

Abstract
Advances in screening and computational methods have enhanced recent efforts to discover/design small-molecule protein inhibitors. One attractive target for inhibition is the myosin family of motor proteins. Myosins function in a wide variety of cellular processes, from intracellular trafficking to cell motility, and are implicated in several human diseases (e.g., cancer, hypertrophic cardiomyopathy, deafness and many neurological disorders). Potent and selective myosin inhibitors are, therefore, not only a tool for understanding myosin function, but are also a resource for developing treatments for diseases involving myosin dysfunction or overactivity. This review will provide a brief overview of the characteristics and scientific/therapeutic applications of the presently identified small-molecule myosin inhibitors before discussing the future of myosin inhibitor and activator design.
The identification and characterization of pharmacological compounds that inhibit the functional activity of one or more specific proteins or processes has been the subject of much scientific investigation. On a basic science level, these membrane-permeable compounds provide the scientific community with a tool for the targeted and functional inhibition of a given protein in the cell; a potent means of evaluating the intracellular functions of that protein [1,2]. From a biomedical standpoint, the characterization of these small-molecule inhibitors affords an opportunity for the development of novel disease treatments centering on the repression of an offensive molecule or the reversal of its downstream effects [3-5].
At present, several complementary methods for obtaining suitable small-molecule inhibitors of specific proteins exist. Traditional methods in inhibitor discovery involve the systematic testing of a series of chemically synthesized or naturally occurring compounds. Advances in robotics and data processing have made it possible to use high-throughput screens to test libraries of thousands or even millions of potential drugs for their ability to inhibit the function of a specific protein in a targeted biochemical or cellular assay [6-8]. These inhibitor discovery processes are complemented by more precise methods in small-molecule inhibitor design. Structure-based methods rely on the use of x-ray crystallographic or NMR-based structures of a protein of interest to design small molecules likely to bind and inhibit protein function [9,10]. Computer-aided inhibitor design uses computational methods to optimize potential inhibitors identified by screening or structure-based methods, to virtually screen for new inhibitors from large libraries and to design potential inhibitors from databases of known protein–ligand interactions [11,12]. In combination, these distinct inhibitor design and discovery processes have resulted in the identification of many potent inhibitors of specific proteins and protein–protein interactions.
One potent protein target for inhibitor design is the myosin family. The myosin family is a divergent collection of actin-based molecular motors that can be divided into more than twenty classes based on phylogenetic analyses of conserved structural domains [13]. The twelve classes of myosins expressed in mammalian cells (I–III, V–VII, IX, X, XV, XVI, XVIII, and XIX) function in a wide variety of critical cellular processes [14]. ‘Conventional’ skeletal myosin IIs generate muscle contraction by sliding along actin filaments in the sarcomeres of muscle cells whereas nonmuscle myosin IIs are involved in a wide range of cellular activities including cell migration and cell division. The remaining, ‘unconventional’ myosins function in such processes as intracellular transport and tethering (e.g., regulation of exocytosis/secretion by myosins 1c/1e, Va/Vb, VI, VII and X), cell division, cell motility, actin cytoskeletal organization and cellular signaling [15]. Myosins have also been implicated in several human diseases, such as hypertrophic cardiomyopathy [16,17], Griscelli syndrome [18], deafness [19,20] and cancer [21,22]. Therefore, inhibitors of specific myosins could act as a valuable tool both in characterizing many intracellular processes and also in developing targeted treatments for diseases involving myosin overproduction/malfunction.
In order to understand the mechanism by which small-molecule myosin inhibitors interfere with myosin function, it is necessary to briefly revisit the basic structural and functional properties of myosin motors. Myosins have a three-part domain structure:
-
■
An N-terminal motor domain containing actin-binding regions and a magnesium adenosine triphosphatase (Mg2+ ATPase) site;
-
■
A central neck or lever-arm region that binds modulatory light chains;
-
■
A C-terminal tail domain that facilitates cargo binding and intracellular targeting [23].
