

Myelofibrotic transformation is a well-recognized complication of essential thrombocythemia (ET) and polycythemia vera (PV).1,2 However, information is scarce on the life expectancy and prognostic factors of patients developing this complication.1,3–5 Prognostic models devised for primary myelofibrosis (PMF) are used to drive treatment decisions in patients with post-ET/PV MF, despite the lack of studies validating the prediction accuracy of such stratification models in this setting. Our aim was to evaluate the performance of the most widely used prognostic model in PMF, i.e. the International Prognostic Scoring System (IPSS),6 in a nationwide series of patients with post-ET/PV MF.
Data from 176 patients diagnosed with post-ET (n=115) or post-PV (n=61) MF in 39 Spanish institutions during January/2000-May/2013 were retrospectively analyzed. Diagnosis was made according to the criteria in use at the time of first observation. Treatment for MF included hydroxycarbamide (n=100) or other cytoreductive therapy (n=25), erythropoiesis-stimulating agents (n=45), JAK inhibitors (n=45), danazol (n=12), immunomodulators (n=12), splenectomy (n=3), splenic irradiation (n=7), and allogeneic transplantation (n=11). Variables taken at MF diagnosis and analyzed for their predictive value on outcomes were gender, previous myeloproliferative neoplasm, splenomegaly, IPSS risk category, each individual factor of the IPSS, thrombocytopenia, JAK2 status, cytogenetics,7 and whether or not the patient was on cytoreductive therapy at MF diagnosis. Multivariate analysis of factors predicting survival was performed by the Cox regression method. Cumulative incidence of acute myeloid leukemia (AML) or AML-unrelated death was analyzed in the framework of competing risks. Multivariate analyses of factors predicting each of the above competing outcomes were performed by the Fine and Gray method.8 Log rank test was used to compare Kaplan-Meier curves. P<0.05 was considered statistically significant.
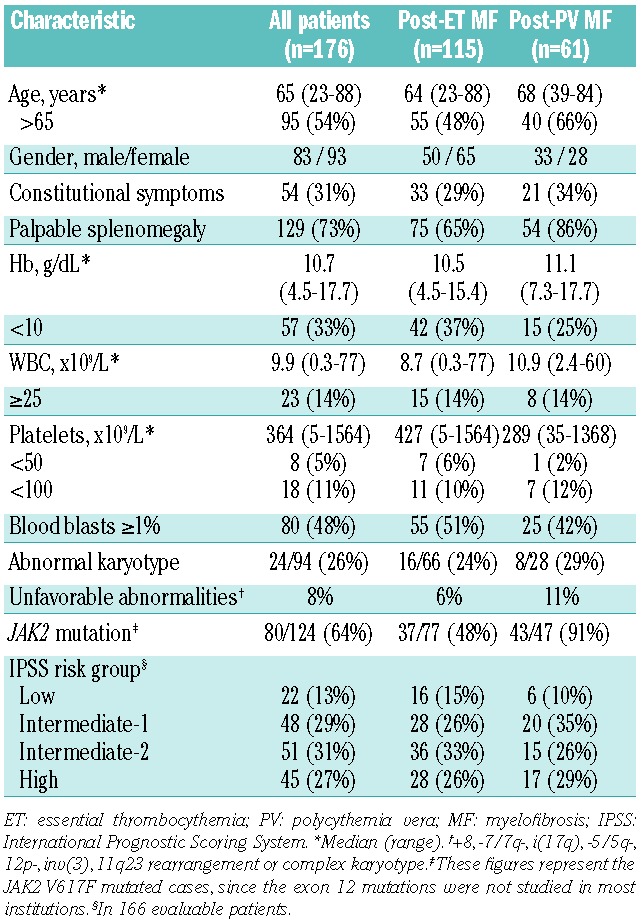
Table 1 shows the patients’ characteristics at MF presentation. Median time from original diagnosis to myelofibrosis was 8.6 years for post-ET MF and 9.8 years for post-PV MF. With a median follow up from post-ET/PV MF diagnosis of 1.8 years, 50 (29%) patients had died, 5 (3%) were lost to follow up, and the remaining were censored alive. Median survival was 8.6 years. Causes of death included progression of MF without AML (n=18), AML (n=11), cardiovascular complications (n=7), transplant-related complications (n=4), infection (n=3), bleeding (n=2), a second malignancy (n=1), or were unknown (n=4).
