

Abstract
Coxsackievirus B (CVB) is a significant pathogen of neonatal diseases with severe systemic involvement and high mortality. Hence, it is essential to develop a CVB-induced acute systemic disease model on newborn mouse and study the injury at the onset phase. In this work, a clinical strain of CVB3, Nancy, and its variant strain, Macocy, were adopted in 24 hour old neonates by oral infection. The pathological changes in the heart, liver and lung tissues were analyzed by pathology assays. In situ end labeling assay for programmed cell death was carried out for liver tissues. The data on fatality and infection rates and pathology scores were analyzed statistically. The genomic sequences of the two strains were aligned. The model represented the manifest clinical syndromes of hepatitis, pneumonia and myocardial injury at the onset phase, in which massive numbers of hepatocytes had undergone programmed cell death. Statistical and pathological analysis indicated that the myocardial injury was mild, whereas the liver and lung were more severe. The fatality rate, infection and pathology of the two CVB strains were the same. Therefore, two nucleotide mutations in the 5’ UTR and four amino acid mutations in polyprotein, which did not alter virulence, were shown. By peroral CVB infection of neonatal mice, we developed an acute systemic disease model for studying visceral pathology and systemic disease. At the onset of acute neonatal systemic disease, the hepatitis and pneumonia may be the dominant reason of death, as the injury of liver and lung is more severe than that of heart.
Keywords: Coxsackievirus B, acute neonatal systemic disease, pathological diversity, mutation sites
Introduction
Coxsackievirus B (CVB) is a human enterovirus, and belongs to the genus Picornaviridae. CVB virions are small, non-enveloped, and contain a single-stranded, positive-sense 7.4-kb genomic RNA. CVB is one of the most significant pathogens causing serious neonatal diseases [1] with a high fatality rate of 11.1% - 40.0% [2]. Meanwhile, CVB induces a high incidence of acute neonatal diseases [3,4], although in adults it usually causes chronic infections, such as dilated cardiomyopathy, chronic pancreatitis and central nervous system infection [5,6].
CVB is often related to myocarditis [1], hepatitis [7] and pneumonia [8] in neonates. The pancreas is the primary target of CVB replication [9] yet the route of infection has been shown to influence the histopathological changes in the pancreases of adolescent mice [10]. After primary proliferation, the virus enters the bloodstream by penetrating the epithelial barrier of the blood vessels, and travels to secondary replication sites in the liver, lungs and other organs. In the first 2 weeks of the neonatal period, CVB-induced disease is typically severe [1]. Lethal CVB infections in neonates are frequently accompanied by signs of systemic involvement [1,11,12]. In particular, the concurrence of myocarditis and hepatitis associated with coagulopathy increases the risk of fatality [7] and has poor outcome [1,13], whereas encephalomyocarditis is commonly associated with hemorrhage hepatitis [12]. Besides, CVB can induce many respiratory symptoms [14,15], which can be fatal for neonates [9]. Most of the experimental CVB in vivo mouse literature focuses on particular organs [16-18]. Even though there are many clinical case reports and epidemiological surveys [19,20], the pathogenesis on systemic involvement needs to be studied further by experimental study, especially the fatal acute neonatal diseases. Thus, the development of an acute systemic disease model on newborn mouse is very important.
Ordinarily, virus intraperitoneal injection was used when establishing the CVB mouse model [21,22]. However this is not a natural route of infection, through which CVB invades. In fact, CVB infection is by fecal-oral transmission, and begins its infecting and proliferating in the intestine. For approaching the natural condition, we used oral infection, which is more significant comparing to the other inoculating routes.
We established a systemic disease model involving hepatitis, pneumonia and myocarditis. Focusing on the onset phase (72 hours after infection) of acute neonatal systemic injury, we studied the probable causes of death from the point of diversity of visceral pathology, and presumed the probable airborne route of CVB3 transmission. Two CVB3 strains, Macocy and Nancy, were used. Macocy is a laboratory variant; Nancy is its parental strain and also a clinical strain. Comparing the sequences of two strains, the mutation sites in the 5’ UTR and polyprotein, which did not change the virulence of CVB3 in the early infection phase, were marked.
Methods
Virus stocks, cell lines and TCID50
The Macocy strain and Nancy strains of CVB3 were used. CVB3/Macocy, as a laboratory variant, was isolated from experimental mouse liver tissue in our lab in 2007. Nancy strain was acquired in 2008 from HuBei Center for Disease Control and Prevention. Vero cell monolayers were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 2 mmol/l L-glutamine and 10% calf serum, at 37°C under 5.0% CO2. After inoculation, the virus was harvested when a cytopathic effect (CPE) appeared. The virus titer was determined by TCID50 with HeLa cells by culturing in 96-well plates. Ten-fold serial dilutions of virus suspensions were produced from 10–1 to 10–10, which were filtered to remove bacteria. The cells were inoculated with virus suspension when monolayers were formed. Every column of 8 wells was added with one dilution of 100 μl/well. The first 10 columns were cultured with virus. The remaining 2 columns were treated as the mock infection by adding DMEM instead. Plates were incubated at 37°C with 5% CO2. When the complete CPE appeared (about 72 hours), the TCID50 was calculated by the Reed–Muench method [23].
Mouse infection
Newborn randomized male and female BALB/c mice, within 24 hours of birth, were bred under specific pathogen-free conditions and in separate rooms for each group. This included the mock-infected control group, and the groups inoculated with strain Macocy or Nancy. Strain Macocy is a natural variant, and Nancy is its parental strain and a clinical pathogen. All the mice were randomly separated into the different groups of 44 mice each. These sucklings were infected immediately after the separation from their dams. The virus-inoculated groups were infected with 5×105 TCID50 of strain Nancy or Macocy diluted in 25 μl DMEM, whereas the mock-infected control group was fed with 25 μl DMEM. The inoculation by feeding was carried out by micropipette with 1 μl virus inoculum in every mouthful. All mice were acquired from the Animal Center of Wuhan University. All the procedures were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee at Wuhan University and approved by the committee. During 72 hours after infection, the sucklings were fed with aseptic milk, the living mice in the three groups were euthanized by isoflurane overdose and dissected. The heart, liver and lung tissues were harvested, washed in PBS, and fixed in 10% formaldehyde.
H&E staining
Formalin-fixed, paraffin-embedded tissues were sliced into sections about 3.5 μm thick. The slices were deparaffinized two times in xylene for 15 minutes each and subsequently rehydrated through sequential concentrations of alcohol and double distilled water (ddH2O), for 3 minutes each time. They were stained with hematoxylin solution for 7 minutes, washed in running water for 2 minutes, differentiated in 1% acid alcohol for 10 seconds, and washed with running water for 10 seconds. The slices were immersed in 0.2% ammonia water for 20 seconds for bluing, washed with running water for 3 minutes, and washed with ddH2O for 2 minutes. For staining, they were immersed in 0.5% eosin solution for 2 minutes. After washing with ddH2O for 10 minutes, they were dehydrated through sequential concentrations of alcohol for 5 minutes each time, and immersed two times in xylene for 5 minutes each. The slices were mounted with xylene-based mounting medium.
