

Abstract
Rationale
AMP-activated protein kinase (AMPK) is a master regulator of cell metabolism and an attractive drug target for cancer, metabolic and cardiovascular diseases. Point mutations in the regulatory γ2-subunit of AMPK (encoded by Prkag2 gene) caused a unique form of human cardiomyopathy characterized by cardiac hypertrophy, ventricular pre-excitation and glycogen storage. Understanding the disease mechanisms of Prkag2 cardiomyopathy is not only beneficial for the patients but also critical to the utility of AMPK as a drug target.
Objective
We sought to identify the pro-growth signaling pathway(s) triggered by Prkag2 mutation and to distinguish it from the secondary response to glycogen storage.
Methods and Results
In a mouse model of N488I mutation of the Prkag2 (R2M), we rescued the glycogen storage phenotype by genetic inhibition of glucose-6-phosphate stimulated glycogen synthase activity. Ablation of glycogen storage eliminated the ventricular pre-excitation but did not affect the excessive cardiac growth in R2M mice. The pro-growth effect in R2M hearts was mediated via increased insulin sensitivity and hyperactivity of Akt, resulting in activation of mTOR and inactivation of FoxO signaling pathways. Consequently, cardiac myocyte proliferation during the postnatal period was enhanced in R2M hearts followed by hypertrophic growth in adult hearts. Inhibition of mTOR activity by rapamycin or restoration of FoxO activity by overexpressing FoxO1 rescued the abnormal cardiac growth.
Conclusions
Our study reveals a novel mechanism for Prkag2 cardiomyopathy independent of glycogen storage. The role of γ2-AMPK in cell growth also has broad implications in cardiac development, growth and regeneration.
Keywords: Mutation in AMPK γ2 subunit, glycogen storage, cardiac hypertrophy, proliferation
INTRODUCTION
AMP-activated protein kinase (AMPK) is known as a fuel gauge and a master regulator of cell metabolism1,2. Activation of AMPK during cellular stress promotes ATP generation via increases of glucose uptake and fatty acid oxidation while it inhibits energy consuming processes such as protein and lipid synthesis. These unique functions of AMPK make it an attractive drug target for metabolic and cardiovascular diseases as well as cancer. AMPK is a heterotrimeric complex composed of a catalytic α-subunit and regulatory β- and γ-subunits with multiple isoforms for each subunit. The γ-subunit is the energy sensor of the complex; competitive binding of ATP against ADP or AMP to the γ-subunit regulates the kinase activity3,4. Two isoforms of the γ-subunit are expressed in the heart, γ1 and γ2. Point mutations in the nucleotide-binding region of the γ2-subunit, encoded by the Prkag2 gene, cause a distinct form of human cardiomyopathy characterized by glycogen storage, pre-excitation arrhythmia and cardiac hypertrophy5–7. Previous studies using mouse models expressing mutant Prkag2 genes in the heart recapitulated the characteristics of human cardiomyopathy and demonstrated that the phenotype was caused by an aberrant increase of kinase activity6,8,9. Metabolic analysis of the mutant mouse hearts show that activation of AMPK in the absence of energy deficit results in global remodeling of the metabolic network in favor of glycogen storage8,10,11. Since the cardiac phenotype of Prkag2 mutation is similar to glycogen storage cardiomyopathy, we sought to determine whether excessive glycogen accumulation is the unifying mechanism responsible for the Prkag2 cardiomyopathy. In a mouse model with cardiac-specific overexpression of the N488I mutant of Prkag2 (R2M), we rescued the glycogen storage phenotype by targeting glucose-6-phosphate stimulated glycogen synthesis via genetic manipulations. Here we show that excessive glycogen accumulation is primarily responsible for ventricular pre-excitation but not cardiac hypertrophy. Rather, the mutation of γ2-AMPK stimulates proliferation and hypertrophy pathways via FoxO and mTOR signaling cascades leading to abnormal cardiac growth.
METHODS
Animal models
Transgenic mice overexpressing N488I AMPK γ2 (R2M), FoxO1 (FO) and harboring a knock-in mutation in GYS1(KI) were generated as described6,12,13. R2M-KI (DM) and R2M-FO (DM-FO) double mutant were generated on an FVB background. Wild type littermates of transgenic mice were used as controls (NTG).
Two weeks old mice were treated with rapamycin (2 mg/kg body weight, i.p.) daily for 4 weeks. Rapamycin (LC Laboratories) was dissolved in DMSO and re-suspended in vehicle (0.2% carboxymethyl cellulose and 0.25% polysorbate-80) before injection. For insulin injection, mice were fasted overnight and anaesthetized with pentobartital (80 mg/kg body weight, i.p.). Heart samples were freeze-clamped 20 minutes after insulin (0.5 U/kg body weight, i.p.). For BrdU labeling experiments, one week old mice were injected with BrdU (50 mg/kg body weight, i.p.) daily for 7 days, and hearts were subsequently harvested and fixed in 10% neutral buffered formalin for immunohistochemistry.
All animal procedures were approved by the institutional IACUC committee at the University of Washington.
Echocardiography and ECG
Murine echocardiography was performed using a Vevo770 high resolution imaging system (VisualSonics Inc.). Electrocardiogram was recorded using implantable wireless monitoring device with DSI mouse ECG transmitter ETA-F10.
Cardiac glycogen synthase activity and glycogen content
Glycogen synthase activity was measured using the method of Thomas et. al.14. Glycogen content was determined a glucose assay kit (Sigma-Aldrich) as described10.
