

Abstract
Trophoblasts play a crucial role in embryo implantation and maintenance of normal pregnancy. Recently, oxidative stress has been considered as one important factor in the pathogenesis of spontaneous abortion and preeclampsia. Many studies have reported that the plasma levels of hydrogen peroxide (H2O2) are significantly increased in women with preeclampsia, but the mechanisms involved in H2O2-induced cell cytotoxicity in trophoblasts are still not completely explained. Our present study was undertaken to provide a united understanding of the role of oxidative stress generated by H2O2 on human trophoblasts and the underlying intracellular signaling pathways. Exposure to H2O2 resulted in a concentration-dependent growth decrease and apoptosis in human trophoblast-like JEG-3 cells. H2O2 treatment also caused intracellular reactive oxygen species (ROS) production and concomitant dissipation of the mitochondrial membrane potential. The three MAPK subfamilies, ERK1/2, JNK and p38 kinase, were all activated under H2O2-induced oxidative stress. Blocking the activation of JNK and p38 kinase increased cell viability and decreased apoptosis induced by H2O2 with their respective inhibitors, SP600125 and SB203580. However, preventing ERK1/2 activation further increased H2O2-induced cell death with U0126, an inhibitor of ERK upstream kinase MEK1/2. Taken together, these findings suggest that the mitochondria-dependent pathways and JNK-p38 kinase pathways are involved in H2O2-induced oxidative damage of human trophoblast-like JEG-3 cells, while ERK1/2 pathway may play an active role in cell survival following oxidant injury.
Keywords: MAPKs, H2O2, apoptosis, ROS, mitochondrial membrane potential, human trophoblasts
Introduction
Trophoblasts are specialized cells of the placenta that exert a crucial role in embryo implantation and maintenance of normal pregnancy. During the early stage of pregnancy, the human cytotrophoblasts proliferate and differentiate into two distinct lineages: the multinucleate syncytiotrophoblast and the invasive extravillous trophoblast. Syncytiotrophoblasts cover the entire surface of the placenta to facilitate fetal-maternal exchanges and secrete numerous hormones, such as human chorionic gonadotrophin, into the maternal circulation, which are required for maintenance and immunological adaptation of pregnancy [1]. Extra-villous trophoblasts grow out from the placenta and penetrate into the decidualized maternal uterus. This process is essential not only for physically attaching the placenta to the mother, but also for remodeling the maternal spiral arteries to allow it to provide an adequate blood supply to the growing fetus as pregnancy progresses [2]. Therefore, factors that impair trophoblast function may result in a range of adverse pregnancy outcomes such as spontaneous abortion, preeclampsia, intrauterine growth restriction and even stillbirth [3].
Oxidative stress, an imbalance between oxidants and antioxidants in favor of oxidants, has been implicated in suboptimal reproductive performance from the earliest stages of development to labor and delivery [4]. The placenta generates reactive oxygen species (ROS) which may contribute to the oxidative stress seen even in normal pregnancy but this is increased in pregnancies complicated by preeclampsia, IUGR and miscarriages [5]. Hydrogen peroxide (H2O2), a stable member of ROS family, is a key terminal metabolite of the cellular oxidative stress cascade that plays an important role in oxidative stress-mediated diseases. H2O2 can diffuse freely through cell membrane, and is also revealed as a component of oxidative ischemia/ reperfusion stress in placenta. It has been reported that the plasma H2O2 levels are significantly higher in women with preeclampsia than those of normal pregnant women [6]. And growing evidence demonstrates that there a correlation between H2O2 and some potential biomarkers of preeclampsia, such as nitric oxide (NO) and soluble TNF-α receptor 2 (sTNF-R2) early in maternal circulation and at term in placenta [7,8], suggesting a direct effect of oxidative stress on placental function. This hypothesis was confirmed by recent in vitro study showing that H2O2 modulates directly the function of placenta. Zhou et al [9] have illustrated that high levels of H2O2 can down-regulate HLA-G expression in trophoblasts during preeclampsia and trophoblasts expressing HLA-G are vulnerable to oxidative stress. Murata et al [10] have proved that H2O2 can induce apoptosis in primary cultured trophoblasts and significantly inhibit the invasion ability, tube-like formation of TCL1 (a human immortalized EVT cell line). In many studies, H2O2 has been used to induce oxidative stress of human trophoblasts [11,12]. But the mechanisms involved in H2O2-induced cell cytotoxicity in trophoblasts are still not completely explained.
Mitogen activated protein kinases (MAPKs) are well-known and evolutionary conserved mediators in signal transduction pathways, which control embryogenesis, gene expression and cell functions [13]. The three well-characterized subfamilies of MAPKs, extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 kinase, are major protein kinases activated by ROS [14]. Shin et al [15] observed increased activation of ERK1/2 and p38 kinase in pre-eclamptic human placentas. Xiong et al [16] found that the phosphorylation of p38 and JNK increased in human placental explants when exposed to various preeclampsia-associated stresses including angiotensin II, hypoxia and inflammatory cytokines. In human choriocarcinoma JAR cells, hydrogen peroxide has been shown to activate JNK, but not p38 and ERK1/2 [12]. Thus it is proposed that MAPKs play a crucial role in the cellular events of human trophoblasts under oxidative stress.