Movement by myosin motors is generated by the energy released from the hydrolysis of ATP by the actin-activated Mg2+ ATPase in the motor domain [24,25]. Briefly, the binding of ATP to an actin-bound myosin motor protein (‘actomyosin complex/rigor state’) causes a major conformational change resulting in dissociation of the myosin motor domain from actin. The dissociated myosin then repositions itself into a ‘cocked’ state and hydrolyzes ATP into ADP and inorganic phosphate (Pi), forming a stable myosin–ADP–Pi intermediate (pre-power stroke state). This intermediate rebinds actin and releases the inorganic phosphate, which triggers the myosin ‘power stroke’ resulting in motor movement along actin. The myosin then releases ADP to form the actomyosin rigor complex and ATP rapidly binds to dissociate the myosin from the actin to start the ATPase cycle again.
Enzymes such as protein kinases can be inhibited by two common types of inhibitors: competitive inhibitors and noncompetitive inhibitors [4]. As the myosin motor domain is characterized functionally by the enzymatic activity of its magnesium ATPase, this division between competitive and noncompetitive inhibitors also underlies the different types of common small-molecule inhibitors possible for myosins. Competitive inhibitors of myosin ATPase activity would bind specifically to the ATP binding pocket in the motor domain, thereby preventing ATP binding and hydrolysis. The cellular applicability of competitive myosin inhibitor use is limited by the fact that myosins bound by competitive inhibitors remain permanently bound to actin (as they do not undergo the ATP binding necessary for release from the rigor state) and so physically impede actin binding by noninhibited myosins. The applicability of competitive myosin inhibitors is further limited by a lack of selectivity, as competive inhibitors structurally similar to ATP could inhibit the activity of nonmyosin ATPases and ATP-binding proteins within the cell. For these reasons, the majority of small-molecule inhibitors of myosin ATPase activity used at present are noncompetitive inhibitors that functionally impede myosin activity by binding to the protein at an allosteric site outside of the ATP binding pocket. This type of inhibition can ablate the functionality of a targeted myosin without blocking actin binding by uninhibited myosins within the cell and without secondary effects on other ATPases/ATP-binding proteins.
Although there have been considerable efforts to develop both specific and universal inhibitors of myosins, the pool of small-molecule myosin inhibitors with the functionality and specificity necessary for practical use is still very small. This review will discuss the development and physical/kinetic characteristics of the main small-molecule myosin inhibitors currently in use, as well as the scientific and therapeutic insights that these inhibitors have provided (Table 1). The potential for using the characteristics of known myosin inhibitors to design new and more specific small-molecule inhibitors will also be explored, as well as the emerging field of myosin activator design.
Table 1.
Summary of the properties of characterized small-molecule inhibitors of myosins.
| Inhibitor name | Molecular formula | Structural formula | Primary myosin target | Binding site | Mechanism | IC50 | Ref. |
|---|---|---|---|---|---|---|---|
| Blebbistatin | C18H16N2O2 |
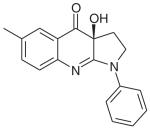
|
Myosin II (nonmuscle, skeletal muscle) | Hydrophobic pocket at apex of 50-kDa cleft in motor domain | Noncompetitive: hinders Pi release | 0.5–5 μM | [26,27] |
| N-benzyl-p-toluene sulphonamide (BTS) | C14H15NSO2 |
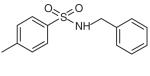
|
Myosin II (fast-twitch skeletal muscle) | Shaw et al. (2003) predicts within 50-kDa cleft in motor domain | Noncompetitive: hinders Pi release and ADP release | ~5 MM | [35,37] |
| 2,3-Butanedione monoxime (BDM) | C4H7NO2 |
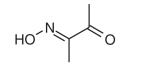
|
Myosin II (skeletal muscle) | Uncharacterized allosteric site in motor domain | Noncompetitive: hinders Pi release | ~5 μM | [39,41] |
| Pentachloropseudilin (PCIP) | C10H4Cl5NO |
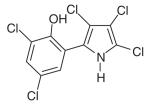
|
Myosin I | Pocket near tip of 50-kDa cleft in motor domain (7.5 Å from blebbistatin-binding pocket) | Noncompetitive: reduces coupling between actin-and nucleotide-binding sites | 1-5 μM | [54] |
| Pentabromopseudilin (PBP) | C10H4Br5NO |
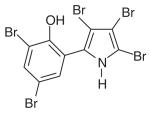
|
Myosin V | Pocket near tip of 50-kDa cleft in motor domain (7.5 Å from blebbistatin-binding pocket) | Noncompetitive: reduces ADP dissociation, ATP binding/ hydrolysis, and coupling between actin, nucleotide-binding sites | 1.2 μM | [55,58] |
| MyoVin-1 | C29H26N6SO3 |
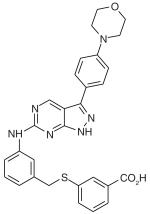
|
Myosin V | Uncharacterized allosteric site in motor domain | Noncompetitive: hinders ADP release | ~6 μM | [60] |
| 2,4,6-Triiodophenol (TIP) | C6H3I3O |
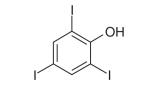
|
Myosin VI | Heissler et al. (2012) predicts PCIP/PBP or blebbistatin-binding sites | Heissler et al. (2012) predicts noncompetitive | ~2 μM | [61] |
Pi: Inorganic phosphate.