Table 1.
Demographic and base-line clinical characteristics of patients at diagnosis of post-ET and post-PV MF.
According to the IPSS, 13% patients were in the low-risk group, 29% in the intermediate-1, 31% in the intermediate-2, and 27% in the high-risk category, and their median survivals were, respectively, not yet reached, 10, 8.5, and 3.1 years. There was no statistically significant difference in survival between the low-risk and the intermediate-1 categories, or between the latter and the intermediate-2, whereas the high-risk group had a significantly poorer survival than the intermediate-2 (P=0.008) (Figure 1). Among factors included in the IPSS, older age, anemia, and circulating blasts retained a univariate association with shorter survival, whereas constitutional symptoms and leukocytosis lacked prognostic value. There was no significant difference in survival between patients with a prior diagnosis of PV or ET. The best predictive model for shorter survival included the following independent variables: age over 65 years (Hazard ratio (HR)=3.6; 95% Confidence Interval (CI):1.8–7.3; P<0.001); Hb <10 g/dL (HR=2.6; 95%CI: 1.4–4.6; P=0.002); platelets <100×109/L (HR=3.5; 95%CI: 1.7–7.3; P=0.001); and being on hydroxycarbamide at MF diagnosis (HR=2.7; 95%CI: 1.5–5.9; P=0.002).
Figure 1.
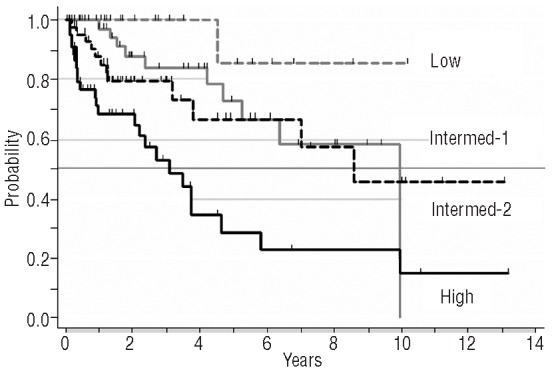
Survival after diagnosis of post-ET/PV myelofibrosis according to the IPSS risk category.
Progression to AML occurred in 12 (6.8%) patients over an observation period of 509 patient-years, an incidence rate of 2.3 cases per 100 patient-years. Thrombocytopenia less than 100×109/L was the only predictor for progression to AML (HR=5.45; 95%CI: 1.51–19.6; P=0.01), whereas age over 65 years (HR=2.58; 95%CI: 1.20–5.55; P=0.01), anemia (HR=2.45; 95%CI: 1.22–4.92; P=0.01), and being on hydroxycarbamide at myelofibrotic transformation (HR=1.96; 95%CI: 0.98–3.90; P=0.05) were associated with AML-unrelated death.
We presume that the lack of prognostic significance of some variables of the IPSS may be due to the effect of the cytoreductive treatment that many patients were receiving at the time of myelofibrotic transformation for the management of ET or PV. This situation differs from that of PMF patients in whom the risk factors at disease diagnosis are usually computed without any myelosuppressive treatment. Other factors could have also influenced our findings. Thus, 39% of patients with constitutional symptoms received JAK inhibitors, whereas this treatment was used in only 19% of those without such symptoms at MF diagnosis. Ruxolitinib has been associated with a reduction in the risk of death compared to conventional therapy,9 which could presumably have blunted the poor prognosis associated with this feature.