The aim of this study is to provide a united understanding of the role of oxidative stress generated by H2O2 on human trophoblasts and the underlying intracellular signaling pathways. Because of ethical reasons, experimental studies of xenobiotics in placenta or fetus can rarely performed in vivo. So in our experiments, we have used a human choriocarcinoma cell line, JEG-3, which has a number of properties characteristic of normal placental trophoblast as an in vitro model system.
Materials and methods
Cell culture
The choriocarcinoma JEG-3 cell, one of the human trophoblast-like cell lines, was obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China) with the original source being the American Type Culture Collection (ATCC). Cells were cultured in DMEM/F12 complete medium supplemented with 10% FBS and maintained in 5% CO2 at 37°C. Cells were detached by routine trypsinization every 3 to 4 days.
Cell viability assay
JEG-3 cells were seeded at a density of approximately 2×104 cells/well in 96-well flat-bottom microplates (Costar, USA). After 12 h, the medium was replaced by phenol red-free DMEM/F12 containing 10% FBS. Meanwhile, H2O2 (0, 100, 250, 500, 750, 1000 μM) or H2O2 (500 μM) with or without U0126 (20 μM), SB203580 (20 μM), SP600125 (10 μM) was added to the corresponding wells, and cultured the cells for another 24 h. Then MTT (Sigma Aldrich St. Louis, MO, USA) was added to each well in 20 ul (5 mg/ml in PBS) and incubated at 37°C for 4 h until purple formazan crystal developed. Subsequently, the MTT-containing medium was removed, 150 ul of DMSO was added and incubated at room temperature for 30 min. The formazan absorbance was measured at a wavelength of 490 nm on an automatic microplate reader (Bio-Rad 3550). The OD values of the treated cells were compared with the values generated from the untreated control cells and reported as the percentage viability of control.
Cell apoptosis assay
JEG-3 cells were cultured in 6-well plates (Costar, USA) and exposed to H2O2 and/or different MAPK inhibitors for 24 h. At the end of exposure, both floating and attached cells were collected by brief trypsinization and washed with PBS twice, then subjected to an Alexa Fluor® 488 annexin V/Dead Cell Apoptosis Kit (Molecular Probes, Inc., UK) following the step-by-step protocol provided by the manufacturer. Samples were incubated at room temperature for 15 min in the dark with Annexin V and PI and analyzed by a BD FACSCalibur flow cytometer for the quantification of apoptotic cells. The apoptosis ratio was calculated as the apoptosis percentage of the treated cells to that of the untreated control.
Cell morphological observation
JEG-3 cells were seeded and cultured in 6-well plates (Costar, USA) until the cell monolayer reaching approximately 50% confluency. Then different concentrations of H2O2 was added and cultured for 24 h. After being washed three times with PBS, cell morphology was observed under phase-contrast microscopy. For fluorescent staining, the cells were then fixed with 4% paraformaldehyde, washed with PBS, and incubated with 4-6-diamidino-2-phenylindole (DAPI) staining solution (Beyotime Company, Hangzhou, China) for 5 min in the dark at room temperature. Fluorescence images were observed by using an Olympus BX51 fluorescence microscope (Tokyo, Japan), and recorded with a high-resolution DP70 Olympus digital camera.
ROS measurement reactive oxygen species
The oxidative fluorescent dye dihydroethidium (DHE, Molecular Probes, Inc., UK) was used to evaluate ROS production in JEG-3 cells after H2O2 treatment. DHE, by virtue of its ability to freely permeate cell membranes, is used extensively to monitor superoxide production. It reacts with superoxide anions and forms a red fluorescent product (2-hydroxyethidium) which binds to DNA in the nucleus [17]. Briefly, after different treatments with H2O2 for 4 h, JEG-3 cells were washed with pre-warmed PBS and incubated with DHE (10 μM) in phenol red-free DMEM/F12 for 30 min at 37°C in the dark. Then cells were washed with pre-warmed phenol red-free medium, images were obtained with a fluorescent microscope, and the signal was quantified using Image ProPlus Software (Olympus, USA). The ROS production was calculated as the fluorescence signal intensities of treated groups to the control.