Immunohistochemistry assay
Monoclonal antibody Mab 948 (Millipore, USA), the CVB3 VP1-specific mouse antibody, was used as the primary antibody. Formalin-fixed, paraffin-embedded tissues were sliced into 3.5-μm sections. The sections were incubated at 56°C for 30 minutes, deparaffinized two times in xylene for 15 minutes each. Slices were rehydrated through sequential concentrations of alcohol and ddH2O. Antigen retrieval was done by boiling in 10 mM citrate buffer at pH 6.0 for 10 minutes in a microwave oven, the slices were cooled in the buffer for at least 1 hour at room temperature, and washed two time in PBS. Moreover, endogenous peroxidase was blocked by 3% hydrogen peroxide in water for 3 minutes at room temperature. After rinsing, slides were incubated with bovine serum albumin (BSA) for 10 minutes to block nonspecific staining. The primary monoclonal antibody was incubated with the slices at 4°C overnight. After extensive washing, slides were incubated at room temperature for 30 minutes with biotin-labeled goat anti-mouse antibody (1:200 final dilution; Vector Laboratories, Burlingame, CA, USA), and for 30 minutes with avidin–biotin peroxidase complexes (1:25 final dilution; Vector Laboratories). Diaminobenzidine (DAB) (0.06%) was used as the final chromogen, and hematoxylin was used as the nuclear counterstain.
In situ end labeling programmed cell death assay
Formalin-fixed, paraffin-embedded tissues were sliced into 3.5-μm-thick sections. The slices were incubated at 56°C for 30 minutes, deparaffinized two times in xylene for 15 minutes each, and rehydrated through sequential concentrations of alcohol and ddH2O. To deactivate endogenous peroxidase, the deparaffinized slices were incubated with 3% H2O2 for 10 minutes at room temperature. The TUNEL staining was processed using a TUNEL cell apoptosis test kit (ZK-8005; Zhongshan Goldenbridge Biotechnology, PRC). Finally, they were slightly re-stained with hematoxylin.
Pathological score
The slides were scored by two individuals separately. So, the pathological changes in 57 virus-inoculated mice (Fifty-seven samples of each tissue) were scored twice, and the total number was 114 for every kind of tissue. Necrosis and inflammatory exudation were noted. The degrees of severity include mild, moderate and severe. The mild level was scored 1-2; moderate, 3-4; severe, 5-6; and no obvious pathology, 0. According to inflammatory exudation, every tissue could acquire an additional score of 1-2, and the greater the exudation, the higher the score acquired. Thus, the score range was 0-8. For lungs, mild pathology was manifested by interstitial tissue thickening, vascular congestion, tissue edema, and alveolar collapse; moderate pathology was manifested by consolidation of the lungs and further deterioration; and severe pathology was manifested by the destruction of tissue structure, and advanced consolidation. For liver, mild pathology was manifested by degeneration of hepatic cells and spotty necrosis; moderate pathology was manifested by focal and moderate bridging necrosis; and severe pathology was manifested by sub-massive and massive necrosis. For heart, mild pathology was manifested by tissue swelling and cell degeneration; moderate pathology was manifested by local necrosis; and severe pathology was manifested by sub-massive and massive necrosis. Each lesion was scored according to the pathological scoring system. The significant pathological lesions criterion was set at a score ≥3. Therefore, the pathological lesion rates were calculated.
Mutation sites alignment and structure generation
The genomic sequences of strain Macocy (GenBank accession number: JQ040513), Nancy (GenBank accession number: JN048468), 0 (GenBank accession number: AY752945) and 28 (GenBank accession number: AY752944) were aligned by BioEdit 7.0.8 software, as described previously [24]. The consensus alignment was compared carefully to identify the nucleotide mutations in 5’ UTR and the amino acid differences in the polyprotein. The sequences are available in GenBank. Protein (VP3, VP1 and 2A) structures of strain Macocy were generated by homology modeling at SWISS-MODEL in project mode, as described previously [25-27].
Statistical analysis
The data were analyzed by SPSS 17.0 for Windows. For multi-comparison among heart, liver and lung for the pathological lesion rates and infection rates, Chi-Square test for 2×4 contingency table method was used with a significance level a=0.05. For pairwise comparison in heart, liver and lung for the pathological lesion rates and infection rates, Chi-Square test for 2×2 contingency-table method was used with the significance level a=0.0083 which was calculated by the Bonferroni method [28], in which for comparing liver and lung for the infection rates, Fisher’s exact test for 2×2 contingency-table method was used. For multi-comparison among heart, liver and lung for the median of pathology score, Kruskal-Wallis test was used with a significance level a=0.05, and for pairwise comparison in heart, liver and lung for the median score, Kruskal-Wallis test was used with the significance level a=0.0042 which was calculated by the previous method [29].
For assessing the statistical significance of fatality rates and infection rates in heart, liver and lung for the Macocy and Nancy groups, Chi-Square test for 2×2 contingency table method was used with significant level a=0.05.
For assessing the difference of fatality rates for group Macocy versus Nancy, Chi-Square test for 2×2 contingency-table method was used with significance level a=0.05. For assessing the difference of infection rates for group Macocy versus Nancy in heart and liver, Chi-Square test for 2×2 contingency-table method was used with significance level a=0.05, and in lung, Fisher’s exact test for 2×2 contingency-table method was used with significance level a=0.05. For assessing the difference of pathological lesion rates for group Macocy versus Nancy in heart, Chi-Square test for 2×2 contingency-table method was used with significance level a=0.05, and in lung and liver, Fisher’s exact test for 2×2 contingency-table method was used with significance level a=0.05.
The sample size per group and the statistical power were calculated by the formulas for Chi-Square test [30,31], and Fisher’s exact test [32] with significance level a=0.05.
Results
Fatality, gross anatomy and general pathology
The fatality rates were 4.55% (2 deaths) in the mock control, 31.82% (14 deaths) in the Nancy group, and 38.64% (17 deaths) in the Macocy group at 72 hours after infection. The fatality rates in both the Macocy (p<0.001) and Nancy (p<0.001) groups were very highly significant [29].
The symptoms observed in the Macocy and Nancy groups were almost identical. After inoculation and during 72 hours incubation, every virus-inoculated mouse had diarrhea with the clinical signs of watery stools and abdominal distension, and developed marasmus. Skin redness was also observed all over the body. The symptoms were the same as the clinical symptoms at the onset of infection [33,34]. When the abdomen was opened by incision, we visually found that all the pancreases had almost been dissolved and had liquefaction necrosis. We also observed intestinal tympanites and localized liquefaction necrosis of abdomen tissue on gross. The lungs, liver and heart had serious infections (Figure 1A) with pathological lesions (Figure 1B). The pathology scores of the different organs are shown in Figure 1C-G.
Figure 1.
