

Abstract
The development of two distinct classes of hepatitis C antiviral agents, direct-acting antivirals (DAAs) and host-targeting antivirals (HTAs), have distinctly impacted the hepatitis C virus (HCV) field by generating higher sustained virological response (SVR) rates within infected patients, via reductions in both adverse side effects and duration of treatment when compared to the old standard of care. Today DAAs are actively incorporated into the standard of care and continue to receive the most advanced clinical trial analysis. With a multitude of innovative and potent second-generation DAA compounds currently being tested in clinical trials, it is clear that the future of DAAs looks very bright. In comparison to the other class of compounds, HTAs have been slightly less impactful, despite the fact that primary treatment regimens for HCV began with the use of an HTA - interferon alpha (IFNα). The compound was advantageous in that it provided a broad-reaching antiviral response; however deleterious side effects and viral/patient resistance has since made the compound outdated. HTA research has since moved onward to target a number of cellular host factors that are required for HCV viral entry and replication such as scavenger receptor-BI (SR-BI), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCoA reductase), cyclophilin A (CypA), fatty acid synthase (FASN) and miRNA-122. The rationale behind pursuing these HTAs is based upon the extremely low mutational rate that occurs within eukaryotic cells, thereby creating a high genetic barrier to drug resistance for anti-HCV compounds, as well as pan-genotypic coverage to all HCV genotypes and serotypes. As the end appears near for HCV, it becomes important to ask if the development of novel HTAs should also be analyzed in combination with other DAAs, in order to address potential hard-to-treat HCV patient populations. Since the treatment regimens for HCV began with the use of a global HTA, could one end the field as well?
Keywords: host-targeting antivirals, alisporivir, HCV, cyclophilin A
Hepatitis C virus (HCV) is a growing global pandemic with an estimated 2–3% of the world’s population thought to be infected with the virus, superseding HIV as the leading cause of death due to an infectious agent in the United States (Lavanchy, 2009). HCV comprises seven genotypes, more than 50 subtypes, and billions of quasi species (Grimm, 2012). It is spread via contaminated blood with 50–80% of infected patients eventually developing chronic hepatitis C, which in turn can lead to liver cirrhosis. After the onset of cirrhosis, hepatocellular carcinoma occurrence becomes the next step in the progression of the disease with annual rates between 1–8% (Ghany et al., 2009; Lauer and Walker, 2001; Poynard et al., 2003). At the moment, no vaccine is available and up until recently the traditional standard of care (SOC) was merely comprised of the combination of injected pegylated-interferon alpha (pegIFN) and oral ribavirin (RBV) administered for up to 48 weeks, which in itself is associated with serious and potentially life-threatening side effects (Ghany et al., 2009). Although this regimen was capable of achieving up to 80% SVR12 (undetectable HCV-RNA in the blood 12 weeks after the completion of a treatment regimen) rates in nongenotype 1 patients, the percentage of genotype 1 patients who reach SVR12 on this treatment was drastically reduced to 40–50%. Given that genotype 1 infections account for approximately 60% of global infections, more effective therapies were developed (Ghany et al., 2009; Magiorkinis et al., 2009).
A better understanding of virus replication coupled with the need to achieve higher SVR rates, particularly in genotype 1 patients, brought about the development of the first set of DAAs. In 2011, the U.S. Food and Drug Administration (FDA) granted approval for the use of two small molecule inhibitors telaprevir (Incivek/Incivo) and boceprevir (Victrelis), whose target is the HCV-encoded protease NS3. Phase III clinical trials achieved SVR rates of up to 75% in treatment-naïve genotype-1-infected patients when combined with pegIFN/RBV. However, major limitations still exist within these DAA regimens, namely: (i) additional side effects; (ii) increased risk of developing drugresistant variants; and (iii) limited to only genotype 1 patients. Bühler and Bartenschlager recently proposed a set of criteria that ideal future therapies of chronic hepatitis C should (i) have to be free of pegIFN in order to reduce side effects; (ii) impose a high barrier of drug resistance; (iii) require only short treatment durations; and (iv) provide more than 90% SVR12 rates (Buhler and Bartenschlager, 2012).
HCV Host-Targeting Antivirals
A number of host-targeting antivirals (HTAs) have been developed and tested to combat HCV. Many are broad target HTAs that work by creating an activated anti-viral state within the host by triggering arms of innate immune response, examples of which include IFNα, interferon lambda (IFNλ), or Toll Like Receptor (TLR) agonists. The targeted group of HTAs are more precise in that they act upon key host enzymes or cellular factors that are required for the HCV lifecycle, such as cyclophilin A (CypA), fatty acid synthase (FASN), and miRNA-122. Generally speaking, the primary advantage of targeting host factors is the extremely low rate of mutation within the host that arises during times of viral or chemotoxic stress, thereby affording a higher barrier to drug resistance while also limiting the potential of viral breakthrough. Since each HTA acts upon a unique step of the virus life cycle, it is also reasonable to believe that these compounds should act synergistically with one another, or even approved DAAs, further expanding the anti-HCV arsenal to address the most difficult to treat cases. Finally, since a number of viruses hijack similar cellular machine it also seems reasonable to develop HTAs that could potentially be used to combat new and emerging infectious diseases.