Mitochondrial membrane potential (MMP) analysis
Alterations in MMP were analyzed by flow cytometry using the mitochondrial membrane potential assay kit with JC-1, which is a marker of mitochondrial activity (Beyotime Company, Hangzhou, China). In normal undamaged nucleate cells, mitochondrion has a high MMP. Breakdown of MMP is often linked to early apoptosis. JC-1 is most widely applied for detecting mitochondrial depolarization occurring in the early stages of apoptosis [18]. JC-1 exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green to red. Cells containing J-aggregates have high MMP, and show red fluorescence (590 nm, FL-2 channel). Cells with low MMP are those in which JC-1 maintains monomeric form, and show green fluorescence (530 nm, FL-1 channel). Briefly, after different treatments with H2O2 for 24 h, JEG-3 cells were collected and incubated with 0.5 ml JC-1 working solution for 20 min at 37°C, then washed, resuspended in medium, and analyzed by flow cytometry. CCCP (carbonylcyanide-p-chlorophenol hydrazone) was used as positive control.Data were revealed as the monomers positive percentage of the treated cells to that of the untreated control.
Western blotting analysis of ERK1/2, JNK and p38 kinase
Following H2O2 (500 μM) treatments for different time points, JEG-3 cell lysates were prepared on ice in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 10 mM NaF, 0.25% sodium deoxycholate, 1 mM EDTA, 1 mM PMSF and phosphatase inhibitors; Roche, USA). The supernatants were obtained by centrifugation at 15 000 ×g for 20 min at 4°C, and detected for protein concentration using the Bradford protein assay kit (Beyotime Company, Hangzhou, China). Protein samples (50 ug) were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes. After blocking, the membrane was probed with specific primary monoclonal rabbit anti-phospho-ERK1/2 (Thr202/Tyr204), anti-phospho-SAPK/JNK (Thr183/Tyr185), anti-phospho-p38 MAP Kinase (Thr180/Tyr182), anti-ERK, anti-SAPK/JNK, polyclonal rabbit anti-p38 MAP Kinase (1:1000; Cell Signaling Technology), and monoclonal mouse anti-GAPDH (1:1000; Santa Cruz, CA, USA) antibodies overnight at 4°C, then followed by incubation with HRP-conjugated secondary antibodies. After extensive washing, proteins of interest were detected by enhanced chemiluminescence system (ECL, Thermo Scientific, UK) and quantified by densitometry using Quantity One (Bio-Rad, USA).
Statistical analysis
Experiments were performed three times independently. Results are expressed as mean ± SEM. Statistical comparisons were performed by one-way analysis of variance (ANOVA) followed by a Dunnett test. Differences were considered as statistically significant at P<0.05.
Results
H2O2 induces cytotoxicity in JEG-3 cells
The growth inhibition of JEG-3 cells by H2O2 was first assessed by MTT assay following treatment of different concentrations of H2O2 for 24 h. As shown in Figure 1A, H2O2 significantly inhibited the viability of JEG-3 cells at the concentrations of 0.5, 0.75 and 1.0 μM for 24 h, respectively (P<0.05). The inhibitory effect of H2O2 on JEG-3 cells was concentration-dependent. At the concentration of 0.5 μM, H2O2 significantly decreased the viability to about 70% after exposing for 24 h, but for the concentration of 1.0 μM, the viability was just approximately 10% of the untreated control.
Figure 1.
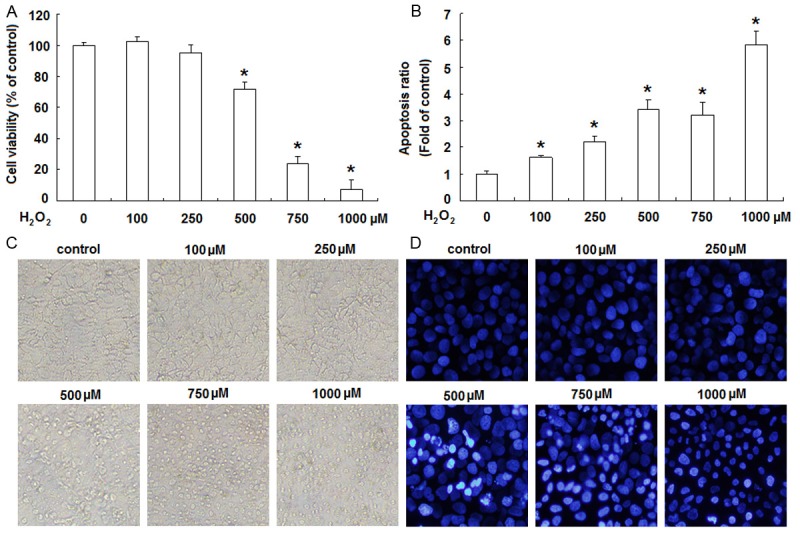
H2O2 induces cytotoxicity in JEG-3 cells. JEG-3 cells were treated with H2O2 at indicated concentrations for 24 h. Subsequently, MTT assay (A) and Annexin V and PI staining (B) were applied to analyze the viability and apoptosis of JEG-3 cells, respectively. Data are presented as the percentage or the fold of untreated control. Error bars represent the standard error of the mean. *P<0.05, versus control. Morphological changes of JEG-3 cells treated with H2O2 for 24 h were visualized by inverted microscope (original magnification ×100) (C) or by fluorescence microscope after DAPI staining (original magnification ×200) (D). Data presented are representative of three individual experiments.