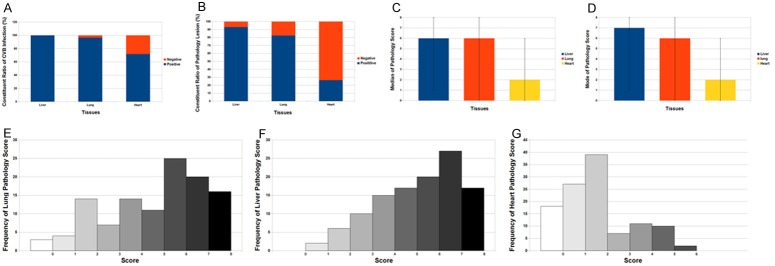
The infection and pathology of the liver, lung and heart respectively. The virus strain for infection was CVB3 Nancy strain and Macocy strain, and at the 72nd hour after infection the tissue was harvested for pathology exam. The two strains are not distinguished in this statistical work. A. The liver, lung and heart were harvested. The infection rates as well as negative percentages of IHC of CVB-infected organs were exhibited on the stacked column. B. The positive lesion rates as well as negative rates of CVB-infected liver, lung and heart were exhibited on the stacked column. C. The median of the pathology scores on liver, lung and heart were shown. The error bars represent the ranges of the score of each tissue. D. The mode of the pathology scores on liver, lung and heart were shown. The error bars represent the ranges of the score of each tissue. E-G. Three bar charts show the frequency distribution of pathology score for lung, liver and heart. The score ranges 0 to 8, and the y-axis shows the frequency of every score.
CVB-induced pneumonitis
In lung tissues, immunohistochemistry showed that 96.30% of tissues were positive for Macocy strain and 96.67% for Nancy strain. This clearly demonstrated that the lungs of infected neonatal mice were severely infected by Nancy (p<0.001) and Macocy (p<0.001) strains. Besides, there was a high positive rate of pathological lesions in the lungs (82.46%) (Figure 1B) and a high positive rate of infection rate (96.49%) for both strains all together (Figure 1A). The median and mode of the lung pathological scores was 6 (Figure 1C, 1D), which represent the upper median level of lung damage with inflammatory infiltration. The score range was 0-8, and 53.5% of samples scored 6-7 (Figure 1E). Diaminobenzidine (DAB) staining demonstrated that the virus was widely spread in the lung tissue (Figure 2F-H). Pathological lesions in the lungs were obvious in the hematoxylin and eosin (H&E)-stained samples (Figure 2B-D). We observed that the alveoli were filled with blood cells (Figure 2B, 2C), which is a proof of severe diffuse alveolar hemorrhage. The septa of dilated pulmonary alveoli were interrupted and alveoli fused into large capsular spaces (Figure 2B-D). Severe pulmonary consolidation was also observed (Figure 2B, 2C). Inflammatory cells and fibrin infiltrated into the pulmonary consolidations regions (Figure 2B, 2C). Compared with normal tissue in the control group, some alveolar septa of infected lungs were obviously widened (Figure 2B-D, 2F). Besides, some lung tissues revealed atelectasis characterized by incomplete expansion of the alveoli (Figure 2G). Others showed emphysema, which was caused by destruction of the alveolar walls (Figure 2H). Based on the pathological analysis, it can be concluded that the mice infected by CVB had severe viral pneumonitis.
Figure 2.
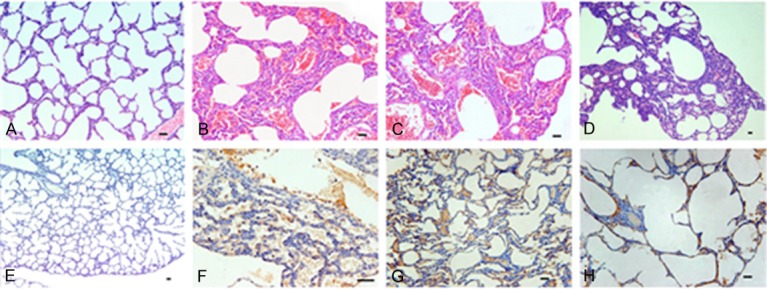
Pathological changes of lung in CVB-infected mice at the disease onset phase. The virus strain for infection was CVB3 Nancy strain and Macocy strain, and at the 72nd hour after infection the tissue was harvested for pathology exam. The two strains are not distinguished in this statistical work. (Images A-D) Display the H&E stained slices, in which (image A) is the mock control, and (images B-D) show the pathology lesions of pulmonary lobes. (Images E-H) Display the IHC staining samples, in which (image E) is the mock control of IHC staining. Brown (images F-H) represents CVB virus which was prevalent with severe pathology in the tissue. All the bars in the pictures represent 50 μm. Magnification, ×120 for (images D, E), magnification, ×240 for (images A-C, G, H); magnification, ×480 for (image F).
CVB-induced hepatitis
Immunohistochemistry showed that 99.99% of tissues were positive for Macocy strain and Nancy strain respectively, which demonstrated that almost all the livers of neonatal mice were infected and damaged (Figure 1A, 1B). Indeed, the apoptosis rates in the Nancy (76.67%) and Macocy (96.30%) groups were very high (Figure 3K). These data suggest that most of the liver tissue was destroyed, accompanied with inflammatory infiltrate. Thus, the pathological assay demonstrated that these mice had acute viral hepatitis.
Figure 3.
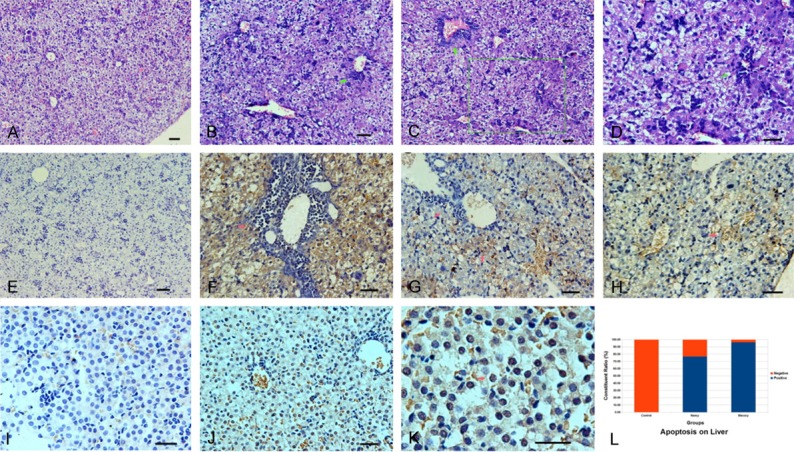
Pathological changes of liver with the apoptosis rates in CVB infected mice at the disease onset phase. The virus strain for infection was CVB3 Nancy strain and Macocy strain, and at the 72nd hour after infection the tissue was harvested for pathology exam. The two strains are not distinguished in this statistical work. (Images A-D) Display the live slices stained with H&E, in which (image A) is the mock control, while (images B-D) show severe pathological changes. The arrows in (images B-D) point the accumulation areas of inflammatory cells. (Image D) is a magnifying photograph of (image C). The area was marked in the (Image C) (green box). (Images E-H) Display the picture of IHC assay, in which (image E) is the mock control. In (images F-H), Brown represents CVB virus which was prevalent with severe hepatocyte lesions, tissue structure damage and inflammatory cell infiltrate in the tissue. The arrow in (image F) and the left arrow in (image G) point two accumulation areas of inflammatory cells. The right arrow in (image G) and the arrow in (image H) point two of a large number of injured cells. (Images I-K) Display CVB-induced hepatocyte apoptosis. The arrow (image K) points one hepatocyte which was undergoing apoptosis. All the bars in the histological pictures represent 50 μm. (Image L) is a stacked column that shows apoptosis rates by CVB3/Nancy strain and Macocy strain as well as negative rates in different groups. Magnification, ×240 for (images A-C, E); magnification, ×480 for (images D, F-J); magnification, ×1200 for (image K).