Glucose uptake and myocardial energetics
31P nuclear magnetic resonance spectroscopy was used to measure glucose uptake rate, ATP, and phosphocreatine with nontracer 2-deoxyglucose as described8,10.
Western blot analysis
Protein samples were prepared from frozen heart samples using a lysis buffer containing protease inhibitors (Sigma). Nuclear and cytosolic fractions were prepared according to instructions of NE-PER extraction kit (Pierce). Tissue lysates were matched for protein concentration and then separated by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane (Bio-Rad laboratories). Membranes were blocked in 5% non-fat milk and incubated with primary antibodies overnight at 4°C. Membranes were incubated with appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) (Pierce) and signal intensities were visualized by Chemiluminescence (Cell Signaling Technology). Films from at least four independent experiments were scanned and densities of the immunoreactive bands were evaluated using NIH Image software.
Immunohistochemistry
Mouse hearts were arrested in diastole with KH buffer containing 30 mM KCl and fixed in 10% neutral buffered formalin6. After stained with appropriate antibodies, the positive signal was detected using confocal microscopy (Zeiss LSM510Meta).
Real time PCR
Real Time PCR was performed using SYBR green (Bio-Rad) as described15.
Statistics
Data are expressed as a mean ± s.d. Comparisons among the groups were performed by 1-way ANOVA and followed by a post hoc Tukey-Kramer test. The comparison between 2 groups was performed using 2 tailed Student’s t tests. A p<0.05 was considered statistically significant.
RESULTS
Rescue of pathological glycogen storage in the R2M heart eliminated ventricular pre-excitation but did not affect cardiac hypertrophy
AMPK is an important regulator of glucose metabolism, and R2M hearts demonstrate significant increases in glucose uptake and intracellular [G-6-P], the major allosteric stimulator for glycogen synthesis10. To determine whether glycogen storage caused by excessive G-6-P accounts for the fundamental disease mechanisms of Prkag2 cardiomyopathy, we sought to block G-6-P stimulated glycogen synthesis in R2M hearts by introducing a knockin mutation (R582A, KI) in the muscle form of glycogen synthase (GSY1). The GYS activity was significantly reduced and the response to G-6-P was eliminated in the heart of KI or DM mice (Figure 1A). Glycogen content was lower in KI heart and glycogen accumulation was markedly reduced (by ~6-fold, p<0.0001) in DM heart (Figure 1B). Reduced glycogen storage was also demonstrated by H&E staining of heart sections in which intracellular vacuoles representing glycogen storage sites in live cells were prominent in R2M hearts but were absent in DM hearts (Figure 1C). The KI did not affect the expression of mutant γ2-AMPK, the AMPK activity, basal glucose uptake rate or the myocardial energetic status of the R2M hearts (Online Figure I).
Figure 1. Targeting muscle form of glycogen synthase (GYS1) rescued glycogen storage and pre-excitation phenotype but not cardiac hypertrophy in R2M hearts.

(A) GYS activity in the absence (□) or presence (■) of glucose-6-phosphate (G6P, 10 mM) in the cardiac tissue homogenates of NTG, R2M, DM, and KI mice. n=3. (B) Cardiac glycogen content in the four groups of mice. n=8. (C) Cardiac sections with Haematoxylin and Eosin (H&E) staining showing the glycogen containing vacuoles in R2M but not in DM mouse heart. n=3. Scale bar, 40 μm. (D) Left, representative electrocardiogram (ECG) in NTG (n=5), R2M (n=6), DM (n=8), and KI (n=5) mice. The shortened PR interval, characteristic of ventricular pre-excitation, was found in R2M but not in DM mice. Right, average PR intervals in the four groups of mice. (E) Heart weight to body weight ratios (HW/BW). n=7–10. (F) Left, representative images of cardiac sections stained with Wheat Germ Agglutinin (WGA). Scale bar, 40 μm. Right, average myocyte cross-sectional area determined from 2 cardiac sections stained with WGA in each heart showing that cross-sectional area of cardiomyocytes is increased in both R2M and DM mice. n=3 each group. All mice used for all experiments were 2 months old.; #p< 0.01 versus NTG; ‡p< 0.01 versus R2M.
Using ambulatory electrocardiogram (ECG) recording, we found that the shortened PR interval present in the ECG of R2M mice, characteristic of ventricular pre-excitation8, was restored in DM mice (Figure 1D). The abnormal annulus fibrosis structure was subsequently improved (Online Figure II), which is in agreement with the previous findings that glycogen causes ventricular preexcitation by disruption of annulus fibrosis in R2M mice. However, in spite of decreased glycogen content, heart weight to body weight ratios (HW/BW) of DM (9.3 ± 1.24 mg/g) remained similar to that of R2M (10.3 ± 1.44 mg/g), and both were significantly greater than that of non-transgenic littermates (NTG, 4.3 ± 0.17 mg/g) at 2 months (Figure 1E). In addition, cross-sectional area of cardiac myocytes was ~2-fold higher in either R2M or DM hearts compared to that in NTG hearts (p<0.01), suggesting marked cellular hypertrophy (Figure 1F). Abnormal cardiac growth in DM mice was further evidenced by increased left ventricular wall thickness assessed by echocardiography (Online Figure III). Noticeably, the reduction of glycogen storage partially improved cardiac function of R2M mice (Fractional shortening, 42 ± 11 in DM vs. 32 ± 7% in R2M and 58 ± 9 % in NTG, p<0.01). These data indicate that the excessive glycogen accumulation is primarily responsible for ventricular pre-excitation and partially accounts for the contractile dysfunction in the heart of R2M mice while the cardiac hypertrophy phenotype is independent of glycogen storage.