Of all of the general HTAs that are targeted to activate the immune response, the one that is the best studied, tested, and understood is IFNα. Treatment of HCV patients with IFNα has been ongoing for the last 25 years (Heim, 2013). The presence of circulating IFNα within a patient leads to receptor binding via the cell, followed by a signaling cascade that promotes the induction of hundreds of antiviral proteins, so-called interferon-stimulated genes (ISGs) (Stetson and Medzhitov, 2006). In an infected cell these ISGs can shut down the host transcription and translation machinery in a myriad of ways, ranging from simple post-translational modifications to proteolytic cleavage of host regulatory elements, collectively serving to create an antiviral state within the cell (Heim, 2013). IFNα has cured many cases of HCV, over a long period of time, during which no other viable alternatives were even available. However the treatment itself has less favorable cure rates in the predominant HCV genotype 1, often leads to the generation of viral escape mutants, and is commonly associated with dramatic side effects (Ghany et al., 2009). Recent cell culture work has gone on to further highlight that HCV is quite capable of creating escape mutants in a few select regions of the viral genome, over a range of IFNα concentrations (Perales et al., 2013). Ongoing clinical trials are predominantly IFNα-free and have shown to afford very promising results with DAA combinations alone (Gilead Sciences. Gilead Reports Interim Data from Phase 2 LONESTAR Study. Press Release. May 2, 2013).
In addition to IFNα, the Type III interferon family member IFNλ1 (IFNlambda) has also been tested, in its pegylated form, on HCV patients with either genotype 1, 2, 3, or 4 and showed promising efficacy in combatting the virus (Muir et al., 2010; Zeuzem et al., 2011). The advantages and rationale behind testing IFNlambda is that: (i) it possesses a receptor specificity distinct from IFNα, (ii) while maintaining antiviral inducing effects comparable to IFNα, and (iii) the receptors are selectively expressed only on specific cell types, which should translate to fewer patient side effects (Donnelly and Kotenko, 2010). Recent work in a cell culture model system has shown that HCV is capable of generating escape mutants to IFN lambda treatment alone (Friborg et al., 2013b), however IFNlambda has also been shown to act synergistically with DAAs (Friborg et al., 2013a). Taken together, it appears that a general antiviral tool such as IFNlambda should continue in development, especially since it could potentially be a frontline broad range antiviral for new and emerging infectious diseases. Continuing on with the theme of broad-range antiviral inducers, work with TLR agonist also shows a marked enhancement of the antiviral response accompanied by HCV elimination. Specifically two separate clinical trials looked at the treatment of HCV patients with either a Toll-like receptor (TLR) 7 agonist, or a TLR 9 agonist, wherein both demonstrated robust activation of the immune response (Boonstra et al., 2012; Rodriguez-Torres et al., 2010). Importantly, the TLR9 agonist IMO-2125, was shown to lower viral loads in the null responder patient population, (Rodriguez-Torres et al., 2010) exemplifying the importance of developing a variety of drug target options.
Phosphatidylinositol 4-kinase III alpha
The advent of cell culture models of subgenomic and then fully infectious HCV enabled the expansion of targeted HTA drugs. Specifically, the identification of many essential host proteins required for the HCV lifecycle were discovered by exhaustive siRNA screens directed against the host (Randall et al., 2007). One of the first host targets that was shown to be essential to HCV replication was phosphatidylinositol 4-kinase III alpha (PI4KIIIα) (Berger et al., 2009). It has recently been shown that PI4KIIIα can modulate the phosphorylation status of NS5A, which in turn modulates the morphology of replication sites, presumably leading to an enhancement in HCV replication (Reiss et al., 2013). Many groups have come out with compounds that target PI4KIIIα and block its downstream phosphorylation activity. However, when Boehringer Ingelheim created conditional PI4KIIIα knockout mice they found that targeting this kinase was lethal, with a complete degeneration of mucosal epithelium in the intestinal track, clearly demonstrating the requirement by the host for this enzyme as well (Vaillancourt et al., 2012). This highlights the key point in specific targeting of host proteins, particularly enzymes, you must know exactly how the drug is working and what are the possible side effects to the host system if you block the activity of said target. Now we will look at more feasible targets.
Scavenger Receptor B1
HCV uses a multitude of cell surface receptors to enter a hepatic cell (Scheel and Rice, 2013). Recently, a number of groups have developed compounds to block one of these receptors, Scavenger Receptor B1 (SR-B1). The concept of blocking a virus prior to entry into the host is not new; however, the stringent requirement of SR-B1 for HCV entry has afforded some potent efficacy in a cell culture system (Syder et al., 2011). There are two different approaches taken in blocking SR-B1: one group of SR-B1 targeted compounds is antibody based, while the other is a small molecule antagonist, ITX 5061 (Scheel and Rice, 2013). Interestingly, monoclonal antibodies directed against SR-B1 have been tested in a humanized mouse model and were seen to block both HCV infection and dissemination (Lacek et al., 2012). ITX 5061 is a compound that is already in the clinical stage as it was shown to increase high-density lipoproteins (HDLs) in both humans and mice by interfering with SR-B1, the main HDL receptor in the liver (Masson et al., 2009). Importantly, ITX 5061 was shown to act synergistically with protease and polymerase DAAs in a fully infectious HCV cell culture system (Zhu et al., 2012). ITX 5061 is currently being tested in mono-infected HCV patients in a proof-of-concept Phase 1 clinical trial, carried out by the AIDS Clinical Trial Group (ACTG). All of this aside, an important caveat to this group of receptor-targeted compounds is the fact that CD-81-independent cell-to-cell transmission has been previously shown by others (Jones et al., 2010; Witteveldt et al., 2009). Therefore it is imperative, before moving too much further, to demonstrate that HCV cannot undergo cell-cell transmission in the absence of SR-B1, or any other receptor that is targeted.