In line with our results, in an Italian series4 of 68 patients with post-PV MF, anemia was the only predictor for survival at disease presentation, whereas age and leukocyte count lacked prognostic significance. In 66 young patients with post-ET/PV MF from the Mayo Clinic,3 anemia was again an independent risk factor for shortened survival, although the strongest adverse factor was the unfavorable cytogenetics. Neither constitutional symptoms nor the leukocyte count predicted for survival.
By multivariate analysis, two variables not included in the IPSS, namely thrombocytopenia and hydroxycarbamide treatment at myelofibrotic transformation, were shown to correlate with survival. The former has been identified as a poor prognostic factor in PMF10,11 and post-PV MF.4 Low platelets are often associated with anemia, making it difficult to qualify thrombocytopenia as an independent prognostic factor, which was the reason why this variable was excluded from the IPSS.6 In our study, thrombocytopenia was an independent predictor for shorter survival probably because it was associated with a higher risk of AML. Such an association has also been reported in PMF.10–13 With regard to the poor prognostic significance of being on hydroxycarbamide at MF diagnosis, we think that some kind of selection bias may be operating here. Indeed, hydroxycarbamide is usually indicated in older patients and in those with more marked myeloproliferative features, and this may have contributed to blur the prognostic significance of such features.
In conclusion, the results from the present study indicate that the IPSS fails to accurately discriminate different prognostic groups in post-ET/PV MF. An alternative tool is, therefore, required for patients’ risk stratification to help physicians in the therapeutic decision-making process.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Cervantes F, Alvarez-Larran A, Talarn C, Gomez M, Montserrat E. Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br J Haematol. 2002;118(3):786–90 [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Larran A, Bellosillo B, Martinez-Aviles L, Saumell S, Salar A, Abella E, et al. Postpolycythaemic myelofibrosis: frequency and risk factors for this complication in 116 patients. Br J Haematol. 2009;146(5):504–9 [DOI] [PubMed] [Google Scholar]
- 3.Dingli D, Schwager SM, Mesa RA, Li CY, Dewald GW, Tefferi A. Presence of unfavorable cytogenetic abnormalities is the strongest predictor of poor survival in secondary myelofibrosis. Cancer. 2006;106(9):1985–9 [DOI] [PubMed] [Google Scholar]
- 4.Passamonti F, Rumi E, Caramella M, Elena C, Arcaini L, Boveri E, et al. A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood. 2008;111(7):3383–7 [DOI] [PubMed] [Google Scholar]
- 5.Guglielmelli P, Barosi G, Pieri L, Antonioli E, Bosi A, Vannucchi AM. JAK2V617F mutational status and allele burden have little influence on clinical phenotype and prognosis in patients with post-polycythemia vera and post-essential thrombocythemia myelofibrosis. Haematologica. 2009;94(1):144–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–901 [DOI] [PubMed] [Google Scholar]
- 7.Caramazza D, Begna KH, Gangat N, Vaidya R, Siragusa S, Van Dyke DL, et al. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients. Leukemia. 2011;25(1):82–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;(94):496–509 [Google Scholar]
- 9.Cervantes F, Vannucchi AM, Kiladjian JJ, Al-Ali HK, Sirulnik A, Stalbovskaya V, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–53 [DOI] [PubMed] [Google Scholar]
- 10.Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–7 [DOI] [PubMed] [Google Scholar]
- 11.Morel P, Duhamel A, Hivert B, Stalniekiewicz L, Demory JL, Dupriez B. Identification during the follow-up of time-dependent prognostic factors for the competing risks of death and blast phase in primary myelofibrosis: a study of 172 patients. Blood. 2010;115(22):4350–5 [DOI] [PubMed] [Google Scholar]
- 12.Huang J, Li CY, Mesa RA, Wu W, Hanson CA, Pardanani A, et al. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer. 2008;112(12):2726–32 [DOI] [PubMed] [Google Scholar]
- 13.Tam CS, Kantarjian H, Cortes J, Lynn A, Pierce S, Zhou L, et al. Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J Clin Oncol. 2009;27(33):5587–93 [DOI] [PMC free article] [PubMed] [Google Scholar]