Small-molecule inhibitors of myosins
Small-molecule inhibitors of myosin II
Blebbistatin
The most well known and widely used of the small-molecule inhibitors specific to myosins is blebbistatin (Table 1). This small-molecule derivative of 1-phenyl-2-pyrrolidinone was identified during a high-throughput screening assay as an inhibitor of the ATPase activity of non-muscle myosin IIa [26]. Structural and kinetic studies of blebbistatin demonstrate that the molecule is a noncompetitive inhibitor that blocks myosin II function by hindering a critical step during its ATPase cycle. Specifically, blebbistatin binds to a hydrophobic pocket at the apex of the 50-kDa cleft in the motor domain of myosin II [27]. During the myosin ATPase cycle, this cleft characteristically closes as a result of structural rearrangements in the nucleotide-binding site mediating the process of Pi release and subsequent ADP release [28]. Blebbistatin binding stabilizes the closed ADP/Pi bound myosin II intermediate state, thus preventing the release of Pi and the associated myosin power stroke (i.e., the force generating step) (Figure 1) [27,29,30]. Blebbistatin is highly suited for studies of the cellular functions of cytoplasmic myosin II, as it blocks myosin II activity in an actin-detached state, thus, preventing artifacts from the formation of strongly bound nonfunctional actomyosin complexes [29].
Figure 1. Mechanisms of small-molecule inhibition of myosin ATPase.

All myosins move or translocate along actin filaments using energy gained from ATP hydrolysis by an actin-activated Mg2+ ATPase. This process occurs in a series of discrete steps and the presently identified myosin inhibitors are characterized by their ability to hinder specific steps. As an overview, the binding of ATP to an actomyosin complex (A) results in dissociation of myosin from actin. ATP is then hydrolyzed to form ADP and inorganic phosphate (Pi) and the motor domain is repositioned into a ‘cocked’ state (B). The myosin–ADP–Pi complex rebinds actin and releases the inorganic phosphate, triggering a ‘power stroke’ of directed myosin (or actin) movement (C). Lastly, the ADP is released (D), leaving a new actomyosin complex [24,25]. All of the presently characterized myosin II inhibitors (blebbistatin, N-benzyl-p-toluene sulphonamide, 2,3-Butanedione monoxime) operate by hindering Pi release during (C) and BTS also hinders ADP release during (D). The myosin V inhibitor Myo-Vin1 hinders ADP release during (D). The myosin V inhibitor pentabromopseudilin has a more global effect on ATPase dynamics, as it has been shown to decrease ATP binding rates (A), ATP hydrolysis rates (B) and ADP dissociation rates (D), as well as reduce the coupling between the actin and nucleotide binding sites in the myosin motor domain (Between [C] and [D]). The myosin I inhibitor pentachloropseudilin also reduces the coupling between the actin and nucleotide binding sites in the motor domain (between [C] and [D]).
BDM: 2,3-Butanedione monoxime; BTS: N-benzyl-p-toluene sulphonamide; PBP: Pentabromopseudilin; PCIP: Pentachloropseudilin; Pi: Inorganic phosphate.