Insulin sensitivity was increased in R2M hearts
It has been shown that AMPK inhibits mammalian target of rapamycin (mTOR) signaling via phosphorylation of Tuberin/TSC (Ser1387) and raptor (Ser792)16,17. Consistent with increased AMPK activity in R2M, we observed increased phosphorylation of TSC and raptor at these sites (Online Figure IV). However, we also found that the phosphorylation level of Akt (S473) was significantly increased in R2M hearts (Figure 2A). Furthermore, a greater phosphorylation of TSC (Ser939, Thr1462) and raptor (Ser863) at the insulin stimulated phosphorylation sites was observed (Figure 2A). The increased insulin signaling pathway was also observed in DM hearts (Online Figure V). To determine whether insulin signaling is enhanced in R2M hearts, we subjected R2M mice to overnight fasting and compared Akt activation to that in fed mice. Fasting completely inhibited the phosphorylation of Akt and its downstream targets, mTOR and ribosomal protein S6, compared to ad lib feeding in both control and R2M mutant hearts (Figure 2B). To determine whether the greater phosphorylation of Akt in fed R2M heart was due to increased sensitivity to insulin we injected a low dose of insulin (0.5 U/kg, i.p.) in fasted mice. R2M hearts responded to insulin injection with a greater increase in the phosphorylation level of Akt (S473) (Figure 2C). It is unlikely that this is due to systemic insulin resistance because the cardiac specific R2M mice did not develop systemic insulin resistance in regard to no difference in body weight, blood glucose level, and glucose tolerance test (Online Figure VI). Rather, this may result from suppressed serine phosphorylation of insulin receptor substrate (IRS) 1 (Figure 3A) and relief of negative feedback of p70S6 kinase on insulin signaling pathway in consistent with a previous finding that AMPK increased sensitivity of glucose transport by insulin via suppression of serine phosphorylation of IRS1 in muscle cells18. In HEK293T cells expressing N488I mutant of γ2-AMPK, phosphorylation of Akt (S473) was also accentuated in response to low concentrations of insulin (0.1 and 1 nM) compared to cells expressing GFP (Figure 2D). Akt phosphorylation level did not differ between the two groups at high concentration of insulin (10 nM). Taken together, these data indicate that mutation of γ2-AMPK increases insulin sensitivity both in vitro and in vivo resulting in enhanced growth signaling mechanisms.
Figure 2. Insulin sensitivity was increased in the hearts of R2M mice.

(A) Left, representative immunoblot of whole heart lysates of NTG and R2M mice probed for phosphorylated Akt at Ser473 (P-Akt (S473)), total Akt (T-Akt), P-TSC2 (S939), P-TSC2 (T1462), T-TSC2, P-raptor (S863), and T-raptor with β-actin as a loading control. Right, quantification of band intensity on the immunoblot. Protein levels were normalized to β-actin and data were expressed as fold change relative to NTG, n=4. (B) Representative immunoblot of whole heart lysates of NTG and R2M mice after ad libitum feeding (Fed) or overnight fasting (Fast) for P-Akt (S473), T-Akt, P-mTOR (S2448), T-mTOR, P-S6 (S235/236), and T-S6. n=4. (C) Top, representative immunoblot of whole heart lysates of NTG and R2M mice 20 min after insulin (0.5 U/kg i.p.) or PBS injection and probed for P-Akt (S473) and T-Akt. Bottom, quantification of band intensity expressed as fold changes over NTG, n=5. (D) Top, representative immunoblot of P-Akt (S473) and T-Akt in HEK293T lysates transfected with mutant (N488I) γ2-AMPK (R2M) or GFP and treated with insulin at the concentrations of 0.1, 1, and 10 nM. Bottom, quantification of band intensity expressed as fold changes compared to GFP, n=3. All animal experiments used 2 month old mice.; #p< 0.01 versus NTG, GFP or PBS; ‡p< 0.01 versus insulin-treated NTG.
Figure 3. The mTOR pathway was activated in the hearts of R2M mice.

(A) Left, representative immunoblot of P-mTOR (S2448), T-mTOR, P-p70S6K (T389), T-p70S6K, P-S6 (S235/236), T-S6, P-4EBP1 (S65), T-4EBP1, P-IRS1 (S636/639), P-IRS1 (S302), P-IRS1 (S1101), T-IRS1, and β-actin in whole heart lysates of 2 month old NTG and R2M mice. Right, quantification of band intensity normalized by corresponding total protein levels, n=4. (B) Representative immunoblot (left) and quantification (right) of band intensity of P-mTOR (S2448), T-mTOR, P-Akt (S473), T-Akt, and β-actin in whole heart lysates of 2 week old NTG and R2M mice. n=4. (C) Representative immunoblot of P-S6 (S235/236), T-S6, P-Akt (S473), and T-Akt, in whole heart lysates of NTG and R2M mice after Rap treatment. (D) Heart weight (HW) to Tibia Length (TL) ratios after rapamycin (Rap, 2 mg/kg body weight, i.p.) or vehicle treatment. The mice were treated for 4 weeks starting at 2 weeks old, n=8; *p<0.05 versus NTG; #p< 0.01 versus vehicle-treated NTG; ‡p< 0.01 versus Rap-treated R2M.