FASN Inhibitors
The FASN gene encodes for a polypeptide composed of seven functional domains in addition to an acyl-carrier protein, which together, catalyze the de novo synthesis of fatty acids (FA) (Kridel et al., 2007). FAs have been previously shown to have a role in the regulation of HCV replication with FASN’s primary products, being 16-carbon FA palmitate, 14-carbon myristate and 18-carbon stearate FAs (Huang et al., 2007; Kapadia and Chisari, 2005). Yang et al. were the first to successfully demonstrate FASN’s role in HCV replication, by showing it to be upregulated in the presence of both the genomic JFH-1 virus (genotype 2a) and subgenomic Con1b, although FASN upregulation was substantially weaker in the subgenomic replicon compared to that of whole virus (Yang et al., 2008). In another study done by Huang et al., FASN was shown to interact with the N-terminal domain of NS5B. It was also found to be associated with detergent-resistant lipid rafts and colocalized with NS5B in active HCV replication complexes. Moreover, FASN was shown to directly increase HCV NS5B RNA-dependent RNA polymerase (RdRp) activity in vitro, suggesting that this interaction plays an important role in modulating HCV replication (Huang et al., 2013).
3-V Biosciences recently presented positive preclinical data with their novel FASN inhibitor, TVB-2640. It was demonstrated that TVB-2640 safely causes FASN inhibition, is rapidly absorbed, is highly bioavailable (~60%) and is well tolerated in both rats and dogs (Evanchik et al., 2012). With FASN inhibitors representing an entirely new category of HTA therapy, combined with a novel mechanism of action and potent antiviral activity, it will be interesting to see data from Phase I/II clinical trials poised to start in 2013. In the case of FASN inhibitors such as 3-V Bioscience’s TVB-2640, the exact mechanism of action (MOA) of the compound is not completely understood with regard to its role in impairing HCV replication. What is known is that FASN is upregulated in the presence of HCV infection, it interacts with the HCV NS5B polymerase and increases HCV’s NS5B polymerase (Huang et al., 2013; Yang et al., 2008). FASN inhibitors are thought to exert their anti-HCV activity by binding to FASN and thus impeding its ability to interact with NS5B. Again a cautious approach must be taken with this group of compounds, as they are targeting a host enzyme that carries out a host need, it is unclear if there is a redundant compensatory activity in the host.
miRNA-122
A discovery in 2005 demonstrated the requirement of the liver-specific microRNA, miR- 122, to HCV replication (Jopling et al., 2005). MicroRNAs (miRNAs) are small, endogenous non-coding RNAs, which regulate post-transcriptional gene expression by binding to partially complementary sites within target messenger RNAs (mRNAs), resulting in translational repression either by transcript cleavage and degradation or suppression of robust translation (Ambros, 2004; Fabian and Sonenberg, 2012; Janssen et al., 2013). While the typical function of most miRNAs is to suppress gene expression, the opposite holds true in the relationship between HCV and miR-122. In this scenario, binding of miR-122 to two sites at the 5’ end of the HCV positive-strand RNA genome does not result in the degradation of the RNA strand, rather it stabilizes the transcript by acting as a cap - which in turn promotes viral replication and stability (Garcia-Sastre and Evans, 2013). As the HCV genome does not contain the traditional cap structure at its 5’ end and undergoes IRES mediated translation via the direct recruitment of ribosomal components, it therefore requires shielding the 5’ end from cytosolic RNA exonuclease digestion (Garneau et al., 2007; Shimakami et al., 2012). In the case of HCV, miR-122 acts to shield viral RNA from Xrn1 degradation and increased RNA stability (Li et al., 2013). This mechanism is also important for transcript fitness and virus survival as the cap also protects aberrant transcripts from triggering RIG-I, an intracellular RNA-triggered innate immune sensor. It is important to note that the miRNA-122-binding sites are conserved among all HCV genotypes and subtypes, thus allowing miRNA-122 to represent a very attractive host target for antiviral therapy (Li et al., 2011).
Miravirsen is the latest HTA from Santaris Pharma to enter into Phase IIa clinical trials. It is a 15-nucleotide locked nucleic acid-modified antisense oligonucleotide complementary sequence to the 5’ region of mature miR-122 (Janssen et al., 2013). Initial studies conducted in chimpanzees with chronic genotype 1 HCV infection provided evidence of miravirsen’s antiviral potency, by demonstrating long-lasting viral suppression without evidence of resistant mutations (Lanford et al., 2010). Importantly, when Phase I clinical trials were conducted in healthy volunteers there were no observable adverse effects (Hildebrandt-Eriksen et al., 2009). Phase IIa studies conducted with miravirsen demonstrated prolonged dose-dependent reductions in HCV RNA levels, without evidence of viral breakthrough mutations. In this study, 36 treatment-naïve chronic HCV genotype 1 patients were randomly assigned to receive five weekly subcutaneous injections of miravirsen at doses of 3, 5, or 7 mg per kilogram of body weight or placebo over a 29-day period. Five patients who received the highest dose (a weekly dose of 7 mg per kilogram) of miravirsen alone achieved undetectable HCV RNA levels, strongly demonstrating the potent efficacy of this drug. Hopes that miravirsen could be utilized as a monotherapy were dashed however, as four of these five patients experienced viral rebound within 14 weeks (Janssen et al., 2013). These results suggest that the given time frame and/or dosage were insufficient to achieve SVR in these patients. Santaris is currently testing the effect of a 12-week regimen in genotype 1 prior null-responder patients (ClinicalTrials.gov number, NCT01727934) (Janssen et al., 2013; Lieberman and Sarnow, 2013). Hopefully this work will further elucidate the true potential of this category of antiviral compounds that can likely be applied to a variety of viral targets.