The high specificity of blebbistatin inhibition further demonstrates the usefulness of the molecule for targeted studies. Blebbistatin inhibits the ATPase activity and in vitro motility of vertebrate nonmuscle myosin IIA and IIB, porcine/rabbit/scallop striated muscle myosin II proteins, and Dictyostelium myosin II, with half-maximal IC50 values between 0.5 and 5 μM [26,31]. However, at comparable concentrations, the molecule does not inhibit the ATPase activity of Acanthamoeba myosin II or that of smooth muscle myosin II, which is highly homologous to vertebrate non-muscle myosin [26,31]. Furthermore, blebbistatin concentrations up to 50 μM do not inhibit the ATPase activity of myosins from other classes tested, including myosins I, V and X [31].
The rapid cell permeability of blebbistatin complements this specificity for myosin II, rendering the molecule highly suited to in vivo studies of myosin II function [26]. The fact that the inhibitory effects of this molecule are reversible further establishes blebbistatin as a powerful tool for characterizing precise cellular events.
The chief limitations in the use of blebbistatin have thus far been the light sensitivity of the molecule and its low solubility in aqueous solutions. Blebbistatin is photoinactivated by prolonged exposure to high levels of blue light and this reaction is cytotoxic, limiting the application of this molecule during live cell microscopy assays [32]. Recent studies indicate, however, that more mild exposure to blue light may permit photoinactivation of blebbistatin without cell death, which raises the possibility of targeted reversal of blebbistatin inhibition in selected cellular areas [33]. Furthermore, an aryl azido derivative of blebbistatin (‘azidoblebbistatin’) that can be covalently crosslinked to myosin has recently been developed [34]. This derivative enhances the inhibitory properties of blebbistatin by removing limitations due to its low water solubility and low binding affinity in vivo.
N-benzyl-p-toluene sulphonamide
The aryl sulphonamide N-benzyl-p-toluene sulphonamide (BTS) was identified in a screen for small-molecule inhibitors of skeletal muscle myosin II (Table 1) [35]. In in vitro studies, the molecule inhibits the actin-activated ATPase activity and gliding motility of skeletal muscle myosin II, with IC50 values of approximately 5 μM [35]. In vivo, BTS can permeate cell membranes to specifically suppress contraction and, hence, force production in skeletal muscle fibers [35,36]. Both in vitro and in vivo inhibition can be reversed by washing out the drug. The molecule is further distinguished as a valuable addition to functional studies of myosin II by its high affinity for myosin and by the fact that an isomer of BTS with no inhibitory effect has been identified for use as a negative control [35].
Amongst different myosin II proteins, the inhibitory effects of BTS selectively target fast-twitch skeletal muscle myosin II. The molecule inhibits fast skeletal muscle myosin II more than 100-times more potently than slow skeletal muscle myosin II, cardiac muscle myosin II or nonmuscle myosin II [35]. Furthermore, it does not inhibit platelet myosin II [35]. The effects of BTS on members of other myosin classes have not yet been examined.
Kinetic analysis of the influence of BTS on the myosin II ATPase cycle suggests a mechanism by which BTS inhibits myosin II [37]. In particular, BTS treatment decreases the rate of Pi release by the myosin II ATPase more than 100-fold in the presence of actin and also decreases the rate of ADP dissociation, thus hindering the release of Pi necessary for force production (Figure 1). The further observation that the myosin II–ADP–Pi intermediate stabilized by BTS treatment has a reduced affinity for actin suggests that BTS suppresses muscle contraction in vivo by hindering both Pi release and actin binding [37]. These results indicating that BTS is a noncompetitive inhibitor of myosin II are supported by the observation that BTS does not bind to the nucleotide-binding site of myosin, since at saturating concentrations of BTS a fluorescent ADP derivative is still able to bind to myosin II [35].
2,3-Butanedione monoxime
The small molecule 2,3-Butanedione monoxime (BDM) was designed as an acetylcholinerase reactivator [38] but was later found to reversibly inhibit muscle contraction in skeletal muscle and to inhibit the ATPase activity of skeletal muscle myosin II (Table 1) [39]. Kinetic studies indicate that BDM, as with BTS and blebbistatin, is a noncompetitive myosin II inhibitor that blocks myosin II function by hindering Pi release and stabilizing the myosin–ADP–Pi intermediate (Figure 1) [40,41]. However, the molecule has a low affinity for myosin and concentrations in the 10-mM range are required to achieve myosin inhibition [42].