Activation of the mTOR pathway partially contributes to cardiac hypertrophy in R2M
Consistent with increased insulin signaling, the phosphorylation level of mTOR was significantly increased in R2M hearts (Figure 2B and Figure 3A). Increased mTOR activity was further evidenced by increased phosphorylation of its downstream targets, p70S6 kinase (by 3.5 fold), ribosomal protein S6 (by 3.7 fold), and eukaryotic translation initiation factor 4E binding protein 4EBP1 (by 3.2 fold). We next sought to rescue the abnormal cardiac growth phenotype in R2M mice by decreasing mTOR activity through treatment with rapamycin (Rap). Two week old mice were treated with either vehicle or rapamycin (2 mg/kg i.p.) daily for 4 weeks. This time point was chosen because activation of Akt and mTOR was first detected in R2M mice at 2 weeks of age (Figure 3B). Rapamycin treatment effectively inhibited mTOR activity, manifested as the loss of S6 phosphorylation (Figure 3C). Rapamycin treatment as expected, did not affect phosphorylation of Akt (S473) in either genotype. After 4 weeks treatment, heart size of rapamycin-treated R2M was reduced by 45% compared to that of vehicle-treated R2M, as measured by heart weight to tibia length ratios (HW/TL). The reduced heart weight was attributed to a decrease in the cardiomyocyte size as the cross-sectional area of myocytes in rapamycin treated R2M hearts was significantly decreased (Online Figure VII). Rapamycin-treated control mice also showed a 20% reduction of HW/TL compared to vehicle-treated control. As a result, HW/TL of R2M-Rap remained ~87% greater than that of NTG-Rap (Figure 3D). Thus, inhibition of mTORC1 pathway by rapamycin only partially reduced the cardiac hypertrophy of R2M hearts.
The FoxO pathway was inactivated in R2M hearts
In searching for additional mechanisms responsible for the abnormal cardiac growth, we found that another downstream target of Akt, FoxO (Forkhead Box O transcription factor), was highly phosphorylated and inactivated in R2M hearts (Figure 4A). In R2M hearts, increased phosphorylation of FoxO3a at multiple Akt target sites was observed, and this was associated with decreased nuclear localization of FoxO3a protein and a lower level of total FoxO3a protein (Figure 4B) suggesting that increased phosphorylation of FoxO3a promoted its export from the nucleus and subsequent degradation in the cytosol. Increased phosphorylation of FoxO1a at Akt target sites was also observed but there was no change in total FoxO1a protein. Inactivation of FoxO was further indicated by changes in the expression of its downstream targets, MURF1 and Cyclin D1 (Figure 4A). Upregulation of Cyclin D1 resulted in a higher protein level in both the nuclear and cytosolic fractions (Figure 4C).
Figure 4. The FoxO pathway was inactivated in the hearts of R2M mice.

(A) Left, representative immunoblot of P-FoxO3a (S318), P-FoxO3a (S253), P-FoxO3a (T32), T-FoxO3a, P-FoxO1a(S256), T-FoxO1, MURF1, and Cyclin D1 in whole heart lysates of NTG and R2M mice with β-actin as a loading control. Right, quantification of band intensity normalized to β-actin, and data are expressed as fold change relative to NTG, n=4. (B) Representative immunoblot of P-FoxO3a (S318), T-FoxO3a in the nuclear and cytosolic fractions of hearts of NTG and R2M mice. Histone H3 was used as a nuclear protein loading control, and GAPDH was used as a cytosolic protein loading control, n=3. (C) Representative immunoblot of cyclin D1 in the nuclear and cytosolic fractions of NTG and R2M hearts, n=4. (D) HW/BW in NTG, R2M, transgenic mice over-expressing wild type of FoxO1 in the hearts (FO), and double mutant over-expressing both N488I mutant form of γ2-AMPK and wild type of FoxO1 in the hearts (DM-FO), n=8. (E) Immunoblot and expression levels of Cyclin D1, P-Akt (S473), T-Akt, P-S6 (S235/236), and T-S6, P-FoxO3a (S318), T-FoxO3a, and T-FoxO1a in whole heart lysates of NTG, R2M, FO and DM-FO mice, n=4. All mice used for all experiments were 2 months old; #p< 0.01 versus NTG; ‡p< 0.01 versus R2M.
To restore FoxO activity we crossed the R2M mice with transgenic mice overexpressing FoxO1a in the heart (FO) since FoxO3a overexpression mice are not viable and FoxO1a have been shown to inhibit cardiac hypertrophy by similar mechanisms as FoxO3a19. Overexpression of FoxO1a in R2M hearts (DM-FO) significantly reduced the heart weight (Figure 4D) and the cardiomyocyte cross-sectional area (236 ± 28 μm2 vs. 328 ± 55 μm2, p<0.01, Online Figure VIII). At the molecular level, the overexpression of FoxO1a decreased phosphorylation of FoxO3a and normalized total Fox3a protein in R2M hearts. The overexpression restored to the normal mRNA level of Cyclin D1 and MURF1 in DM-FO hearts (Figure 4E and Online Figure IX). The overexpression dramatically down-regulated cyclin D1 in R2M hearts, whereas it did not affect phosphorylation levels of Akt and S6 (Figure 4E). When administrated rapamycin, the DM-FO mice showed a fully normalized heart size compared to NTG (Online Figure VIII). These data indicate that changes in mTOR and FoxO signaling pathway are sufficient to account for to the excessive cardiac growth in R2M.