In order to quantitatively evaluate the pro-apoptotic effects of H2O2 on JEG-3 cells, annexin V and PI double staining was performed followed by flow cytometric analysis. As shown in Figure 1B, H2O2 induced a moderate to strong apoptotic death in a concentration-dependent manner. Following the treatment with H2O2 at the concentrations of 0.5 μM for 24 h, the apoptosis ratio increased approximately 3.52 fold compared to untreated control.
The cytotoxicity of H2O2 on JEG-3 cells was also confirmed by the morphological study. When the concentration of H2O2 was less than 250 μM, there were no obvious morphological changes in JEG-3 cells. From the concentration of 500 μM, H2O2 induced pronounced cell damage as displayed by cell rounded-up, shrinkage and gradual detachment from culture dishes under phase-contrast microscopy (Figure 1C). And we further visualized the nuclear morphology by DAPI staining under fluorescence microscope. The nuclei of H2O2-untreated JEG-3 cells were stained uniformly, but the H2O2-treated cells exhibited chromatin condensation and nuclear fragmentation in a concentration-dependent manner (Figure 1D).
H2O2 causes intracellular ROS production and MMP loss in JEG-3 cells
ROS play an important role in apoptosis induction under both physiologic and pathologic conditions [19]. So we next investigated the intracellularROS generation in JEG-3 cells after H2O2 treatment using the fluorescence dye DHE. JEG-3 cells were exposed to H2O2 for 4 h, and then incubated with DHE (10 μM) in phenol red-free DMEM/F12 for 30 min in the dark. The fluorescence images depicted a gradually rise in ROS level of JEG-3 cells (Figure 2A). Quantification of the fluorescence intensity showed that ROS generation was increased in a concentration-dependent manner after H2O2 treatment (Figure 2B).
Figure 2.

H2O2 causes intracellular ROS production and MMP loss in JEG-3 cells. JEG-3 cells were treated with H2O2 at indicated concentrations for 4 h and stained with DHE for 30 min to measure ROS production by fluorescence microscope. (A) Representative fluorescence images (original magnification ×200), (B) quantitative presentation of ROS production by the fluorescence signal intensities of treated cells to the untreated control. After treatment with H2O2 for 24 h, JEG-3 cells were stained with JC-1 to evaluate the mitochondrial membrane potential (MMP). (C) Representative flow cytometry plots, (D) quantitative presentation of JC-1 monomers positive ratio by the percentage of the treated cells to the untreated control. Error bars represent the standard error of the mean from three individual experiments. *P<0.05, versus control.
Breakdown of MMP is the marker of mitochondria dysfunction and has been shown to associate with cell apoptosis [20]. To determine whether H2O2-induced apoptosis in JEG-3 cells involves mitochondrial disruption, we measured the fluorescence emission shift (red to green) by a JC-1 dye kit to examine the depolarization of mitochondrial membrane. JEG-3 cells were treated with different concentrations of H2O2 for 24 h, thereafter stained with JC-1 dye and analyzed by flow cytometry. From the concentration of 0.5 μM, H2O2 treatment induced a noticeable increase in green fluorescence intensity (Figure 2C). Quantification of JC-1 monomer positive cells (lower rectangle) demonstrated a significant concentration-dependent increase following H2O2 treatment compared to the untreated control (Figure 2D).
H2O2 stimulates the activation of ERK1/2, p38MAPK and JNK in JEG-3 cells
To study the signaling mechanisms, we examined the involvement of MAPKs in the effects of H2O2 on JEG-3 cells. Treatment with H2O2 (500 μM) of JEG-3 cells resulted in a rapid increase of Thr202/Tyr204 phosphorylation of ERK1/2. This phenomenon was time dependent, being maximal at 15 min and reversing to baseline after 60 min (Figure 3A and 3B). The phosphorylation of p38 (Thr180/Tyr182) and JNK (Thr183/Tyr185) was also raised in a similar time-dependent manner (Figure 3A, 3C and 3D). Total immunoreactive ERK, p38 and JNK did not alter evidently during this time frame (Figure 3A). Immunoblots with anti-GAPDH antibody confirmed equal protein loading. These data demonstrate that H2O2 treatment can activate these three MAPK pathways.
Figure 3.

H2O2 stimulates the activation of MAPKs in JEG-3 cells. JEG-3 cells were serum starved for 12 h, and then stimulated with H2O2 (500 μM) for the indicated time points. The phosphorylation of ERK, p38MAPK and JNK were evaluated by western blot analysis. GAPDH was used as a loading control. (A) Typical blots; (B-D) densitometric analysis, the phospho/total MAPK is normalized to 1 in untreated control. Data presented are the representative of three independent experiments. Error bars represent the standard error of the mean. *P<0.05, versus control.