CVB-induced myocardial infection
Seventy-two hours after virus inoculation, CVB virus was detected in cardiac muscle fibers with a positive rate of 66.67% in the Nancy group and 77.78% in the Macocy group (Figures 1A, 4E, 4F). There was a very highly significant difference in infection rate between the control group and the Nancy (p<0.001) and Macocy (p<0.001) groups with an infection rate of 71.93% for both strains all together (Figure 1A). However, for both infection groups heart pathology lesions were not obvious generally compared with the control group, with a low rate of only 26.32% all together (Figures 1B, 4A-C). In fact, both the mode and median score for heart pathology was 2 (Figure 1C, 1D), which meant that most cardiac muscle was not seriously injured, and cellular necrosis was scarcely observed. The scores for the heart ranged from 0 to 6 (Figure 1G). Nearly 75% of heart samples had a score of 0-2, whereas <5% had a score >6 (Figure 1G).
Figure 4.
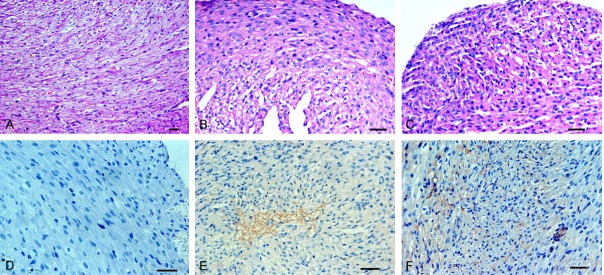
Pathological changes of heart in CVB infected mice at the disease onset phase. The virus strain for infection was CVB3 Nancy strain and Macocy strain, and at the 72nd hour after infection the tissue was harvested for pathology exam. The two strains are not distinguished in this statistical work. (Images A-C) Display the slices stained with H&E on heart, in which (image A) shows the normal state as a mock control, while (B and C) does not displays the evident pathology of CVB-infected heart. (Images D-F) Display the result of IHC assay, in while (C) is a mock control. Brown (images E and F) shows the CVB virus in heart. The arrow (image F) points the accumulation areas of inflammatory cells. All the bars in the histological pictures represent 50 μm. Magnification, ×240 for (image A); magnification, ×480 for (image B-F).
Diversity of systemic infection by statistical comparison
Heart, lungs and liver had various degrees of infections (Figure 1A) and pathology lesion (Figure 1B) at the 72nd hours after infection. In the two groups of neonatal mice, pneumonia, hepatitis and myocardial injury were concurrent; the diffuse pneumonia and hepatitis were fulminant and severe, while the myocardial injury was mild. For judging the different severities of these diseases, we first compared the infection rates of the three tissues, and found that they differed significantly (p<0.001). Based on prerequisite multiple comparisons, the infection rates were compared between every combination of two types of tissues. There were no significant differences in the comparisons between liver and lung (p=0.154), whereas there were significant differences in the other comparisons for heart versus liver (p<0.001) and heart versus lung (p<0.001). Thus, the infection rate of liver (99.99%) was significantly greater than that of heart (71.93%), while that of lung (96.49%) was significantly greater than that of heart (71.93%). Subsequently, we compared the pathological lesion rates among the three tissues, and found that they were significantly different (p<0.001). Based on these prerequisite multiple comparisons, we compared the pathological lesion rates between every combination of two tissues. There was no significant difference in the comparisons for liver versus lung (p<0.087), whereas there were significant differences in the other comparisons for heart versus liver (p<0.001) and heart versus lung (p<0.001,). Therefore, the lesion rate of liver (92.98%) and lung (82.46%) was significantly greater than that of heart (26.32%). Also, the median scores among the three tissues were compared, and they were significantly different (p<0.001). Based on these prerequisite multiple comparisons, the median scores between every combination of two tissues were compared. There was no significant difference in the comparisons for liver versus lung (p=0.179), while there were significant differences in the other comparisons for heart versus liver (p<0.001) and heart versus lung (p<0.001). Thus, the median score for liver (6) and lung (6) was significantly greater than that for heart (2) (Table 1).
Table 1.
The median of score, pathological lesion rates and infection rates for multi-comparison and pairwise-comparison
| Median Score | Pathological Lesion Rate (%) | Infection Rate (%) | |
|
| |||
| Heart | 2 | (15/57) 26.32 | (41/57) 71.93 |
| Liver | 6 | (53/57) 92.98 | (57/57) 99.99 |
| Lung | 6 | (47/57) 82.46 | (55/57) 96.49 |
| Total | — | (141/228) 61.84 | (200/228) 87.72 |
|
| |||
| Hypothesis Test | Kruskal-Wallis | Chi-Square | Chi-Square |
|
| |||
| p-value (multi-comparison)a | <0.001 | <0.001 | <0.001 |
| p-value (heart:liver)b | <0.001 | <0.001 | <0.001 |
| p-value (heart:lung)b | <0.001 | <0.001 | <0.001 |
| p-value (liver:lung)b | 0.179 | 0.087 | 0.154c |
p-value for the multi-comparison among heart, liver and lung.
p-value for the pairwise-comparisons between heart and liver, heart and lung and liver and lung respectively.
It was calculated by Fisher’s exact test.
No difference between Macocy and Nancy strains
There was no difference in fatality rates, infection rates and pathological lesion rates between the Macocy and Nancy strains. The fatality rates of mice at 72 hours were compared in the Macocy and Nancy groups, and there was no significant difference (p=0.503). When we compared the infection rates in the Macocy and Nancy groups, there was no significant difference in heart (p=0.351), liver (the infection rates of liver in both groups are >99.9%) and lung (p=1.000). The pathological lesion rates in the Macocy and Nancy groups did not differ significantly in heart (p=0.590), liver (p=1.000) and lung (p=1.000), with a=0.05. From the three-ply comparisons with pathological observation, we found that the virulence of stain Macocy was similar to that of Nancy in heart, liver and lung. Since we confirmed that there was no difference between these two experimental groups, the two groups formed a mutual control group for the experiment.
The sample size for statistical tests
We calculated a series of sample sizes for the statistical tests. As the sample size varied, so too did the power values. In terms of the statistical test for infection rates, from the variation tendency of sample size, it was apparent that even if the sample size was 1, there would be a chance to find a significant difference for CVB3 infecting these tissues (power >0.593) (Figure 5A). In terms of pairwise comparisons for the infection rates and pathological lesions for heart, liver and lung tissues, from the variation tendency of sample size, we found that the minimum acceptable sample size was approximately 40, as the power values for statistical tests with significance were ≥0.654 then. Meanwhile it cannot get an ideal sample size for those tests without significance, as all the power values were very small (power ≤0.353) (Figure 5B). In fact, 57 was the real size that we adopted in the experiment. In terms of infection and pathological lesions comparison tests for the Nancy versus Macocy groups, from the variation tendency of sample size, it was apparent that we cannot get a sample size for these tests without significance. Even if the sample sizes increased to nearly 100, there would be little chance of finding a significant difference (power ≤0.412) (Figure 5C).
Figure 5.