Cardiomyocyte proliferation was increased in R2M hearts during postnatal cardiac growth
Since the upregulation of cyclin D1 suggests a potential increase of cell proliferation in R2M, we determined if cellular hyperplasia contributes to the abnormal cardiac growth in R2M mice. Measurements of heart weight at multiple ages showed that increases in HW/BW started between 1–2 weeks, progressed through the postnatal period and leveled off after weaning (Figure 5A). At 2 weeks when HW/BW increased by 50% in R2M, cardiomyocyte size did not change in terms of the length and width (Figure 5B). Rather, protein level of proliferating cell nuclear antigen (PCNA), a cell proliferation marker, was elevated during the first three weeks after birth in R2M hearts while its level declined sharply in NTG hearts after 1 week (Figure 5C). In 2-week old R2M, mRNA levels were increased for cyclin D1 (1.7 fold), D2 (1.7 fold), and E (2.3 fold) and so were protein levels of cyclin D1 (4.1 fold) and D2 (1.7 fold) (Figure 5, D and E). We subsequently performed immunohistological analysis of 2-week old hearts using antibodies against proliferation markers, Ki67 and phospho-Histone 3 (PH3). R2M hearts contained more Ki67+ or PH3+ cells, suggesting an increased cell proliferation (Figure 6A). The total number of cells (based on DAPI staining) or cardiomyocytes (based on GATA4 staining) per cross section of the heart was also increased in R2M (Figure 6B). There was no difference in the distribution of mono- vs. multi-nucleated cardiomyocytes between the two groups (Online Figure X). BrdU labeling between one and two weeks after birth demonstrated that there was a modest increase in the percentage of all cell types that incorporated BrdU in R2M hearts compared to age matched NTG (25 vs. 30%). However, the fraction of cardiomyocytes in R2M hearts that incorporated BrdU was increased by 2-fold indicating that a higher rate of cardiomyocyte proliferation during this period (Figure 6C). Comparing hearts at 2 weeks with that at 2 months, we found that the number of PH3+ cells decreased substantially in the adult hearts in both genotypes (Figure 7A–C). However, at either age, the number of proliferating cells was 3–5 fold higher in R2M hearts compared to age matched NTG. Importantly, the PH3+ cardiomyocyte was absent in NTG hearts but present albeit at a much lower frequency in adult R2M hearts. Overexpression of FoxO1a in R2M eliminated the proliferating cardiac myocytes in the adult heart (Figure 7C). Taken together, these findings show that increased heart size in R2M resulted from increases of both cell number and cell size demonstrating the mutation of γ2-AMPK stimulates cardiac growth by promoting cardiomyocyte proliferation and enhancing hypertrophic growth.
Figure 5. Expression of cell proliferation markers was increased in the hearts of R2M mice during postnatal growth.
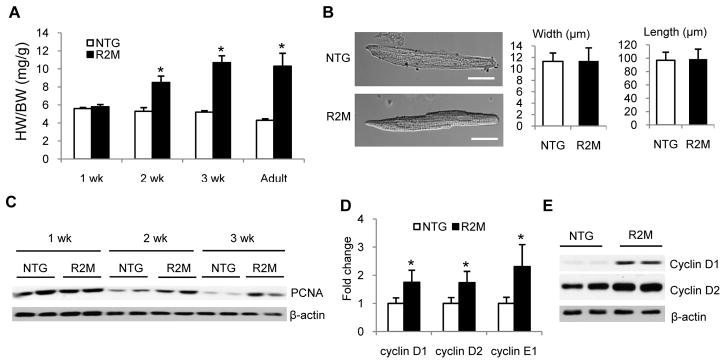
(A) HW/BW of R2M mice at 1, 2, 3 weeks (wk), and 2 months (adult). (B) Left, typical cardiomyocytes from NTG or R2M mouse hearts at 2 weeks. Right, average length and width of cardiomyocytes isolated from 2 week old NTG and R2M hearts. A total of 100–120 cardiomyocytes were counted in each heart, n=3 for each genotype. Scale bar, 20 μm.(C) Representative immunoblot of PCNA in whole heart lysates of NTG and R2M mice at 1, 2 and 3 weeks. (D) mRNA and (E) protein levels of cyclin D1, D2 and E in whole heart lysates of NTG and R2M mice at 2 weeks. n=4; *p< 0.05 versus NTG.
Figure 6. Cell proliferation was increased in the hearts of R2M mice during postnatal growth.

(A) Left, representative confocal image of 2 week old NTG and R2M hearts stained for phosphorylated Histone 3 (PH3, red, upper), Ki67 (green, lower), and DAPI (blue), and merged image (merge). Right, quantification of cells stained positive for PH3 or Ki67 in one cross section of the hearts from 2 week old mice, n=6 for each genotype. Scale bar, 50 μm.(B) Top, representative confocal image of a 2-week old R2M heart stained for alpha-sarcomeric actin (α-SA, red), GATA4 (green) and DAPI (blue), and merged image (merge). Bottom, quantification of all nuclei (DAPI+) and cardiomyocyte nuclei (GATA4+α-SA+) in each cross-section of the hearts, n=4 for each gentoype. Scale bar, 10 μm. (C) Top, BrdU was injected into 1-week-old pups for 7 days. Hearts were harvested at 2 weeks. Heart sections were stained for GATA4 (green), BrdU (red) and DAPI (blue). Arrow indicates cardiomyocyte with BrdU incorporation. Bottom, percentage of proliferative cells (BrdU+/DAPI+) and percentage of proliferative cardiomyocyte (GATA4+BrdU+/GATA4+) in the 2-week old heart. Thirty random fields were counted in each section, n=3. Scale bar, 10 μm; *p< 0.05 versus NTG.
Figure 7. Proliferating cardiomyocytes in NTG and R2M hearts at 2 weeks and 2 months of age.