The primary limitation to the general use of BDM is the conflict in the field as to the specificity of its inhibitory function. Although all parties agree that BDM is a distinct (although low affinity) inhibitor of skeletal muscle myosin II, the influence of BDM on other myosins is a matter of contention. One study indicates that BDM also inhibits the ATPase activity of nonmuscle myosin II, myosin V and myosin VI, but does not inhibit kinesin ATPase activity [43]. These findings have spurred the use of BDM as a ‘general’ inhibitor of all myosin ATPase activity in cell biological studies [44-47]. More recent kinetic studies, however, raised the contradictory observations that BDM does not inhibit myosin Ic, V, VI [48] or nonmuscle myosin II [35]. These conflicting data regarding the specificity of BDM must be resolved before the molecule can be used with any confidence in cell biological or in vitro studies [49].
In addition to questions regarding the myosin specificity of BDM, this molecule has also been demonstrated to affect many proteins and processes independent of myosin ATPase activity. In addition to its original roles as an acetylcholinerase reactivator, BDM has been shown to inhibit myosin light chain kinase [50] and to facilitate neurotransmitter release [42]. BDM affects both voltage and ligand activated ion channels in muscle and nerve cells [42], protein phosphorylation in cardiomyocytes [51] and calcium regulation [52,53]. This wide range of effects for BDM separate from its inhibition of myosins, as well as its low binding affinity, must be seriously taken into consideration if BDM is used to assess myosin II function in vivo.
Small-molecule inhibitors of myosin I
Pentachloropseudilin
The small molecule pentachloropseudilin (PCIP) has been identified as an inhibitor of myosin I function (Table 1) [54]. This highly halogenated natural antibiotic reversibly inhibits both the ATPase activity and in vitro motility of myosins 1c and 1b, with an IC50 value between 1 and 5 μM. Concentrations of PCIP in this range have also been shown to directly inhibit the cellular functions of myosin Ic in vivo. In particular, treatment of HeLa cells with 1-μM PCIP results in abnormalities in lysosomal morphology and distribution that mimic the phenotype observed when myosin Ic is ‘knocked-down’ by RNAi [54].
Structural studies of the binding of PCIP to a model myosin (Dictyostelium discoideum myosin II) indicate that the compound binds to an allosteric pocket in the myosin motor domain and thus acts as a noncompetitive inhibitor of motor activity [54]. The PCIP-binding pocket is located near actin-binding residues at the tip of the 50-kDa cleft in the myosin motor domain, approximately 16 Å from the nucleotide binding site and 7.5 Å from the hydrophobic pocket bound by blebbistatin. PCIP binding to this allosteric pocket reduces ATPase activity via a conserved communication pathway between the allosteric and nucleotide binding sites that results in local structural changes in the myosin, reduction of coupling between the actin and nucleotide binding sites, and global changes in myosin dynamics (Figure 1) [54,55]. Complementary computational studies indicate that the specific preference of PCIP for the binding pocket in class I myosins may stem from the presence of a relatively higher number of polar/charged residues within the binding site of this myosin class [55].
It is important to note that although the potent inhibition of myosin I proteins by PCIP has been characterized in greatest detail, the drug also inhibits the ATPase activity of myosin Vb and nonmuscle myosin II at higher concentrations (IC50 >90 μM) [54]. This lack of specificity at higher concentrations must be taken into account when designing in vivo experiments using PCIP.
Small-molecule inhibitors of myosin V
Pentabromopseudilin
The small molecule pentabromopseudilin (PBP) is an antibiotic originally isolated from Pseudomonas bromoutilis that belongs to the same alkaloid family of pseudilins as the myosin I inhibitor PCIP (Table 1) [56,57]. It is a potent and reversible inhibitor of the ATPase activity and in vitro motility of myosin Va (IC50 = 1.2 μM) [58]. This general inhibition of myosin V activity has been reinforced by cellular studies of the phenotypic effects of PBP treatment in vivo. For example, yeast cells treated with 500-nM PBP show a similar mitochondrial fragmentation phenotype to mutant yeast strains with defects in the expression of the class V myosin myo2p [58].
PBP binds to the same allosteric binding pocket in the motor domain as PCIP [58] and the polarity of this binding site similarly targets its myosin isoform specificity. In particular, PBP most potently inhibits myosin V due to its affinity for the increased hydrophobicity of the binding pocket in this class of myosins [55]. PBP has been shown to inhibit the ATPase activity of this myosin by increasing its affinity for ADP, decreasing ATP binding and hydrolysis rates, and reducing coupling between the actin- and nucleotide-binding sites in the motor domain (Figure 1). These changes occur via a conserved communication pathway between the allosteric and nucleotide-binding sites [55,58].
