- 24.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129(1):93–100 [DOI] [PubMed] [Google Scholar]
- 25.Plaimauer B, Kremer Hovinga JA, Juno C, Wolfsegger MJ, Skalicky S, Schmidt M, et al. Recombinant ADAMTS13 normalizes von Willebrand factor-cleaving activity in plasma of acquired TTP patients by overriding inhibitory antibodies. J Thromb Haemost. 2011;9(5):936–44 [DOI] [PubMed] [Google Scholar]
- 26.Scheiflinger F, Knoebl P, Trattner B, Plaimauer B, Mohr G, Dockal M, et al. Nonneutralizing IgM and IgG antibodies to von Willebrand factor-cleaving protease (ADAMTS-13) in a patient with thrombotic thrombocytopenic purpura. Blood. 2003;102(9):3241–3 [DOI] [PubMed] [Google Scholar]
- 27.El Shikh ME, Pitzalis C. Follicular dendritic cells in health and disease. Front Immunol. 2012;3(292):1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–77 [DOI] [PubMed] [Google Scholar]
- 29.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–25 [DOI] [PubMed] [Google Scholar]
- 30.Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J Immunol. 2013;190(8):4315–23 [DOI] [PubMed] [Google Scholar]
- 31.van der Neut KM, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317(5844):1554–7 [DOI] [PubMed] [Google Scholar]
- 32.van der Zee JS, van SP, Aalberse RC. Serologic aspects of IgG4 antibodies. II. IgG4 antibodies form small, nonprecipitating immune complexes due to functional monovalency. J Immunol. 1986;137(11):3566–71 [PubMed] [Google Scholar]
- 33.Luken BM, Turenhout EA, Kaijen PH, Greuter MJ, Pos W, van Mourik JA, et al. Amino acid regions 572–579 and 657–666 of the spacer domain of ADAMTS13 provide a common antigenic core required for binding of antibodies in patients with acquired TTP. Thromb Haemost. 2006;96(3):295–301 [DOI] [PubMed] [Google Scholar]
- 34.Zheng XL, Wu HM, Shang D, Falls E, Skipwith CG, Cataland SR, et al. Multiple domains of ADAMTS13 are targeted by autoantibodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura. Haematologica. 2010;95(9):1555–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shmagel KV, Chereshnev VA. Molecular bases of immune complex pathology. Biochemistry (Mosc.). 2009;74(5):469–79 [DOI] [PubMed] [Google Scholar]
- 36.Hussain A, Pankhurst T, Goodall M, Colman R, Jefferis R, Savage CO, et al. Chimeric IgG4 PR3-ANCA induces selective inflammatory responses from neutrophils through engagement of Fcgamma receptors. Immunology. 2009;128(2):236–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filippone EJ. Idiopathic membranous nephropathy and IgG4: an interesting relationship. Clin Nephrol. 2013;February 5 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mihai S, Chiriac MT, Herrero-Gonzalez JE, Goodall M, Jefferis R, Savage CO, et al. IgG4 autoantibodies induce dermal-epidermal separation. J Cell Mol Med. 2007;11(5):1117–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15(Suppl 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elliott MA, Heit JA, Pruthi RK, Gastineau DA, Winters JL, Hook CC. Rituximab for refractory and or relapsing thrombotic thrombocytopenic purpura related to immune-mediated severe ADAMTS13-deficiency: a report of four cases and a systematic review of the literature. Eur J Haematol. 2009;83(4):365–72 [DOI] [PubMed] [Google Scholar]
- 41.Westwood JP, Webster H, McGuckin S, McDonald V, Machin SJ, Scully M. Rituximab for thrombotic thrombocytopenic purpura: benefit of early administration during acute episodes and use of prophylaxis to prevent relapse. J Thromb Haemost. 2013;11(3):481–90 [DOI] [PubMed] [Google Scholar]
- 42.Schaller M, Studt JD, Voorberg J, Kremer Hovinga JA. Acquired thrombotic thrombocytopenic purpura. Development of an autoimmune response. Hamostaseologie. 2013;33(2):121–30 [DOI] [PubMed] [Google Scholar]
- 43.Goyal J, Adamski J, Lima JL, Marques MB. Relapses of thrombotic thrombocytopenic purpura after treatment with rituximab. J Clin Apher. 2013;28(6):390–4 [DOI] [PubMed] [Google Scholar]
- 44.Feys HB, Vandeputte N, Palla R, Peyvandi F, Peerlinck K, Deckmyn H, et al. Inactivation of ADAMTS13 by plasmin as a potential cause of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2010;8(9):2053–62 [DOI] [PubMed] [Google Scholar]