(A) Representative confocal image of 2-week old heart section showing a proliferating cardiomyocyte stained positive for PH3 (red), GATA4 (green), and DAPI (blue) in R2M (lower panel) and a proliferating non-cardiomyocyte stained positive for PH3 and DAPI but not for GATA4 in NTG (upper panel). (B) Representative confocal image of 2-month old heart sections showing a proliferating cardiomyocyte stained positive for PH3 (green), alpha-sarcomeric actin (α-SA, red), and DAPI (blue) in R2M (lower panel) and a proliferating non-cardiomyocytes stained positive for PH3 and DAPI but not α-SA in NTG (upper panel). (C) Quantification of all proliferating cells per section (PH3+) and proliferating cardiomyocytes (PH3+GATA4+ or PH3+α-SA+) in 2-week and 2-month old hearts. Cells were counted in one cross-section of the heart, n=3 for each genotype; #p< 0.01 versus NTG; ‡p< 0.01 versus R2M.
DISCUSSION
The study demonstrates that point mutation of Prkag2 causes glycogen storage and cardiac hypertrophy via distinct mechanisms. By abrogating G6P-stimulated activity of GYS1, we have rescued pathological glycogen storage and consequently the pre-excitation phenotype in the mouse model of Prkag2 cardiomyopathy. However, the effects of Prkag2 mutation on cardiac growth are independent of glycogen storage or pre-excitation and cannot be rescued by reducing glycogen content of the heart. The mutation, via enhanced insulin sensitivity and activation of Akt, stimulates cardiomyocyte proliferation during postnatal growth by downregulating FoxO signaling and promoting cardiac hypertrophy via mTOR activation in the developed hearts. These findings elucidate important mechanisms underlying Prkag2 cardiomyopathy, and furthermore, reveal a novel function of γ2-AMPK that is critical for the utilization of AMPK as a therapeutic target.
Although the most common clinical complaint of Prkag2 cardiomyopathy is pre-excitation arrhythmia, the unique characteristics of the disease, ventricular pre-excitation associated with severe cardiac hypertrophy and glycogen storage, distinguish it from Wolff-Parkinson-White syndrome with structurally normal hearts5,6,20,21. Rather, the cardiomyopathy phenotype appears to be similar to glycogen storage diseases caused by mutation in lysosomal associated membrane protein 2 and alpha galactosidase A20,22–24. Different from most forms of glycogen storage disease, mouse hearts expressing mutant Prkag2 are able to use glycogen while faithfully recapitulating the human cardiomyopathy phenotypes suggesting that the disease mechanism is attributable to an imbalance of glycogen synthesis and utilization8,11. Elimination of excessive glycogen accumulation by targeting the G6P-stimulated activity of GYS1, shown here, corroborates the prior metabolic flux analysis and provides definitive evidence that enhanced glycogen synthesis due to high intracellular glucose and G-6-P is responsible for the glycogen storage in Prkag2 cardiomyopathy10. Furthermore, it provides an opportunity to distinguish the direct consequence of Prkag2 mutation from changes secondary to glycogen storage. The rescue of pre-excitation in the double mutant heart shows that the arrhythmia is a consequence of glycogen storage rather than a direct effect of AMPK on ion channels as previously speculated25–27. It is also noteworthy that the reduction of glycogen storage is associated with improvement of cardiac function suggesting that strategies of preventing/reducing glycogen storage are of therapeutic value. The mechanisms by which glycogen causes cardiac dysfunction remain unclear. We speculate the possible mechanisms for that. First of all, preexcitation can induce cardiac dysfunction. The accessory pathway causing pre-excitation has been implicated as the cause of cardiac dysfunction. Many reports have shown that normalization of preexcitation with depletion of the accessory pathway can restore cardiac function28,29. Second, extraordinary glycogen accumulation displaces contractile elements, distorts the overall cell morphology, and causes cardiac dysfunction30,31. Third, when accumulated glycogen is utilized and metabolized, glycogen metabolism increases lactate production, lowers pH, and hampers contractile ability due to its action on sarcomeric protein. The improved cardiac function by lowering glycogen content may result from any one of the above mentioned mechanisms.
Surprisingly, cardiac hypertrophy remains even after glycogen storage is prevented and ventricular pre-excitation rescued in the double mutant heart, indicating that the abnormal cardiac growth were regulated by separate mechanisms. Previous studies have shown that the Prkag2 cardiomyopathy is dependent on the kinase activity of AMPK8 but the cardiac hypertrophy phenotype is in contrary to the classic paradigm of AMPK function in cell growth thus presenting a puzzle for the field. A number of studies have shown that AMPK inhibits protein synthesis and cell growth under stress conditions by inhibiting mTOR signaling via phosphorylation of TSC2 and Raptor16,17. Consistent with this notion, activation of AMPK by AICAR in cultured neonatal cardiac myocytes prevents agonist-induced hypertrophy32. In the Prkag2 mutant heart, however, increased phosphorylation of TSC2 and Raptor is observed both at the AMPK and the Akt sites reflecting increases both in AMPK activity and insulin sensitivity of these hearts. This is unique to the Prkag2 mutant heart in which the activation of AMPK occurs under nutrient rich and energy sufficient conditions10,11 in contrast to stress-induced activation of AMPK which is invariantly associated with energy deficit. The resultant activation of the mTOR pathway suggests that the insulin stimulated phosphorylation cascade overrides the inhibitory regulation of AMPK on mTOR activity leading to a pro-growth phenotype in the mutant heart. Similarly, increased Akt activity and p70S6K phosphorylation have been reported in another mouse model harboring T400N point mutation of Prkag2 with increases of AMPK activity in the heart33.