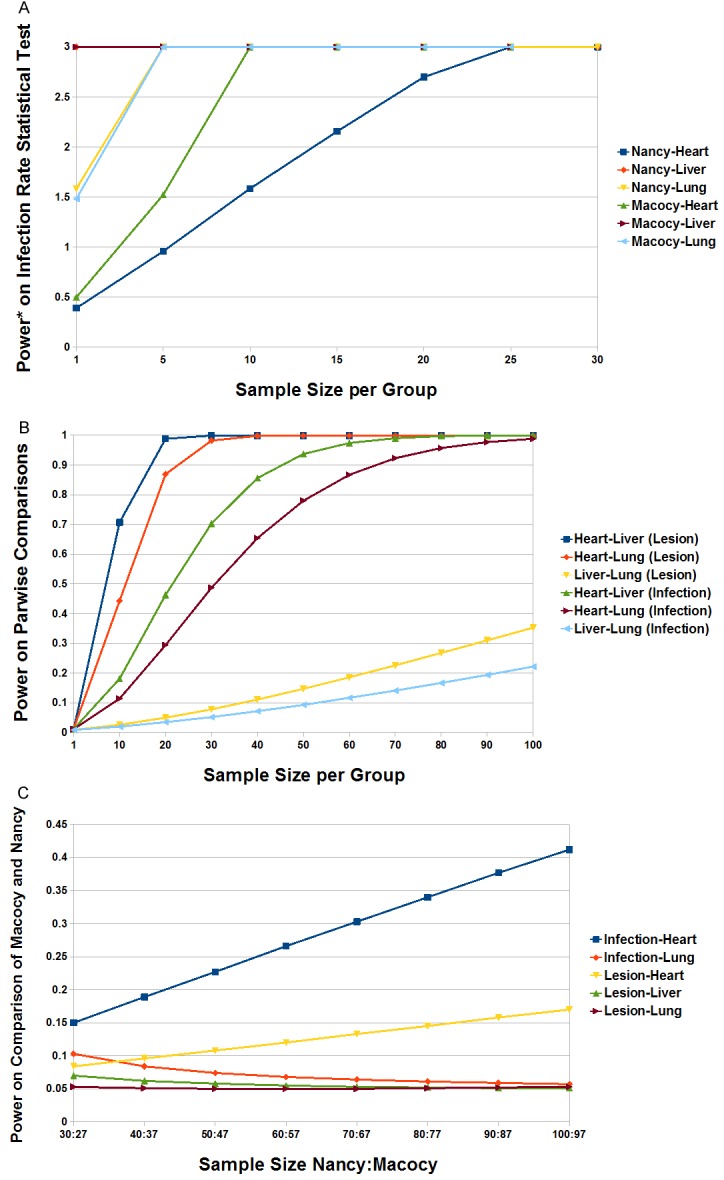
The sample size in statistical tests. A. The trend of sample size for the infection of heart, liver and lung of group Macocy and Nancy was shown by the six color lines, with the arising of the power value. The power* represents the mathematically changed power by log101/(1-x), in which x = power and power* = log101/(1-x). For instance, power* of 0.39 equals power of 0.59, power* of 1.52 equals power of 0.97 and so on. B. The trend of sample size for comparing the lesions of heart-liver, heart-lung, and liver-lung, as well as the infection of heart-liver, heart-lung, and liver-lung was shown by the six color lines, with the power value arising. C. The trend of sample size for comparing the infection and lesions respectively for group Macocy versus Nancy on heart, lung and liver was shown by the five color lines, with the power value arising.
Sequence mutation
The 5’ UTR sequences and polyprotein sequences of strain Macocy, Nancy, 28 and 0 were aligned. Strain Macocy is a variant of Nancy; therefore, we focused on the mutation sites of Macocy comparing strain Nancy. In 5’ UTR of strain Macocy, 408G and 581T were unique, but the other mutation sites (125A, 529T, 578A and 586T) were the same with strain 28. In the polyprotein of strain Macocy, there were five mutations. 563I mutation was the same as in strain 28. But the other four mutations (566K, 829R, 861V and 1486Y) were unique to Macocy. There was a site 234U in the 5’ UTR of strain Macocy, which was the same as in Macocy, Nancy and 28, but in strain 0 it was 234C (Table 2). The mature protein structures (VP3, VP1 and 2A) of strain Macocy were generated by homology modeling, and the unique mutation sites are shown in Figure 6.
Table 2.
Mutation sites in 5’ UTR and polyprotein comparing strain Macocy, Nancy, 28 and 0
| Strain | Nucleotide sites (nt)c | Amino acid sites (aa)d | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||
| 5’ UTR (1-742) | VP3 (333-570) | VP1 (571-854) | 2A (855-1001) | 3A (1430-1518) | ||||||||
|
|
|
|||||||||||
| 125 | 234 | 408 | 529 | 578 | 581 | 586 | 231 (563) | 234 (566) | 259 (829) | 7 (861) | 57 (1486) | |
| Macocya | A | U | G | T | A | T | T | I | K | R | V | Y |
| Nancy | G | U | A | C | G | C | C | T | E | K | A | H |
| 28 | A | U | A | T | A | C | T | I | E | K | A | H |
| 0b | A | C | A | T | A | C | T | I | E | K | A | H |
Positions in bold are unique in strain Macocy comparing the other cadiovirulence strain-Nancy and 28.
The position in bold are unique in strain 0 comparing the cardiovirulence strains.
Positions are numbered from the start of the nucleotide sequence.
Position numbers out of the brackets are started from the N-terminal of each mature protein sequence, and position numbers in the brackets are started from the start coden for each potein sequence.
Figure 6.
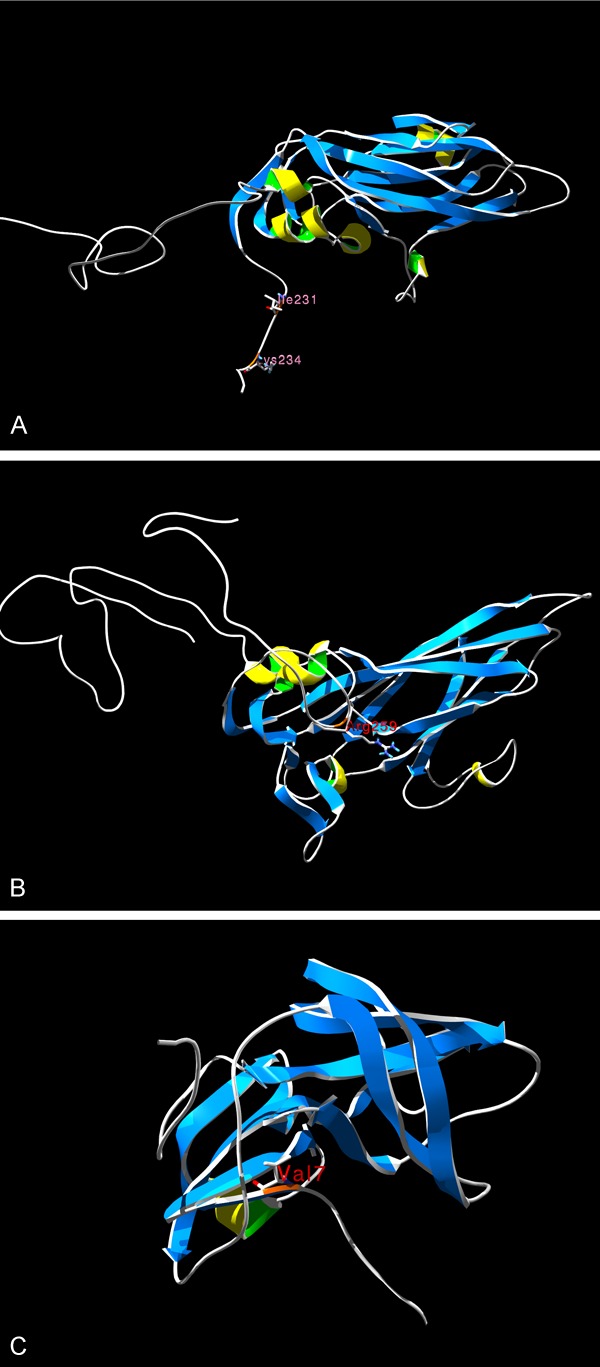
The proteins’ structure of CVB3/Macocy strain with mutation sites. The orange marks the mutation sites of strain Macocy comparing its parental strain Nancy. Picture A. Shows the VP3 protein structure of CVB3/Macocy strain, and two mutation sites (Ile231, Lys234) were marked. Picture B. Shows the VP1 protein structure of CVB3/Macocy strain, and one mutation site (Arg259) was marked. Picture C. Shows the 2A protein structure of CVB3/Macocy, strain and one mutation site (Val7) was marked.