Roles of MAPKs in H2O2-induced cytotoxicity of JEG-3 cells
To further elucidate the roles of the three MAPK pathways in the cytotoxicity of H2O2 on JEG-3 cells, we investigated the effects of U0126 (an inhibitor of ERK upstream kinase MEK1/2), SB203580 (a p38 MAPK inhibitor) and SP600125 (a JNK inhibitor) on the viability and apoptosis of JEG-3 cells with the presence of H2O2. Interestingly, the cell viability was increased with SB203580 and SP600125, but further decreased with U0126 compared to H2O2-treated group (Figure 4A). And similarly, H2O2-induced apoptosis of JEG-3 cells was alleviated by SB203580 and SP600125, but further intensified by U0126 (Figure 4B).
Figure 4.
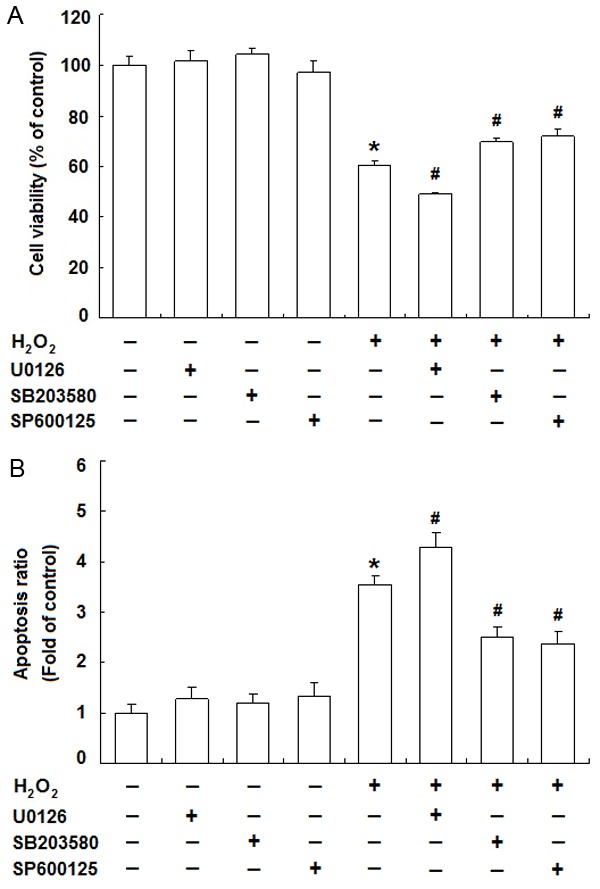
Roles of MAPKs in H2O2-induced cytotoxicity of JEG-3 cells. JEG-3 cells were treated with H2O2 (500 μM) for 24 h with or without U0126 (20 μM), SB203580 (20 μM) or SP600125 (10 μM). Thereafter, MTT assay was used to measure the cell viability (A) and Annexin V and PI staining was for apoptosis analysis by flow cytometry (B). Data are presented as the percentage or the fold of control. Error bars represent the standard error of the mean from three individual experiments. *P<0.05, versus control; #P<0.05, versus H2O2-treated group.
Discussion
Oxidative stress induced-damage has been linked to the pathophysiology of a number of disease states involving cancer, diabetes mellitus, cardiovascular disease, neurological disorders, ischemia/reperfusion injury and inflammatory diseases, and ageing [21]. It occurs when the production of free radicals exceeds the capacity of antioxidant defenses. Free radicals can be classified as reactive oxygen species or reactive nitrogen species. Hydrogen peroxide, a representative ROS member, is a metabolite generated by a variety of enzyme-catalyzed redox reactions from nearly all sources of oxidative stress. Increasing evidence has demonstrated that elevated level of H2O2 may contribute to the occurrence and development of Alzheimer’s disease thyroid diseases [22,23].
Recently, oxidative stress has been suggested to play a critical role in the pathogenesis of spontaneous abortion and preeclampsia [24]. Sugino et al [25] reported that the decrease in superoxide dismutase (SOD, an antioxidant enzyme) expression and the increase in lipid peroxide in the decidua could be involved in the termination of spontaneous abortion. Jauniaux et al [26] found that the immunoreactivity for heat shock protein 70 (a marker for cellular stress) and nitrotyrosine residues (a marker of protein oxidative damage) was greater in samples from peripheral than from central regions of normal placentas, and from missed miscarriage compared to controls.
Poranen’s investigation showed lipid peroxidation was increased and the activity of antioxidant enzymes SOD and glucose 6-phosphate-dehydrogenase (G6PD) was decreased in preeclamptic placenta [27]. Hilali’s study demonstrated that oxidative stress and DNA damage were elevated in mildly pre-eclamptic patients and their offspring, indicating that increased oxidative stress may be important in inducing DNA damage in pre-eclamptic patients [28]. And it has been reported that plasma H2O2 levels are increased in women with preeclampsia [6-9] and high levels of H2O2 can directly reduce the viability and damage the function of human extravillous trophoblast cells [10]. These results agree with our present study, in which we demonstrated that H2O2 treatment decreased the viability and triggered the apoptosis of human trophoblast-like cell line JEG-3 cells in a concentration-dependent manner. And this cytotoxicity of H2O2 on JEG-3 cells was displayed as the apoptosis characterization by cell rounded-up and shrinkage, chromatin condensation and nuclear fragmentation.