It has recently been shown that γ2-subunit is abundantly expressed in the fetal heart; its expression falls sharply after birth but reappears in cardiac hypertrophy and failure34,35. Our results show that activation of γ2-containing AMPK in the post-natal period results in failure to arrest cardiomyocyte proliferation during maturation, hence revealing a novel function of γ2-AMPK in cell cycle progression of post mitotic myocytes. In non-myocytes, activated AMPK complex has been shown to localize in the mitotic apparatus in human cancer cells36, and AMPK phosphorylates proteins involved in cell cycle and promotes cell cycle progression in human endothelial cells37. Loss of AMPK function delays cell cycle progression in Drosophila as well as in mammalian cells38–40. It remains to be determined whether the function of AMPK in the proliferation of those cell types is dependent on the isoform composition, the energy status, or both. Since the human phenotype of Prkag2 mutations is restricted to the heart in spite of the ubiquitous expression in adult tissues, it is possible that unique functions of γ2-subunit or expressions of unique splice variant(s) of the protein exist in the heart as previously suggested35.
In summary, we show that mutation of the γ2-subunit of the AMPK accentuates insulin signaling, increases cell proliferation during the postnatal growth period and stimulates myocyte hypertrophy in adulthood, leading to abnormal cardiac growth in Prkag2 cardiomyopathy. These changes are attributable to mechanisms independent of glycogen accumulation in the heart, thus providing new insights to the pathogenesis of Prka2g cardiomyopathy. The findings also underscore the importance of isoform-specific function of AMPK that is critical for the pharmacological targeting of the AMPK.
Supplementary Material
Novelty and Significance.
What Is Known?
AMP-activated protein kinase (AMPK) is an energy sensor and master regulator of cell metabolism, growth and survival.
Point mutations in the regulatory γ2 subunit of AMPK, encoded by PRKAG2, cause human cardiomyopathy characterized by myocyte hypertrophy, glycogen storage, and ventricular pre-excitation.
Prkag2 mutation results in aberrant AMPK activity leading to metabolic remodeling and glycogen storage in the heart.
What New Information Does This Article Contribute?
Pathological glycogen storage could be rescued by genetically targeting glycogen synthesis in a mouse model for PRKAG2 cardiomyopathy (R2M).
Reduced glycogen content in the R2M heart normalizes ventricular pre-excitation but does not affect cardiac hypertrophy.
Increased insulin sensitivity in R2M leads to activation of mammalian target of rapamycin (mTOR) and inactivation of forkhead box O transcription factor (FoxO) pathways thus, causing abnormal cardiac growth by increasing both the number and the size of cardiomyocytes.
Activation of AMPK during stress conditions maintains energy homeostasis by stimulating substrate metabolism for ATP production and by suppression of growth and promotion of autophagy. These functions make AMPK an attractive drug target. Interestingly, point mutations in γ2 regulatory subunit increased AMPK activity in the absence of energy deficit but caused cardiomyopathy with glycogen storage, arrhythmia and excessive cardiac growth in humans. These observations raise the concern about using AMPK activation as a therapeutic approach and also the question of isoform-specific function of γ2-AMPK. We show that the mutation of γ2-AMPK accentuates insulin signaling, increases cell proliferation during the postnatal growth period and stimulates myocyte hypertrophy in adulthood, leading to abnormal cardiac growth. These changes were independent of glycogen accumulation in the heart.. The findings provide new insights into the pathogenesis of PRKG2 cardiomyopathy and o underscore the importance of isoform-specific function of AMPK, critical for the pharmacological targeting of AMPK.
Acknowledgments
We thank J. Sadoshima (University of Medicine & Dentistry of New Jersey) for the FoxO1 transgenic mice. The vector containing PRKAG2N488I cDNA was provided by M. Arad (Sheba Medical Center).
SOURCES OF FUNDING
This study was supported in part by grants from the National Institutes of Health: R01 HL067970 and HL088634 (R.T.), HL0962842 (S.K.); American Heart Association Postdoctoral Fellowship 10POST4170097 (M.K.); British Heart Foundation PG/09/059 (K.S and R.W.H); British Medical Research Council (K.S.).
Nonstandard Abbreviations and Acronyms
- AMPK
AMP-activated protein kinase
- GYS
glycogen synthase
- ECG
electrocardiogram
- Akt
ν-akt murine thymoma viral oncogene homolog
- mTOR
mammalian target of rapamycin
- FoxO
forkhead box O transcription factor
- BrdU
5-bromo-2-deoxyuridine
Footnotes
DISCLOSURES
None.