Discussion
An acute systemic disease model by CVB on neonatal mice was established. The pathology assays, the pathological scoring, the statistical tests and the mutual control groups demonstrated the reliability of the model.
It was difficult to collect the pancreatic tissue which showed extensive necrosis, the probable mechanism being pancreas autodigestion. In the acute pancreatitis stage, the pancreatic digestive enzymes are activated in pancreatic gland, and the autodigestion happens. Pancreatitis can be used as an ideal criterion to establish whether mice are infected by CVB, because it can be caused by any CVB strain in mice [9]. Combined with statistical tests, our pathological assay showed that the inoculated mice suffered from pancreatitis, hepatitis, pneumonitis and mild myocardial injury simultaneously at 72 hours, which is the onset of acute infection. Indeed, the validity of infection rates of the four tissues must be statistically tested, even though pathological judgment could exclude false–positive results.
The fatality rate of the infected neonatal mice (31.82 - 38.64%) is similar to the neonatal humans (11.1% - 40.0%). Pneumonitis, hepatitis and myocardial injury were observed, and the pancreas was badly damaged. This is very similar to the situation in humans. The infection in newborn mice are more severe that in newborn humans. Considering good medical care in newborn humans, it was reasonable that newborn mice had more severe acute disease than newborn humans. Also, the separation of newborn mice from their mother would create stress, which would affect the immune system. This may be another factor that contributed more severe infection in neonatal mice. However, the circumstance without medical care, under which mice stayed, helped revealing more details of the disease. Concerning the 2 deaths in the control group, it was a natural fatality in neonatal mice. The neonatal mice were raised separated with their mothers, which may increase the fatality rate. The human and mouse model systems differ due to the medical care and environment in which the human neonates are treated. Natural death rate was 4.5%, which was statistically low as compared to the infected groups.
There is no doubt that CVB can induce myocarditis considering that there were enormous clinical and laboratory report on the relationship between CVB and myocarditis [35-37]. However, several epidemiological studies, which are about acute enterovirus-induced clinical features on neonates, seldom refer to myocarditis [33,38]. Meanwhile, there are 1.5% of all enteroviral infections presenting cardiovascular symptoms. Although the cardiovascular symptoms are mostly induced by CVB, the incidence for CVB infection is only 3.5% [39,40]. In other words, these reports are in conflict with the thought that CVB has an ability of infecting neonates with high fatality rate. In our research, the neonatal mouse model indeed showed that the myocardium was infected by CVB strains but the pathological changes were mild (Figure 4B, 4C, 4E and 4F), while lung, liver and other organs were severely infected and injured. It is noticeable that this myocardial injury was a kind of mild myocarditis, although it was not evident generally with large sample size.
For comparing the severity of this systemic disease, the infection rates, pathological lesion rates and scores of each organ were considered. The pathological diversity of this systemic disease revealed that the severity of pneumonia, hepatitis and myocardial injury was different at the acute onset stage, when hepatitis and pneumonia were more severe diseases than myocardial injury. It can be presumed that the large fatality rates of the experimental mice (31.82% in Nancy group and 38.64% in Macocy group) mainly came from suffering from severe pneumonia and hepatitis, or the combination of muti-organs diseases.
The report of CVB-induced hepatitis can be traced back to the era of coxsackievirus just found. After that, it has paid enough attention to CVB-induced encephalitis and myocarditis, but not to CVB-induced hepatitis. Clinically, about half of the infants with neonatal enterovirus infection have the signs of hepatitis with jaundice [41]. And some of the patients with hepatitis will progress to fulminant hepatitis with the symptoms marked by serum aminotransferase activity abnormal, jaundice and coagulopathy in the acute CVB infection [42,43]. Lin TY et al reported that 42 (30%) neonates among the 146 cases had developed the hepatitis with coagulopathy and 10 (24%) of the 42 patients had fatal outcomes [7]. This result is in accord with another enterovirus-induced disease study which reported that there was a mortality rate of 31% on neonates’ fulminant hepatitis with coagulopathy [13]. Our mouse model also showed the severe acute viral hepatitis with massive virus-induced hepatocyte apoptosis (Figure 3B-D, 3F-H and 3J-L).
In both the clinical survey and our results, acute CVB infection on neonate could induce severe pneumonia, hepatitis. Our model showed the rates for hepatitis were higher than that for the myocardial injury with very high significance. This result is in accord with the epidemiological surveys and clinical reports that infants with CVB infection could suffer from high possibility on hepatitis but not myocarditis [7,33,38-41,44]. Indeed, severe hepatitis in neonates is an important lethal factor [7,42,45]. At the same time, the pathological test with statistical analysis shows the rates for pneumonia are higher than that for myocardial injury with high significance too. In fact, there is a report about CVB inducing neonatal death because of pneumonia [8]; while there is another 401 cases viral pneumonia diagnosis including mycoplasma reveals that 9.2% pathogenic organism responsible for it is CVB [46]. Actually, even though the pneumonia is not the hot spot of CVB infection research, there is a report that the CVB can induce many respiratory symptoms [14,15], which could be fatal for neonates.
Based on the surveys of others and our mouse model study, it can be indicated that CVB-induced hepatitis and pneumonia play a far more important role than myocarditis on high infancy fatality rate, although myocarditis could be concurred with hepatitis.
Clinical reports have revealed that CVB infection can cause respiratory symptoms [14,15]. We found severe pneumonitis in our combined disease model. Coxsackievirus A21 can be transmitted effectively from person to person by aerosols [47]. Due to CVB virions are very stable for at least 24 hours under conditions similar to a household environment [48], CVB may infect host cells if it is active. It is tempting to speculate that CVB can be transmitted by air in addition to the classical fecal–oral route and by vertical transmission from mother to infant.