Cyclophilin A
Cyclosporine A (CsA), an immunosuppressant drug used in organ transplantation to prevent rejection, was the first cyclophilin inhibitor (CypI) shown to have anti-HCV activity in vitro (Goto et al., 2006; Ishii et al., 2006; Nakagawa et al., 2004; Watashi et al., 2003). CsA exerts its immunosuppressive activity by binding to its intracellular partner, CypA, forming a ternary complex with calcineurin that triggers a reduction in the stimulation of the growth and differentiation of T cells (Borel, 2002). The clinical effectiveness of CsA was first demonstrated when it was shown to be more effective in combination with pegIFN than pegIFN alone (Inoue et al., 2003; Inoue and Yoshiba, 2005). Subsequent in vitro studies done in hepatoma cell lines attributed this effect to CsA’s ability to prevent HCV RNA replication and protein synthesis (Goto et al., 2006; Ishii et al., 2006; Nakagawa et al., 2004; Watashi et al., 2003). This discovery led to the development of a second generation of CypIs, which lack the ability to bind calcineurin and in turn are devoid of the immunosuppressive properties of CsA.
Nonimmunosuppressive CypIs derived from CsA include NIM-811, SCY-635, EDP-546 and alisporivir (ALV) (previously known as Debio-025) (Hopkins et al., 2010; Ma et al., 2006; Owens CM, 2013. Cyclophilin Inhibitor EDP-546 is a Potential Cornerstone Drug for Use in Combination with NS5A and Protease Inhibitors Due to its High Barrier to HCV Resistance.; Paeshuyse et al., 2006).
A number of CypIs are currently under development and at various stages of preclinical and clinical phases. Among these, SCY-635, currently in Phase II clinical trials has demonstrated clinical efficacy in genotype 1 patients by reducing mean viral load of 2.2 log10 after 15 days of treatment with 300 mg TID (three times a day) of SCY-635 alone (Hopkins et al., 2012; Hopkins et al., 2010). Other CsA-derived CypIs such as EDP-546 (Jiang et al., 2012; Owens CM, 2013. Cyclophilin Inhibitor EDP-546 is a Potential Cornerstone Drug for Use in Combination with NS5A and Protease Inhibitors Due to its High Barrier to HCV Resistance.) together with another class of CypI, sanglifehrins, a group of naturally occurring cyclophilin-binding polyketides, which are structurally distinct from cyclosporines, such as BC556, are currently being tested in preclinical studies and have shown promising in vitro anti-HCV activities (Ahmed-Belkacem et al., 2012; Gregory et al., 2011; Moss et al., 2012).
ALV was the first oral nonimmunosuppressive CypI to enter into clinical trials, and to date, is the most advanced with the largest clinical database of HCV genotype 1, 2, 3 and 4 patients. ALV was initially developed for HIV-1 and was discovered in a Phase I clinical trial with 23 HIV-1 patients, including 19 co-infected with HCV, to be incredibly effective against HCV, while having only minimal effects against HIV. ALV decreased HCV RNA by 3.63 log10 in patients treated with 1200 mg ALV BID (twice daily) for 15 days vs. 0.73 log10 in the placebo group (Flisiak et al., 2008; Lin and Gallay, 2013). This study demonstrated for the first time the clinical efficacy of CypIs in HCV-infected patients, paving the way for HTA treatments.
Subsequent Phase IIa clinical trials with ALV in combination with pegIFN and RBV demonstrated that very impressive mean viral load reductions could be achieved in treatment-naïve HCV-infected patients after only four weeks, with 4.6 log10 and 5.9 log10 viral load reductions attained in genotypes 1/4 and genotypes 2/3, respectively (Flisiak et al., 2009; Gallay and Lin, 2013; Lin and Gallay, 2013). Based on these results, the Phase IIb study, known as ESSENTIAL, focused on treatment-naïve genotype 1 HCV patients who were treated with ALV or placebo in combination with pegIFN and RBV for 24–48 weeks (Flisiak et al., 2011). A loading dose of 600 mg BID ALV was given during the first week of treatment with 600 mg QD for the remaining weeks. 76% of patients on ALV triple therapy successfully attained SVR compared to 55% on placebo triple therapy, with the rate of viral breakthrough being much lower in the ALV-treated group, thereby confirming its high barrier to resistance. In another Phase II study known as FUNDAMENTAL, pegIFN null-responder genotype 1 HCV-infected patients were treated for 24 weeks with either ALV 400 mg BID with pegIFN and RBV or pegIFN and RBV alone. Remarkably, 75.4% of patients on ALV triple therapy attained SVR12 compared to 8.9% in the control group (Alberti et al., 2012).
The VITAL-1 study, an open label Phase IIb clinical trial, was set up to evaluate the safety and efficacy of ALV administered alone, with pegIFN, or with pegIFN and RBV in 340 treatment-naïve genotype 2/3 HCV patients. Within this study one third of the patients were infected with genotype 2 and the remainder with genotype 3. Patients were randomly assigned to 1 of 5 treatment arms: 1000 mg QD ALV monotherapy, 600 mg QD ALV plus RBV, 800 mg QD ALV plus RBV, 600 mg QD ALV plus pegIFN, or standard therapy using pegIFN/RBV. All patients were started with a loading dose of 600 mg BID ALV for 4 weeks before beginning their assigned regimen. Those who achieved RVR stayed on the assigned regimen through week 24. Those who did not switched to 600 mg ALV plus pegIFN/RBV and continued for the same duration. At the conclusion of this study, it was determined that ALV improved SVR24 rates to 80–85% compared to 58% for pegIFN and RBV treatment. Overall, ALV was well tolerated within patients with very low relapse rates occurring within patients treated with ALV/RBV (between 4–9%). Also it is worth noting that genotype 3 patients across all the pegIFN-free arms responded the same or better as genotype 2 patients to ALV, emphasizing the major advantage ALV may have in treating genotype 3 infected patients (Pawlotsky et al., 2012).
Phase III clinical trials began in early 2011 to study the combination of ALV with pegIFN and RBV in treatment-naïve genotype 1 HCV patients. Unfortunately, this study was put on partial clinical hold after six cases of acute pancreatitis were reported, including one death in early 2012, in patients receiving ALV with pegIFN and RBV. Acute pancreatitis is a known adverse event with pegIFN/RBV therapy and no cases of pancreatitis were reported with ALV, pegIFN-free therapies in the VITAL-1 study (Lin and Gallay, 2013).