References
- 1.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 2006;21:48–60. doi: 10.1152/physiol.00044.2005. [DOI] [PubMed] [Google Scholar]
- 3.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- 6.Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gollob MH, Green MS, Tang AS, Roberts R. PRKAG2 cardiac syndrome: familial ventricular preexcitation, conduction system disease, and cardiac hypertrophy. Curr Opin Cardiol. 2002;17:229–234. doi: 10.1097/00001573-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, Perez-Atayde AR, Stapleton D, Bali D, Xing Y, Tian R, Goodyear LJ, Berul CI, Ingwall JS, Seidman CE, Seidman JG. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112:3140–3148. doi: 10.1161/CIRCULATIONAHA.105.550806. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee SK, Ramani R, Saba S, Rager J, Tian R, Mathier MA, Ahmad F. A PRKAG2 mutation causes biphasic changes in myocardial AMPK activity and does not protect against ischemia. Biochem Biophys Res Commun. 2007;360:381–387. doi: 10.1016/j.bbrc.2007.06.067. [DOI] [PubMed] [Google Scholar]
- 10.Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, Seidman CE, Seidman JG, Ingwall JS, Balschi JA, Tian R. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest. 2007;117:1432–1439. doi: 10.1172/JCI30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zou L, Shen M, Arad M, He H, Lofgren B, Ingwall JS, Seidman CE, Seidman JG, Tian R. N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ Res. 2005;97:323–328. doi: 10.1161/01.RES.0000179035.20319.c2. [DOI] [PubMed] [Google Scholar]
- 12.Bouskila M, Hunter RW, Ibrahim AF, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J, Sakamoto K. Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 2010;12:456–466. doi: 10.1016/j.cmet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Hariharan N, Ikeda Y, Hong C, Alcendor RR, Usui S, Gao S, Maejima Y, Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One. 2013;8:e51632. doi: 10.1371/journal.pone.0051632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas JA, Schlender KK, Larner J. A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal Biochem. 1968;25:486–499. doi: 10.1016/0003-2697(68)90127-9. [DOI] [PubMed] [Google Scholar]
- 15.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541–1548. doi: 10.1161/CIRCRESAHA.109.212753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 17.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju JS, Gitcho MA, Casmaer CA, Patil PB, Han DG, Spencer SA, Fisher JS. Potentiation of insulin-stimulated glucose transport by the AMP-activated protein kinase. Am J Physiol Cell Physiol. 2007;292:C564–572. doi: 10.1152/ajpcell.00269.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, Cheng J, Lu G, Morris DJ, Castrillon DH, Gerard RD, Rothermel BA, Hill JA. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114:1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arad M, Maron BJ, Gorham JM, Johnson WH, Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 21.Gollob MH, Seger JJ, Gollob TN, Tapscott T, Gonzales O, Bachinski L, Roberts R. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 2001;104:3030–3033. doi: 10.1161/hc5001.102111. [DOI] [PubMed] [Google Scholar]
- 22.Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–57. doi: 10.1212/wnl.31.1.51. [DOI] [PubMed] [Google Scholar]
- 23.Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995;333:288–293. doi: 10.1056/NEJM199508033330504. [DOI] [PubMed] [Google Scholar]
- 24.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 25.Light PE, Wallace CH, Dyck JR. Constitutively active adenosine monophosphate-activated protein kinase regulates voltage-gated sodium channels in ventricular myocytes. Circulation. 2003;107:1962–1965. doi: 10.1161/01.CIR.0000069269.60167.02. [DOI] [PubMed] [Google Scholar]
- 26.Wyatt CN, Mustard KJ, Pearson SA, Dallas ML, Atkinson L, Kumar P, Peers C, Hardie DG, Evans AM. AMP-activated protein kinase mediates carotid body excitation by hypoxia. J Biol Chem. 2007;282:8092–8098. doi: 10.1074/jbc.M608742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King JD, Jr, Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, Hallows KR. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol. 2009;297:C94–101. doi: 10.1152/ajpcell.00677.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shan Q, Jin Y, Cao K. Reversible left ventricular dyssynchrony and dysfunction resulting from right ventricular pre-excitation. Europace. 2007;9:697–701. doi: 10.1093/europace/eum138. [DOI] [PubMed] [Google Scholar]
- 29.Shankar PB, Shanthi C, Cherian KM. Pre-excitation induced left ventricular dysfunction: A less known cause of cardiomyopathy in children. Ann Pediatr Cardiol. 2013;6:77–79. doi: 10.4103/0974-2069.107241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewandowska E, Wierzba-Bobrowicz T, Rola R, Modzelewska J, Stepien T, Lugowska A, Pasennik E, Ryglewicz D. Pathology of skeletal muscle cells in adult-onset glycogenosis type II (Pompe disease): ultrastructural study. Folia Neuropathol. 2008;46:123–133. [PubMed] [Google Scholar]
- 31.Yi H, Thurberg BL, Curtis S, Austin S, Fyfe J, Koeberl DD, Kishnani PS, Sun B. Characterization of a canine model of glycogen storage disease type IIIa. Dis Model Mech. 2012;5:804–811. doi: 10.1242/dmm.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 33.Banerjee SK, McGaffin KR, Huang XN, Ahmad F. Activation of cardiac hypertrophic signaling pathways in a transgenic mouse with the human PRKAG2 Thr400Asn mutation. Biochim Biophys Acta. 2010;1802:284–291. doi: 10.1016/j.bbadis.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Kim M, Shen M, Ngoy S, Karamanlidis G, Liao R, Tian R. AMPK isoform expression in the normal and failing hearts. J Mol Cell Cardiol. 2012;52:1066–1073. doi: 10.1016/j.yjmcc.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pinter K, Grignani RT, Czibik G, Farza H, Watkins H, Redwood C. Embryonic expression of AMPK gamma subunits and the identification of a novel gamma2 transcript variant in adult heart. J Mol Cell Cardiol. 2012;53:342–349. doi: 10.1016/j.yjmcc.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle. 2009;8:788–791. doi: 10.4161/cc.8.5.7787. [DOI] [PubMed] [Google Scholar]
- 37.Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, Gygi SP, Cantley LC, Yaffe MB, Shokat KM, Brunet A. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell. 2011;44:878–892. doi: 10.1016/j.molcel.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thaiparambil JT, Eggers CM, Marcus AI. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol. 2012;32:3203–3217. doi: 10.1128/MCB.00418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, Chung J. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 40.Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16:256–270. doi: 10.1016/j.devcel.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.