The unique sites in the 5’ UTR (408T and 581G) and polyprotein (566K, 829R, 861V and 1486Y) of strain Macocy did not change the virulence of CVB3 in the early phase of infection. By sequence identity comparison, we found that strain 0 and 28 are the most two similar strains with Macocy with sequence identity of 99.69% and 99.73% respectively. Thus, we chose these two strains and Nancy strains to analyze the genome difference. Based on the pathological analysis of the early phase of acute neonatal infection, it was evident that the virulence of the strain Macocy was similar to that of strain Nancy, so the six unique sites did not change the virulence. We aligned the 5’ UTR and polyprotein of strains Macocy, Nancy, 28 and 0, in which strains Nancy and 28 are cardiovirulent, and strain 0 is not cardiovirulent [49]. It was remarkable that the 234C site in avirulent strain 0, which is a crucial site relating to non-cardiovirulence [49], was different from the site in strain Macocy, Nancy and 28 (all of the tree strains were 234U). That is to say, 234C site determined the avirulence of CVB3, while 234U site related to the cardiovirulence. In the secondary structure of 5’ UTR, the two mutation sites 408G and 581T are not present in the paired region [50]. Mutation in VP1 is near to the C terminus of the protein and there is no secondary structure. Mutations in VP3 also occurred at the C terminus of this protein and there is no secondary structure either. Therefore, mutations in this area should not damage the whole structure of the protein and affect its function. In 2A protease, the mutation site Val7 was present in β-strands. However, this change does not alter the hydrophobic nature of this site. Furthermore, some CVB4 virulent strains also have Val at this site. Thus, in CVB3, Val7 mutation in 2A protease does not affect its function [51].
Our scoring system was based on the classical pathological changes in tissue injury. The pathological score can be used to quantify the pathological changes, by which we confirmed the CVB-induced injury in liver, lung and heart tissues. The pathological scoring is a standard criterion to qualify the degrees of tissues lesions and makes the analysis more accurate and objective. As the scores are non-parametric data, Kruskal-Wallis test was chosen.
The statistical tests showed that there were no significant differences between strains Macocy and Nancy when comparing the fatality rates, immunohistochemical staining and pathological lesions. Meanwhile, the CVB3-induced symptoms in both groups of mice were identical. Thus, the CVB-induced pathological changes were confirmed by the mutual control groups, since the benefit of two identical mutual control groups was for well demonstrating the reliability of the model.
The statistical power is a parameter for calculating the significant sample size for every statistical test. Larger power value reveals a greater chance of finding a significant difference, whereas very small power value reveals little chance of finding a significant difference. The power value is positively relative to the sample size for a hypothesis test. Hence, we can get the smallest sample size by which a significant experimental result can be obtained.
Acknowledgements
This study was supported by the grant (2009ZX09301-014-1) awarded by National Mega Project on Major Drug Development (People’s Republic of China).
Disclosure of Conflict of interests
The authors declare that they have no competing interests.
References
- 1.Kaplan MH, Klein SW, McPhee J, Harper RG. Group B coxsackievirus infections in infants younger than three months of age: a serious childhood illness. Rev Infect Dis. 1983;5:1019–1032. doi: 10.1093/clinids/5.6.1019. [DOI] [PubMed] [Google Scholar]
- 2.Khetsuriani N, Lamonte A, Oberste MS, Pallansch M. Neonatal enterovirus infections reported to the national enterovirus surveillance system in the United States 1983-2003. Pediatr Infect Dis J. 2006;25:889–893. doi: 10.1097/01.inf.0000237798.07462.32. [DOI] [PubMed] [Google Scholar]
- 3.Khetsuriani N, Lamonte-Fowlkes A, Oberst S, Pallansch MA. Centers for Disease Control and Prevention: Enterovirus surveillance--United States, 1970-2005. MMWR Surveill Summ. 2006;55:1–20. [PubMed] [Google Scholar]
- 4.Kibrick S. Viral infections of the fetus and newborn. In: Pollard M, editor. Perspectives in virology II. Minneapolis: Burgess Publishing Co; 1961. pp. 140–157. [Google Scholar]
- 5.Chapman NM, Kim KS. Persistent coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol. 2008;323:275–292. doi: 10.1007/978-3-540-75546-3_13. [DOI] [PubMed] [Google Scholar]
- 6.Feuer R, Ruller CM, An N, Tabor-Godwin JM, Rhoades RE, Maciejewski S, Pagarigan RR, Cornell CT, Crocker SJ, Kiosses WB, Pham-Mitchell N, Campbell IL, Whitton JL. Viral persistence and chronic immunopathology in the adult central nervous system following Coxsackievirus infection during the neonatal period. J Virol. 2009;83:9356–9369. doi: 10.1128/JVI.02382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin TY, Kao HT, Hsieh SH, Huang YC, Chiu CH, Chou YH, Yang PH, Lin RI, Tsao KC, Hsu KH, Chang LY. Neonatal enterovirus infections: emphasis on risk factors of severe and fatal infections. Pediatr Infect Dis J. 2003;22:889–894. doi: 10.1097/01.inf.0000091294.63706.f3. [DOI] [PubMed] [Google Scholar]
- 8.Flewett TH. Histological study of two cases of Coxsackie B virus pneumonia in children. J Clin Pathol. 1965;18:743–746. doi: 10.1136/jcp.18.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tracy S, Gauntt C. Group B Coxsackievirus Virulence. Curr Top Microbiol Immunol. 2008;323:49–63. doi: 10.1007/978-3-540-75546-3_3. [DOI] [PubMed] [Google Scholar]
- 10.Bopegamage S, Kovacova J, Vargova A, Motusova J, Petrovicova A, Benkovicova M, Gomolcak P, Bakkers J, van Kuppeveld F, Melchers WJ, Galama JM. Coxsackie B virus infection of mice: inoculation by the oral route protects the pancreas from damage, but not from infection. J Gen Virol. 2005;86:3271–3280. doi: 10.1099/vir.0.81249-0. [DOI] [PubMed] [Google Scholar]
- 11.Modlin JF. Perinatal echovirus and group B coxsackievirus infections. Clin Perinatol. 1988;15:233–246. [PubMed] [Google Scholar]
- 12.Romero JR. Pediatric Group B Coxsackievirus Infections. In: Tracy S, Oberste MS, Drescher KM, editors. Group B Coxsackieviruses. Heidelberg: Springer-Verlag; 2008. pp. 223–240. [Google Scholar]
- 13.Abzug MJ. Prognosis for neonates with enterovirus hepatitis and coagulopathy. Pediatr Infect Dis J. 2001;20:758–763. doi: 10.1097/00006454-200108000-00008. [DOI] [PubMed] [Google Scholar]
- 14.Eckert HL, Portnoy B, Salvatore MA, Ressler R. Group B Coxsackie virus infection in infants with acute lower respiratory disease. Pediatrics. 1967;39:526–531. [PubMed] [Google Scholar]