ROS can modulate various physiological cell functions, whereas excess ROS induce oxidative modification of cellular macromolecules, inhibit protein function, and ultimately result in cell death either by apoptosis or necrosis [29]. In the present study, we also analyzed the intracellular ROS production in JEG-3 cells after H2O2 treatment. The results of DHE staining indicated that H2O2 induced ROS production in a concentration-dependent manner, suggesting that H2O2-mediated cytotoxicity was correlated with the increased levels of intracellular ROS in JEG-3 cells. It is a well proven fact that mitochondria of living cells play a main role in the formation of free radicals and dissipation of the mitochondrial membrane potential (MMP) is a key event in initiation of apoptosis signaling pathways [30]. To evaluate the role of mitochondria in H2O2-induced apoptosis in JEG-3 cells, we used a MMP sensitive JC-1 dye to examine the depolarization of mitochondrial membrane. The flow cytometric analysis showed that H2O2 led to a significant disruption of MMP as evidenced by the increase in JC-1 monomer positive cells. These data indicate that the involvement of mitochondria-dependent pathways in H2O2-indued oxidative injury of JEG-3 cells, and further examination should be done to clarify the detailed signaling pathways.
Reactive oxygen species have been considered as a “second messenger” in intracellular signaling cascades that control cell growth, proliferation, migration, and apoptosis [31]. However, growing evidence implicates alterations in redox signaling as a contributor to many disease processes [32]. MAPK signaling pathways are well known to be involved in diverse physiological processes, including the morphological and functional differentiation of villous trophoblast, and have been proved to be critical for induction of oxidative stress responses [14,33]. So to elucidate the potential mechanisms leading to the oxidative stress by H2O2 of JEG-3 cells, we explored the role of MAPK signal cascades, including ERK1/2, JNK and p38 kinase.
Our results indicated that the phosphorylation of these three MAPK subfamilies was all increased under H2O2-induced oxidative stress in JEG-3 cells. To further investigate the roles of MAPK pathways in the oxidative stress induced by H2O2, we blocked ERK1/2, JNK and p38 kinase pathways with their respective inhibitors U0126, SP600125 and SB203580, and analyzed the viability and apoptosis of JEG-3 cells with the presence of H2O2. Interestingly, SB203580 and SP600125 treatment increased cell viability and decreased apoptosis, while U0126 treatment further decreased cell viability and increased apoptosis induced by H2O2. These results suggest that H2O2-induced oxidative injury may involve only p38 and JNK activation, but not ERK activation of JEG-3 cells.
Generally, JNK and p38 kinase are classified together as stress-responsive kinases, which are critical mediators of oxidative stress-induced apoptosis [14]. Wu et al [34] have shown that the activation of JNK may participate in H2O2-induced apoptosis by mediating the level of Mammalian Ste20-like protein kinase 3 (Mst3) in the 3A-sub-E human trophoblast cell line. Shen et al [35] have indicated that Aflatoxin G1, one of the most common contaminants in food, induces oxidative DNA damage and triggers apoptosis through ROS-mediated JNK and p38 kinase pathways in A549 human alveolar basal epithelial cells. Consistently with these studies, our results demonstrated that the phosphorylation of JNK and p38 kinase was all increased under H2O2-induced oxidative stress in JEG-3 cells. And the respective inhibitors SP600125 and SB203580 of JNK and p38 kinase increased cell viability and decreased apoptosis of JEG-3 cells with the presence of H2O2, suggesting that JNK and p38 kinase pathways are involved in H2O2-induced oxidative injury of JEG-3 cells.
It is well known that ERK1/2 play an important role in stimulating cell survival and cell cycle progression. Many studies have shown that ERK1/2 have proliferative and protective effects on cells exposed to oxidative stress [36,37]. Our study is also in line with their known pro-survival role, because H2O2 stimulated the activation of ERK1/2 and U0126, a specific inhibitor of ERK upstream kinase MEK1/2, further intensified the cell oxidative damage induced by H2O2 in JEG-3 cells. However, it was recently reported that ERK1/2 also have pro-apoptotic roles. H2O2-induced apoptosis has been shown to be dependent on ERK1/2 in human glioma cells and gingival fibroblasts [38,39]. These data suggest that Whether ERK1/2 promote cell survival or death is probably in large part cell type specific.