The potential for the targeted use of PBP as a myosin Va inhibitor in vivo is limited by the fact that this molecule has a wide range of effects on other myosins and myosin-dependent processes. At higher concentrations than those required for myosin Va inhibition, PBP inhibits the ATPase activity of myosin Vb (IC50 = ~20 μM), non-muscle myosin II (IC50 = ~25 μM) and myosin Ie (IC50 = ~50 μM) [58]. Treatment with PBP also reduces the isometric tension development and unloaded shortening velocity of skeletal muscle myosin II (IC50 = ~25 μM) [58].
MyoVin-1
The chemically synthesized compound MyoVin-1 was designed as an inhibitor of myosin V ATPase activity (Table 1). This pyrazolopyrimidine-based compound was synthesized based on ‘privileged’ chemical scaffolds determined from a collection of known kinase inhibitors [59,60]. The relative selectivity of MyoVin-1 for the inhibition of myosin V ATPase activity (IC50 = ~6 μM) is reinforced by the demonstration that 50 μM myoVin-1 does not significantly inhibit the ATPase activity of myosin VI or nonmuscle myosin II [60]. Similarly, 100-μM myoVin-1 does not significantly inhibit a collection of representative kinases (e.g., CHK1, PLK1, Abl kinase, p42 MAP kinase, casein kinase II, and Aurora kinase) [60]. Kinetic studies indicate that myoVin-1 reduces myosin V ATPase activity by specifically inhibiting ADP release from the actomyosin complex (Figure 1) [60].
Small-molecule inhibitors of myosin VI
2,4,6-Triiodophenol
A recent series of computational and experimental binding studies have identified the halogenated phenol 2,4,6-triiodophenol (TIP) as an inhibitor of the ATPase activity of myosin VI (Table 1) [61]. Live cell studies demonstrate that TIP treatment produces the same characteristic reduction in vesicle secretion levels observed after siRNA knockdown of myosin VI, with an IC50 of approximately 2 μM [61]. Complementary studies of the specificity of TIP demonstrate that 50 μM drug treatment does not affect the ATPase activities of myosin Id, nonmuscle myosin IIc or cardiac myosin II [61]. Additional studies will be necessary to characterize the binding sites and mechanism of TIP inhibition of the myosin ATPase, but it has been suggested that the molecule may bind to the PCIP/PBP- or blebbistatin-binding sites and act as a noncompetitive inhibitor [61].
Applications of small-molecule myosin inhibitors
Small-molecule inhibitors of myosins are valuable both as scientific tools for understanding myosin-dependent biological processes and also as therapeutic agents for treating myosin-related diseases. The following sections will examine the contributions of the presently available small-molecule myosin inhibitors to both of these areas.
Scientific insight from inhibitor use
The most straightforward application of small-molecule inhibitors of myosins is the inhibition of a given myosin as a means of characterizing its function within the cell. The use of inhibitors in this manner is more time-efficient than siRNA knockdown in culture cells (e.g., inhibition of myosin VI in HeLa cells via 25 min incubation with TIP produces the same secretion defect as a 5-day myosin VI siRNA knockdown protocol [61]). In addition, small-molecule inhibition of myosins is more widely applicable than siRNA (e.g., small-molecule inhibitors can be used in oocytes, which are known for their resistance to siRNA treatment due to their long-lasting storage proteins). The reversible nature and relatively inexpensive price of established small-molecule myosin inhibitors further confer advantages over traditional gene knockout technology in a whole organism. It is important to consider, however, that many of the presently characterized myosin inhibitors do not have the level of specificity necessary to achieve the removal of a certain myosin with the same selectively as, for example, gene knockout technology. As such, the off-target effects of specific small-molecule myosin inhibitors on other myosins or nonmyosins within the cell (e.g., the pleiotropic effects of BDM) must be taken into consideration when characterizing myosin function using these inhibitors.