- 15.Smyth RL, Openshaw PJ. Bronchiolitis. Lancet. 2006;368:312–322. doi: 10.1016/S0140-6736(06)69077-6. [DOI] [PubMed] [Google Scholar]
- 16.Yun SH, Lee WG, Kim YC, Ju ES, Lim BK, Choi JO, Kim DK, Jeon ES. Antiviral activity of coxsackievirus B3 3C protease inhibitor in experimental murine myocarditis. J Infect Dis. 2012;205:491–497. doi: 10.1093/infdis/jir745. [DOI] [PubMed] [Google Scholar]
- 17.Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minkowitz S, Berkovich S. Hepatitis produced by coxsackievirus B1 in adult mice. Arch Pathol. 1970;89:427–433. [PubMed] [Google Scholar]
- 19.Cheng LL, Ng PC, Chan PK, Wong HL, Cheng FW, Tang JW. Probable Intrafamilial Transmission of Coxsackievirus B3 With Vertical Transmission, Severe Early-Onset Neonatal Hepatitis, and Prolonged Viral RNA Shedding. Pediatrics. 2006;118:e929–933. doi: 10.1542/peds.2006-0554. [DOI] [PubMed] [Google Scholar]
- 20.Porres ER, Werthammer J, Moss N, Bernstein JM, Belshe RB. Fatal coxsackievirus B4 infection in a neonate. South Med J. 1985;78:1254–1256. doi: 10.1097/00007611-198510000-00028. [DOI] [PubMed] [Google Scholar]
- 21.Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. 2007;41:118–122. doi: 10.1016/j.ymeth.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vargová A, Bopegamage S, Borsanyiová M, Petrovicová A, Benkovicová M. Coxsackievirus infection of mice. II. Viral kinetics and histopathological changes in mice experimentally infected with coxsackievirus B3 by intraperitoneal route. Acta Virol. 2003;47:253–257. [PubMed] [Google Scholar]
- 23.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. J Virol Methods. 1938;27:493–497. [Google Scholar]
- 24.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 25.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 26.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guex N, Peitsc MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 28.Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosner B. In: Fundamentals of Biostatistics. 5th ed. Rosner B, editor. Pacific Grove: Duxbury; 2000. p. 555. [Google Scholar]
- 30.Diegert C, Diegert KV. Note on Inversion of Casagrande-Pike-Smith Approximate Sample-Size Formula for Fisher-Irwin Test on 2×2 Tables. Biometrics. 1981;37:595. [Google Scholar]
- 31.Fleiss JL, Tytun A, Ury HK. A Simple Approximation for Calculating Sample Sizes for Comparing Independent Proportions. Biometrics. 1980;36:343–346. [PubMed] [Google Scholar]
- 32.Walters DE. In defense of the arc sine approximation. Journal of the Royal Statistical Society. 1979;28:219–222. [Google Scholar]
- 33.Abzug MJ, Levin MJ, Rotbart HA. Profile of enterovirus disease in the first two weeks of life. Pediatr Infect Dis J. 1993;12:820–824. doi: 10.1097/00006454-199310000-00005. [DOI] [PubMed] [Google Scholar]
- 34.Lake AM, Lauer BA, Clark JC, Wesenberg RL, McIntosh K. Enterovirus infections in neonates. J Pediatr. 1976;89:787–791. doi: 10.1016/s0022-3476(76)80808-6. [DOI] [PubMed] [Google Scholar]
- 35.Kim KS, Hufnagel G, Chapman NM, Tracy S. The group B coxsackieviruses and myocarditis. Rev Med Virol. 2001;11:355–368. doi: 10.1002/rmv.326. [DOI] [PubMed] [Google Scholar]
- 36.Abelmann WH. Virus and the heart. Circulation. 1971;44:950–956. doi: 10.1161/01.cir.44.5.950. [DOI] [PubMed] [Google Scholar]
- 37.Seko Y, Shinkai Y, Kawasaki A, Yagita H, Okumura K, Yazaki Y. Evidence of perforin-mediated cardiac myocyte injury in acute murine myocarditis caused by Coxsackie virus B3. J Pathol. 1993;170:53–58. doi: 10.1002/path.1711700109. [DOI] [PubMed] [Google Scholar]
- 38.Verboon-Maciolek MA, Krediet TG, van Loon AM, Kaan J, Galama JM, Gerards LJ, Fleer A. Epidemiological survey of neonatal non-polio enterovirus infection in the Netherlands. J Med Virol. 2002;66:241–245. doi: 10.1002/jmv.2136. [DOI] [PubMed] [Google Scholar]
- 39.Grist NR, Bell EJ, Assaad F. Enteroviruses in human disease. Prog Med Virol. 1978;24:114–157. [PubMed] [Google Scholar]
- 40.Whitton JL. Immunopathology during coxsackievirus infection. Springer Semin Immunopathol. 2002;24:201–213. doi: 10.1007/s00281-002-0100-4. [DOI] [PubMed] [Google Scholar]
- 41.Tebruegge M, Curtis N. Enterovirus infections in neonates. Semin Fetal Neonatal Med. 2009;14:222–227. doi: 10.1016/j.siny.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 42.Wang SM, Liu CC, Yang YJ, Yang HB, Lin CH, Wang JR. Fatal coxsackievirus B infection in early infancy characterized by fulminant hepatitis. J Infect. 1998;37:270–273. doi: 10.1016/s0163-4453(98)92076-x. [DOI] [PubMed] [Google Scholar]
- 43.Clavell M, Barkemeyer B, Martinez B, Craver R, Correa H, Gohd R, Schmidt-Sommerfeld E. Severe hepatitis in a newborn with coxsackievirus B5 infection. Clin Pediatr (Phila) 1999;38:739–741. doi: 10.1177/000992289903801208. [DOI] [PubMed] [Google Scholar]
- 44.Toshima H, Ohkita Y, Shingu M. Clinical features of acute coxsackie B viral myocarditis. Jpn Circ J. 1979;43:441–444. doi: 10.1253/jcj.43.441. [DOI] [PubMed] [Google Scholar]
- 45.Read RB, Ede RJ, Morgan-Capner P, Moscoso G, Portmann B, Williams R. Myocarditis and fulminant hepatic failure from coxsackievirus B infection. Postgrad Med J. 1985;61:749–752. doi: 10.1136/pgmj.61.718.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann H, Heinz F, Stary A. Aetiological studies on viral pneumonia (author’s transl) Wien Klin Wochenschr. 1976;88:412–415. [PubMed] [Google Scholar]
- 47.Couch RB, Douglas RG Jr, Lindgren KM, Gerone PJ, Knight V. Airborne transmission of respiratory infection with coxsackievirus A type 21. Am J Epidemiol. 1970;91:78–86. doi: 10.1093/oxfordjournals.aje.a121115. [DOI] [PubMed] [Google Scholar]
- 48.McGeady ML, Siak JS, Crowell RL. Survival of coxsackievirus B3 under diverse environmental conditions. Appl Environ Microbiol. 1979;37:972–977. doi: 10.1128/aem.37.5.972-977.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu Z, Chapman NM, Hufnagel G, Tracy S, Romero JR, Barry WH, Zhao L, Currey K, Shapiro B. The cardiovirulent phenotype of coxsackievirus B3 is determined at a single site in the genomic 5’ nontranslated region. J Virol. 1995;69:4607–4618. doi: 10.1128/jvi.69.8.4607-4618.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey JM, Tapprich WE. Structure of the 5’ nontranslated region of the coxsackievirus b3 genome: Chemical modification and comparative sequence analysis. J Virol. 2007;81:650–668. doi: 10.1128/JVI.01327-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baxter NJ, Roetzer A, Liebig HD, Sedelnikova SE, Hounslow AM, Skern T, Waltho JP. Structure and dynamics of coxsackievirus B4 2A proteinase, an enyzme involved in the etiology of heart disease. J Virol. 2006;80:1451–1462. doi: 10.1128/JVI.80.3.1451-1462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]