The exact MOA of CypIs is also not yet fully understood, but a number of critical discoveries are helping to unravel their role in the inhibition of HCV replication: (i) CypA governs HCV replication via its peptidyl-prolyl isomerase hydrophobic pocket (Chatterji et al., 2009; Liu et al., 2009); (ii) CypIs inhibit peptidyl-prolyl isomerase activity by binding to the enzymatic pocket of CypA (Ke et al., 1991; Zydowsky et al., 1992); and (iii) the HCV NS5A protein is the viral ligand for CypA (Chatterji et al., 2010; Coelmont et al., 2010; Fernandes et al., 2010; Hanoulle et al., 2009; Waller et al., 2010). It is believed that the main antiviral mechanism of action of CypIs is the disruption of CypA-NS5A interactions that regulate multiple phases of HCV replication (Gallay and Lin, 2013; Owens CM, 2013. Cyclophilin Inhibitor EDP-546 is a Potential Cornerstone Drug for Use in Combination with NS5A and Protease Inhibitors Due to its High Barrier to HCV Resistance.).
Host Targeting Antivirals a Beginning and an End
PegIFN- and RBV-free treatment regimens for HCV infection are currently being tested in the clinic and will likely be within the grasp of patients in the coming years. Thus, as we bid farewell to the first generation of HTAs we must look toward the future generation of HTA products that are in the pipeline for a number of companies. DAAs are undeniably effective against HCV and as patient-based treatment progresses we will likely see subsets of individuals that prove to be difficult to treat. It is these populations of patients that HTAs should be directly tested, in conjunction with DAAs. Given the distinct advantages that HTAs possess over DAAs - high barrier to resistance, broad pangenotypic coverage, and potential synergistic activity with DAAs - it is easy to envision in the near future that this class of drugs, in combination with next generation DAAs, will afford a novel approach to combat the most difficult of HCV cases. Moreover it is reasonable to see this class of compounds be tested against a variety of other pathogens.
Figure 1.

Mechanism of action of host-targeting antivirals against hepatitis C. A number of host pathways are exploited by HCV during its life-cycle. Adapted from (Baugh et al., 2012) and (Santaris, 2010).
Table 1.
Some of the host-targeting antivirals currently under clinical development for the treatment of hepatitis C.
| Compound | Sponsor | Host Target | Structure | HCV EC50 | Tested Genotype Coverage |
Status | Reference |
|---|---|---|---|---|---|---|---|
| NIM811 | Novartis | Cyclophilin A | 120 nM | N/A | Terminated | Lawitz et al. (2011) | |
| Alisporivir | Novartis | Cyclophilin A | 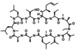 |
45 nM | GT1/2/3/4 | Phase III | Pawlotsky el al. (2012) |
| SCY-635 | Scynexis | Cyclophilin A | 100 nM | GT1 | Phase IIa | Hopkins el al. (2012) | |
| EDP-546 | Enanta | Cyclophilin A | Unpublished | 67 nM | N/A | Preclinical | Jiang et al. (2012) |
| NVPO18 | Neurovive | Cyclophilin A | 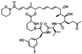 |
38 nM | N/A | Preclinical | Moss et al. (2012) |
| Miravirsen | Santaris Pharma | miR-122 | Unpublished | 671 nM | GT1 | Phase II | Janssen et al. (2013) |
| TVB-2640 | 3-V Biosciences | FASN | Unpublished | <100 nM | GT1/2 | Phase I | Kemble et al. (2012) |
GALLAY Highlights 9-23-13.
Host-targeting agents (HTAs) typically create an activated antiviral state within the host.
HTAs act directly upon cellular receptors, enzymes, and cofactors for hepatitis C virus (HCV) infection.
Alisporivir targets cyclophilin A and is the most advanced HTA against HCV.
HTAs combined with direct-acting antivirals may be needed for hard-to-treat HCV patient populations.
Further development of HTAs could address new and emerging infections.