In conclusion, we have shown that the mitochondria-dependent pathways and MAPK pathways are involved in H2O2-indued oxidative injury of human trophoblast-like JEG-3 cells. We also provide evidence that JNK-p38 kinase pathways play a critical role in pro-apoptotic effect while ERK pathway in protective effect following oxidant injury. Our study may help to establish the novel policy to maintain proper oxidative balance in the placenta which is necessary for successful pregnancy.
Acknowledgements
This work was supported by National Nature Science Foundation of China (NSFC) 81300505 (to C-L Tang), Youth Foundation of Shanghai Municipal Health Bureau 2012Y032 (to C-L Tang), and Program for Creative Talents Education of Key Discipline of Fudan University (to C-L Tang); NSFC30910103909 and NSFC 31270969 (to D-J Li); the Foundations from Shanghai Science and Technology Committee 134119a4300 (to L-P Jin).
Disclosure of conflict of interest
None.
References
- 1.Bansal AS, Bora SA, Saso S, Smith JR, Johnson MR, Thum MY. Mechanism of human chorionic gonadotrophin-mediated immunomodulation in pregnancy. Expert Rev Clin Immunol. 2012;8:747–753. doi: 10.1586/eci.12.77. [DOI] [PubMed] [Google Scholar]
- 2.Harris LK. Trophoblast-vascular cell interactions in early pregnancy: how to remodel a vessel. Placenta. 2010;31:S93–S98. doi: 10.1016/j.placenta.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Lunghi L, Ferretti ME, Medici S, Biondi C, Vesce F. Control of human trophoblast function. Reprod Biol Endocrinol. 2007;5:6. doi: 10.1186/1477-7827-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poston L, Igosheva N, Mistry HD, Seed PT, Shennan AH, Rana S, Karumanchi SA, Chappell LC. Role of oxidative stress and antioxidant supplementation in pregnancy disorders. Am J Clin Nutr. 2011;94:1980S–1985S. doi: 10.3945/ajcn.110.001156. [DOI] [PubMed] [Google Scholar]
- 5.Myatt L. Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta. 2010;31:S66–S69. doi: 10.1016/j.placenta.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kharfi A, Giguère Y, De Grandpré P, Moutquin JM, Forest JC. Human chorionic gonadotropin (hCG) may be a marker of systemic oxidative stress in normotensive and preeclamptic term pregnancies. Clin Biochem. 2005;38:717–721. doi: 10.1016/j.clinbiochem.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Aris A, Benali S, Ouellet A, Moutquin JM, Leblanc S. Potential biomarkers of preeclampsia: inverse correlation between hydrogen peroxide and nitric oxide early in maternal circulation and at term in placenta of women with preeclampsia. Placenta. 2009;30:342–347. doi: 10.1016/j.placenta.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Leblanc S, Ouellet A, Giguère Y, Forest JC, Moutquin JM, Aris A. A positive correlation between hydrogen peroxide and soluble TNF-alpha receptor 2 early in maternal blood and at term in placenta of pregnant women with preeclampsia. Hypertens Pregnancy. 2012;31:357–366. doi: 10.3109/10641955.2010.525281. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Zhang GY, Wang J, Lu SL, Cao J, Sun LZ. A novel bridge between oxidative stress and immunity: the interaction between hydrogen peroxide and human leukocyte antigen G in placental trophoblasts during preeclampsia. Am J Obstet Gynecol. 2012;206:447, e7–e16. doi: 10.1016/j.ajog.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Murata M, Fukushima K, Takao T, Seki H, Takeda S, Wake N. Oxidative stress produced by xanthine oxidase induces apoptosis in human extravillous trophoblast cells. J Reprod Dev. 2013;59:7–13. doi: 10.1262/jrd.2012-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moll SJ, Jones CJ, Crocker IP, Baker PN, Heazell AE. Epidermal growth factor rescues trophoblast apoptosis induced by reactive oxygen species. Apoptosis. 2007;12:1611–1622. doi: 10.1007/s10495-007-0092-6. [DOI] [PubMed] [Google Scholar]
- 12.Boronkai A, Brubel R, Racz B, Tamas A, Kiss P, Horvath G, Lubics A, Szigeti A, Bellyei S, Toth G, Lakatos A, Reglodi D. Effects of pituitary adenylatecyclase activating polypeptide on the survival and signal transduction pathways in human choriocarcinoma cells. Ann N Y Acad Sci. 2009;1163:353–357. doi: 10.1111/j.1749-6632.2008.03630.x. [DOI] [PubMed] [Google Scholar]
- 13.Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005;118:3569–3572. doi: 10.1242/jcs.02470. [DOI] [PubMed] [Google Scholar]
- 14.Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal. 2011;15:205–218. doi: 10.1089/ars.2010.3733. [DOI] [PubMed] [Google Scholar]