Acknowledgements
We acknowledge financial support from the U.S. Public Health Service grant no. AI087746 (P.A.G.). This is publication no. 24052 from the Department of Immunology & Microbial Science, The Scripps Research Institute, La Jolla, CA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed-Belkacem A, Colliandre L, Ahnou N, Lerat N, Bessin Y, Barthe P, Bourget W, Douguet D, Guichou JF, Pawlotsky JM. New Cyclophilin Inhibitors Unrelated to Cyclosporine Potently Inhibit HCV Replication and Revert HCV-induced Mitochondrial Dysfunction. Hepatology. 2012;56:1067A–1068A. [Google Scholar]
- Alberti A, Chuang WL, Flisiak R, Mazzella G, Horban A, Goeser T, Calistru P, Buti M, Davis G, Gong Y, Avila C, Kao JH. Alisporivir (Alv) Plus Peg-Interferon/Ribavirin (Pr) in Hcv G1 Treatment-Experienced Patients Achieves Primary Endpoint with Superior Efficacy at Treatment Week 12 Compared to Retreatment with Pr. Journal of hepatology. 2012;56:S553–S554. [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Baugh J, Gallay P. Cyclophilin involvement in the replication of hepatitis C virus and other viruses. Biol. Chem. 2012;393:579–587. doi: 10.1515/hsz-2012-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. P Natl Acad Sci USA. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonstra A, Liu BS, Groothuismink ZMA, Bergmann JF, de Bruijne J, Hotho DM, Hansen BE, van Vliet AA, de Rooij JV, Fletcher SP, Bauman LA, Rahimy M, Appleman JR, Freddo JL, Reesink HW, de Knegt RJ, Janssen HLA. Potent immune activation in chronic hepatitis C patients upon administration of an oral inducer of endogenous interferons that acts via Toll-like receptor 7. Antivir Ther. 2012;17:657–667. doi: 10.3851/IMP2023. [DOI] [PubMed] [Google Scholar]
- Borel JF. History of the discovery of cyclosporin and of its early pharmacological development. Wiener klinische Wochenschrift. 2002;114:433–437. [PubMed] [Google Scholar]
- Buhler S, Bartenschlager R. New targets for antiviral therapy of chronic hepatitis C. Liver international : official journal of the International Association for the Study of the Liver. 2012;32(Suppl 1):9–16. doi: 10.1111/j.1478-3231.2011.02701.x. [DOI] [PubMed] [Google Scholar]
- Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, Sakamoto N, Vuagniaux G, Parkinson T, Gallay P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. The Journal of biological chemistry. 2009;284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji U, Lim P, Bobardt MD, Wieland S, Cordek DG, Vuagniaux G, Chisari F, Cameron CE, Targett-Adams P, Parkinson T, Gallay PA. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. Journal of hepatology. 2010;53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, Lim P, Vliegen I, Paeshuyse J, Vuagniaux G, Vandamme AM, Bartenschlager R, Gallay P, Lippens G, Neyts J. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. Plos One. 2010;5:e13687. doi: 10.1371/journal.pone.0013687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly RP, Kotenko SV. Interferon-Lambda: A New Addition to an Old Family. J Interf Cytok Res. 2010;30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanchik M, Cai HY, Feng QS, Hu L, Johnson R, Kemble G, Kosaka Y, Lai J, Oslob JD, Sivaraja M, Tep S, Yan HB, Zaharia CA, McDowell R. TVB-2640, a Novel Anti-HCV Agent, Safely Causes Sustained Host-Target Inhibition in Vivo. Hepatology. 2012;56:1066A–1067A. [Google Scholar]
- Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nature structural & molecular biology. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- Fernandes F, Ansari IU, Striker R. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. Plos One. 2010;5:e9815. doi: 10.1371/journal.pone.0009815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flisiak R, Feinman SV, Jablkowski M, Horban A, Kryczka W, Pawlowska M, Heathcote JE, Mazzella G, Vandelli C, Nicolas-Metral V, Grosgurin P, Liz JS, Scalfaro P, Porchet H, Crabbe R. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009;49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, Siwak E, Cielniak I, Higersberger J, Kierkus J, Aeschlimann C, Grosgurin P, Nicolas-Metral V, Dumont JM, Porchet H, Crabbe R, Scalfaro P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47:817–826. doi: 10.1002/hep.22131. [DOI] [PubMed] [Google Scholar]
- Flisiak R, Pawlotsky JM, Crabbe R, Calistru PI, Kryczka W, Haussinger D, Mazzella G, Romero-Gomez M, Purcea D, Vuagniaux G, Bao W, Avila C, Zeuzem S. Once Daily Alisporivir (Deb025) Plus Pegifnalfa2a/Ribavirin Results in Superior Sustained Virologic Response (Svr24) in Chronic Hepatitis C Genotype 1 Treatment Naive Patients. Journal of hepatology. 2011;54:S2–S2. [Google Scholar]
- Friborg J, Levine S, Chen CQ, Sheaffer AK, Chaniewski S, Voss S, Lemm JA, McPhee F. Combinations of Lambda Interferon with Direct-Acting Antiviral Agents Are Highly Efficient in Suppressing Hepatitis C Virus Replication. Antimicrob Agents Ch. 2013a;57:1312–1322. doi: 10.1128/AAC.02239-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friborg J, Lin B, Chen C, McPhee F. Isolation and characterization of interferon lambda-resistant hepatitis C virus replicon cell lines. Virology. 2013b;444:384–393. doi: 10.1016/j.virol.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug design, development and therapy. 2013;7:105–115. doi: 10.2147/DDDT.S30946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Evans MJ. miR-122 is more than a shield for the hepatitis C virus genome. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1571–1572. doi: 10.1073/pnas.1220841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nature reviews. Molecular cell biology. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Ghany MG, Strader DB, Thomas DL, Seeff LB American Association for the Study of Liver, D. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilead Sciences. Gilead Reports Interim Data from Phase 2 LONESTAR Study. Press Release. 2013 May 2; [Google Scholar]
- Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun. 2006;343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Bobardt M, Obeid S, Chatterji U, Coates NJ, Foster T, Gallay P, Leyssen P, Moss SJ, Neyts J, Nur-e-Alam M, Paeshuyse J, Piraee M, Suthar D, Warneck T, Zhang MQ, Wilkinson B. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob Agents Chemother. 2011;55:1975–1981. doi: 10.1128/AAC.01627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D. All for one, one for all: new combinatorial RNAi therapies combat hepatitis C virus evolution. Molecular therapy : the journal of the American Society of Gene Therapy. 2012;20:1661–1663. doi: 10.1038/mt.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. The Journal of biological chemistry. 2009;284:13589–13601. doi: 10.1074/jbc.M809244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim MH. 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat Rev Immunol. 2013;13:535–542. doi: 10.1038/nri3463. [DOI] [PubMed] [Google Scholar]
- Hildebrandt-Eriksen ES, Bagger YZ, Knudsen TB, Petri A, Persson R, Boergesen HM, McHulchison JG, Levin AA. A Unique Therapy for Hcv Inhibits Microrna-122 in Humans and Results in Hcv Rna Suppression in Chronically Infected Chimpanzees: Results from Primate and First-in-Human Studies. Hepatology. 2009;50:12A–12A. [Google Scholar]
- Hopkins S, DiMassimo B, Rusnak P, Heuman D, Lalezari J, Sluder A, Scorneaux B, Mosier S, Kowalczyk P, Ribeill Y, Baugh J, Gallay P. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. Journal of hepatology. 2012;57:47–54. doi: 10.1016/j.jhep.2012.02.024. [DOI] [PubMed] [Google Scholar]
- Hopkins S, Scorneaux B, Huang Z, Murray MG, Wring S, Smitley C, Harris R, Erdmann F, Fischer G, Ribeill Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob Agents Chemother. 2010;54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Tseng CP, Liao MH, Lu SC, Yeh WZ, Sakamoto N, Chen CM, Cheng JC. Hepatitis C Virus Replication Is Modulated by the Interaction of Nonstructural Protein NS5B and Fatty Acid Synthase. Journal of virology. 2013;87:4994–5004. doi: 10.1128/JVI.02526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sekiyama K, Yamada M, Watanabe T, Yasuda H, Yoshiba M. Combined interferon alpha2b and cyclosporin A in the treatment of chronic hepatitis C: controlled trial. Journal of gastroenterology. 2003;38:567–572. doi: 10.1007/s00535-002-1104-5. [DOI] [PubMed] [Google Scholar]
- Inoue K, Yoshiba M. Interferon combined with cyclosporine treatment as an effective countermeasure against hepatitis C virus recurrence in liver transplant patients with end-stage hepatitis C virus related disease. Transplantation proceedings. 2005;37:1233–1234. doi: 10.1016/j.transproceed.2004.11.041. [DOI] [PubMed] [Google Scholar]
- Ishii N, Watashi K, Hishiki T, Goto K, Inoue D, Hijikata M, Wakita T, Kato N, Shimotohno K. Diverse effects of cyclosporine on hepatitis C virus strain replication. Journal of virology. 2006;80:4510–4520. doi: 10.1128/JVI.80.9.4510-4520.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. Treatment of HCV infection by targeting microRNA. The New England journal of medicine. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- Jiang LJ, Liu S, Phan TU, Polemeropoulos AJ, Rhodin MH, Owens CM, Luo X, Hoang K, Long J, Gao X, Wang GQ, Or YS. EDP-546, a Potent and Novel Cyclophilin Inhibitor with Favorable Preclinical Pharmacokinetic and Safety Profiles. Hepatology. 2012;56:1076A–1076A. [Google Scholar]
- Jones CT, Catanese MT, Law LMJ, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol. 2010;28 doi: 10.1038/nbt.1604. 167-U116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke HM, Zydowsky LD, Liu J, Walsh CT. Crystal structure of recombinant human T-cell cyclophilin A at 2.5 A resolution. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9483–9487. doi: 10.1073/pnas.88.21.9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel SJ, Lowther WT, Pemble CWt. Fatty acid synthase inhibitors: new directions for oncology. Expert opinion on investigational drugs. 2007;16:1817–1829. doi: 10.1517/13543784.16.11.1817. [DOI] [PubMed] [Google Scholar]
- Lacek K, Vercauteren K, Grzyb K, Naddeo M, Verhoye L, Slowikowski MP, Fafi-Kremer S, Patel AH, Baumert TF, Folgori A, Leroux-Roels G, Cortese R, Meuleman P, Nicosia A. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. Journal of hepatology. 2012;57:17–23. doi: 10.1016/j.jhep.2012.02.018. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer GM, Walker BD. Hepatitis C virus infection. The New England journal of medicine. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. The global burden of hepatitis C. Liver international : official journal of the International Association for the Study of the Liver. 2009;29(Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR-122 and the 5' exonuclease Xrn1 in regulation of hepatitis C virus replication. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YP, Gottwein JM, Scheel TK, Jensen TB, Bukh J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5' UTR. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4991–4996. doi: 10.1073/pnas.1016606108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J, Sarnow P. Micromanaging hepatitis C virus. The New England journal of medicine. 2013;368:1741–1743. doi: 10.1056/NEJMe1301348. [DOI] [PubMed] [Google Scholar]
- Lin K, Gallay P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral research. 2013 doi: 10.1016/j.antiviral.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. Journal of virology. 2009;83:6554–6565. doi: 10.1128/JVI.02550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Boerner JE, TiongYip C, Weidmann B, Ryder NS, Cooreman MP, Lin K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50:2976–2982. doi: 10.1128/AAC.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiorkinis G, Magiorkinis E, Paraskevis D, Ho SY, Shapiro B, Pybus OG, Allain JP, Hatzakis A. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS medicine. 2009;6:e1000198. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson D, Koseki M, Ishibashi M, Larson CJ, Miller SG, King BD, Tall AR. Increased HDL Cholesterol and ApoA-I in Humans and Mice Treated With a Novel SR-BI Inhibitor. Arterioscl Throm Vas. 2009;29:U2054–U2170. doi: 10.1161/ATVBAHA.109.191320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Garcia-Rivera JA, Leyssen P, Coates N, Foster T, Kendrew S, Nur-E-Alam M, Stanley AE, Suthar D, Warneck T, Neyts J, Gallay P, Wilkinson B, Gregory MA. Bc556, a Potent, Pan-Genotypic, High Barrier to Resistance, Second Generation Cyclophilin Inhibitor for Treatment of Chronic Hcv Infection. Journal of hepatology. 2012;56:S328–S328. [Google Scholar]