- 15.Shin JK, Jeong YT, Jo HC, Kang MY, Chang IS, Baek JC, Park JK, Lee SA, Lee JH, Choi WS, Paik WY. Increased interaction between heat shock protein 27 and mitogen-activated protein kinase (p38 and extracellular signal-regulated kinase) in preeclamptic placentas. J Obstet Gynaecol Res. 2009;35:888–894. doi: 10.1111/j.1447-0756.2009.01053.x. [DOI] [PubMed] [Google Scholar]
- 16.Xiong Y, Liebermann DA, Holtzman EJ, Jeronis S, Hoffman B, Geifman-Holtzman O. Preeclampsia-associated stresses activate Gadd45a signaling and sFlt-1 in placental explants. J Cell Physiol. 2013;228:362–370. doi: 10.1002/jcp.24139. [DOI] [PubMed] [Google Scholar]
- 17.Sun Q, Yue P, Ying Z, Cardounel AJ, Brook RD, Devlin R, Hwang JS, Zweier JL, Chen LC, Rajagopalan S. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol. 2008;28:1760–1766. doi: 10.1161/ATVBAHA.108.166967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal C, Singh RP, Agarwal R. Grape seed extract induces apoptotic death of human prostate carcinoma DU145 cells via caspases activation accompanied by dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis. 2002;23:1869–1876. doi: 10.1093/carcin/23.11.1869. [DOI] [PubMed] [Google Scholar]
- 19.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 20.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (ΔΨm) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 21.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Milton NG. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: implications for treatment. Drugs Aging. 2004;21:81–100. doi: 10.2165/00002512-200421020-00002. [DOI] [PubMed] [Google Scholar]
- 23.Ohye H, Sugawara M. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp Biol Med (Maywood) 2010;23:424–433. doi: 10.1258/ebm.2009.009241. [DOI] [PubMed] [Google Scholar]
- 24.Poston L, Raijmakers MT. Trophoblast Oxidative Stress, Antioxidants and Pregnancy outcome--a review. Placenta. 2004;25:S72–S78. doi: 10.1016/j.placenta.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Sugino N, Nakata M, Kashida S, Karube A, Takiguchi S, Kato H. Decreased superoxide dismutase expression and increased concentrations of lipid peroxide and prostaglandin F(2 alpha) in the decidua of failed pregnancy. Mol Hum Reprod. 2000;6:642–647. doi: 10.1093/molehr/6.7.642. [DOI] [PubMed] [Google Scholar]
- 26.Jauniaux E, Hempstock J, Greenwold N, Burton GJ. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am J Pathol. 2003;162:115–125. doi: 10.1016/S0002-9440(10)63803-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poranen AK, Ekblad U, Uotila P, Ahotupa M. Lipid peroxidation and antioxidants in normal and pre-eclamptic pregnancies. Placenta. 1996;17:401–405. doi: 10.1016/s0143-4004(96)90021-1. [DOI] [PubMed] [Google Scholar]
- 28.Hilali N, Kocyigit A, Demir M, Camuzcuoglu A, Incebiyik A, Camuzcuoglu H, Vural M, Taskin A. DNA damage and oxidative stress in patients with mild preeclampsia and offspring. Eur J Obstet Gynecol Reprod Biol. 2013;170:377–380. doi: 10.1016/j.ejogrb.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 29.Kannan K, Jain SK. oxidative stress and apoptosis. Pathophysiology. 2000;7:153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 30.Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 31.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 32.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaillancourt C, Lanoix D, Le Bellego F, Daoud G, Lafond J. Involvement of MAPK signalling in human villous trophoblast differentiation. Mini Rev Med Chem. 2009;9:962–973. doi: 10.2174/138955709788681663. [DOI] [PubMed] [Google Scholar]
- 34.Wu HY, Lin CY, Lin TY, Chen TC, Yuan CJ. Mammalian Ste20-like protein kinase 3 mediates trophoblast apoptosis in spontaneous delivery. Apoptosis. 2008;13:283–294. doi: 10.1007/s10495-007-0161-x. [DOI] [PubMed] [Google Scholar]
- 35.Shen H, Liu J, Wang Y, Lian H, Wang J, Xing L, Yan X, Wang J, Zhang X. Aflatoxin G1-induced oxidative stress causes DNA damage and triggers apoptosis through MAPK signaling pathway in A549 cells. Food Chem Toxicol. 2013;62:661–669. doi: 10.1016/j.fct.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 36.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 37.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 38.Lee WC, Choi CH, Cha SH, Oh HL, Kim YK. Role of ERK in hydrogen peroxide-induced cell death of human glioma cells. Neurochem Res. 2005;30:263–270. doi: 10.1007/s11064-005-2449-y. [DOI] [PubMed] [Google Scholar]
- 39.Gutiérrez-Venegas G, Arreguín-Cano JA, Arroyo-Cruz R, Villeda-Navarro M, Méndez-Mejía JA. Activation of ERK1/2 by protein kinase C-alpha in response to hydrogen peroxide-induced cell death in human gingival fibroblasts. Toxicol In Vitro. 2010;24:319–326. doi: 10.1016/j.tiv.2009.08.007. [DOI] [PubMed] [Google Scholar]