- Muir AJ, Shiffinan ML, Zaman A, Yoffe B, de la Torre A, Flamm S, Gordon SC, Marotta P, Vierling JM, Lopez-Talavera JC, Byrnes-Blake K, Fontana D, Freeman J, Gray T, Hausman D, Hunder NN, Lawitz E. Phase 1b Study of Pegylated Interferon Lambda 1 With or Without Ribavirin in Patients with Chronic Genotype 1 Hepatitis C Virus Infection. Hepatology. 2010;52:822–832. doi: 10.1002/hep.23743. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Sakamoto N, Enomoto N, Tanabe Y, Kanazawa N, Koyama T, Kurosaki M, Maekawa S, Yamashiro T, Chen CH, Itsui Y, Kakinuma S, Watanabe M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochemical and biophysical research communications. 2004;313:42–47. doi: 10.1016/j.bbrc.2003.11.080. [DOI] [PubMed] [Google Scholar]
- Owens CM, R M, Polemeropoulos A, Long J, Wang G, Jiang L, Or YS. Cyclophilin Inhibitor EDP-546 is a Potential Cornerstone Drug for Use in Combination with NS5A and Protease Inhibitors Due to its High Barrier to HCV Resistance. Cyclophilin Inhibitor EDP-546 is a Potential Cornerstone Drug for Use in Combination with NS5A and Protease Inhibitors Due to its High Barrier to HCV Resistance; 48th Annual Meeting of the European Association for the Study of the Liver (EASL 2013); Amsterdam. 2013. Abstract 1213. [Google Scholar]
- Paeshuyse J, Kaul A, De Clercq E, Rosenwirth B, Dumont JM, Scalfaro P, Bartenschlager R, Neyts J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43:761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- Pawlotsky JM, Sarin SK, Foster GR, Peng CY, Rasenack J, Flisiak R, Piratvisuth T, Wedemeyer H, Chuang WL, Zhang W, Naoumov NV. Alisporivir plus Ribavirin achieves high rates of sustained HCV clearance (SVR24) as interferon (IFN)-free or IFN-add-on regimen in treatment-naive patients with HCV GT2 or GT3: Final results from VITAL-1 study. Hepatology. 2012;56:309A–310A. [Google Scholar]
- Perales C, Beach NM, Gallego I, Soria ME, Quer J, Esteban JI, Rice C, Domingo E, Sheldon J. Response of Hepatitis C Virus to Long-Term Passage in the Presence of Alpha Interferon: Multiple Mutations and a Common Phenotype. Journal of virology. 2013;87:7593–7607. doi: 10.1128/JVI.02824-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynard T, Yuen MF, Ratziu V, Lai CL. Viral hepatitis C. Lancet. 2003;362:2095–2100. doi: 10.1016/s0140-6736(03)15109-4. [DOI] [PubMed] [Google Scholar]
- Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. Cellular cofactors affecting hepatitis C virus infection and replication. P Natl Acad Sci USA. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. The Lipid Kinase Phosphatidylinositol-4 Kinase III Alpha Regulates the Phosphorylation Status of Hepatitis C Virus NS5A. Plos Pathog. 2013;9 doi: 10.1371/journal.ppat.1003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Torres M, Ghalib RH, Gordon SC, Lawitz E, Patel K, Pruitt R, Sheikh AM, Arbeit RD, Bexon AS, Kandimalla ER, Precopio M, Sullivan T, Muir AJ, McHutchison JG. Imo-2125, a Tlr9 Agonist, Induces Immune Responses Which Correlate with Reductions in Viral Load in Null Responder Hcv Patients. Hepatology. 2010;52:336A–336A. [Google Scholar]
- Scheel TKH, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. Journal of hepatology. 2011;54:48–55. doi: 10.1016/j.jhep.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Vaillancourt FH, Brault M, Pilote L, Uyttersprot N, Gaillard ET, Stoltz JH, Knight BL, Pantages L, McFarland M, Breitfelder S, Chiu TT, Mahrouche L, Faucher AM, Cartier M, Cordingley MG, Bethell RC, Jiang HP, White PW, Kukolj G. Evaluation of Phosphatidylinositol-4-Kinase III alpha as a Hepatitis C Virus Drug Target. Journal of virology. 2012;86:11595–11607. doi: 10.1128/JVI.01320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller H, Chatterji U, Gallay P, Parkinson T, Targett-Adams P. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J Virol Methods. 2010;165:202–210. doi: 10.1016/j.jviromet.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AGN, Keck ZY, Foung SKH, Pietschmann T, Rice CM, Patel AH. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol. 2009;90:48–58. doi: 10.1099/vir.0.006700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology. 2008;48:1396–1403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuzem S, Arora S, Bacon B, Box T, Charlton M, Diago M, Dieterich D, Mur RE, Everson G, Fallon M, Ferenci P, Flisiak R, George J, Ghalib R, Gitlin N, Gladysz A, Gordon S, Greenbloom S, Hassanein T, Jacobson I, Jeffers L, Kowdley K, Lawitz E, Lee S, Leggett B, Lueth S, Nelson D, Pockros P, Rodriguez-Torres M, Rustgi V, Serfaty L, Sherman M, Shiffman M, Sola R, Sulkowski M, Vargas H, Vierling J, Yoffe B, Ishak L, Fontana D, Xu D, Lester J, Gray T, Horga A, Hillson J, Ramos E, Lopez-Talavera JC, Muir A, Grp ES. PEGYLATED INTERFERON-LAMBDA (PEGIFN-lambda) SHOWS SUPERIOR VIRAL RESPONSE WITH IMPROVED SAFETY AND TOLERABILITYVERSUS PEGIFN alpha 2A IN HCV PATIENTS (G1/2/3/4): EMERGE PHASE IIB THROUGH WEEK 12. Journal of hepatology. 2011;54:S538–S539. [Google Scholar]
- Zhu HH, Wong-Staal F, Lee H, Syder A, McKelvy J, Schooley RT, Wyles DL. Evaluation of ITX 5061, a Scavenger Receptor B1 Antagonist: Resistance Selection and Activity in Combination With Other Hepatitis C Virus Antivirals. Journal of Infectious Diseases. 2012;205:656–662. doi: 10.1093/infdis/jir802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, Walsh CT. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1:1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]