The presently characterized small-molecule inhibitors of myosins have provided a great deal of scientific insight when used to inhibit myosin function. Initial studies using blebbistatin demonstrated that the position of the cytokinetic cleavage furrow is maintained by signals from microtubules controlling nonmuscle myosin II localization [26]. Targeted inhibition of myosin II with blebbistatin has also revealed that nonmuscle myosin II plays a role in fear memory consolidation in the lateral amygdala [62], that nonmuscle myosin IIA and IIB regulate the migration of rat hepatic stellate cells [63] and that nonmuscle myosin II is required for the ligand-induced internalization of the EGFR [64]. In addition, depletion of myosin ATPase activity in plant cells using BDM demonstrates that chloroplast actin filaments reorganize in a manner independent of chloroplast movement [65] and that the actomyosin complex mediates early transduction events during the gravitropic response of snapdragon spikes [66]. Treatment of muscle fibers with myosin II inhibitors has been used to further characterize muscle contraction. For example, blebbistatin treatment was used to demonstrate the dynamic and variable range of mitochondrial ADP-stimulated respiratory kinetics in skeletal muscle [67], whereas BDM treatment was used to show that fibronectin/integrin interactions are modulated by cardiomyocyte contractile status [68] and BTS treatment was used to demonstrate the structural orientation and contractile properties of skeletal muscle in zebrafish larvae [69]. These studies represent only a few of the many examples of the insight into biological processes provided by small-molecule myosin inhibitors.
In addition to their role as a tool for probing myosin-dependent biological processes, small-molecule myosin inhibitors have been used on a more general level as a means of improving laboratory techniques in areas such as cell culture and imaging. Blebbistatin or BDM treatment extends the culture lifetime of mouse cardiac myocytes, a cell line that is notoriously difficult to maintain in primary culture and very valuable for the characterization of heart disorders [70,71]. Furthermore, selective treatment with blebbistatin has been demonstrated as a tool to synchronize mammalian culture cells during different stages of mitotic division (metaphase, anaphase, telophase), a task that proved difficult in the past due to the short duration of anaphase [72]. In addition, both BDM and blebbistatin have been used to eliminate the ‘motion artifacts’ caused by muscle contractions during live optical imaging of cardiac electrical activity [73,74].
Therapeutic applications
Small-molecule inhibitors of myosins can also be applied as leads for the development of therapeutic treatments for many serious diseases associated with myosin function. For example, the anti-inflammatory effects of blebbistatin-based myosin II inhibition have been shown to ameliorate progressive renal disease in a rat model [75]. Furthermore, the fact that blebbistatin is phototoxic to human cancer cells under exposure to blue light [76] and blocks the invasiveness of both MCF-7 breast cancer cells [77] and pancreatic adenocarcinoma cells [78] targets this molecule as a lead for anticancer agent development. Since myosin II inhibition with blebbistatin or BDM increases the outflow of aqueous humor, these inhibitors could contribute to the development of treatments to reduce intraocular pressure in glaucoma patients [79,80]. In addition, analysis of BDM could assist with the development of therapeutics for diarrhea and toxoplasmosis in the human population, as this drug has been shown to inhibit the motility and invasiveness of both the apicomplexan Cryptosporidium parvum that causes human diarrheal disease [81] and the protozoa Toxoplasma gondii that causes toxoplasmosis [82].
On a more practical therapeutic level, treatment with BDM has been highlighted as a means of increasing the storage lifetime and efficiency of heart transplantation, as BDM treatment reduces stress and metabolic deregulation in donor hearts [46,83].
This overview of the numerous contributions of the few well-characterized small-molecule myosin inhibitors to the understanding of biological processes and the development of disease treatments reinforces the importance of the targeted application of more recently developed myosin inhibitors, as well as the development and application of new inhibitors.
Future perspective
As the widespread scientific and therapeutic applications of small-molecule myosin inhibitors have become increasingly evident, the desire to develop a wider array of inhibitors has been an object of increasing scientific focus. The goals of such inhibitor development include both the discovery of small-molecule inhibitors for previously untargeted myosins, as well as the development of more potent, less toxic and more selective inhibitors for already targeted myosins. These development methods are increasingly geared toward the computer-aided design of new and improved small-molecule inhibitors, as the field has expanded from screening methodology (e.g., the screen-based discovery of BDM, BTS and blebbistatin in the 1990s and early 2000s [26,35,38]) to include more targeted computational methods (e.g., the recent characterization of TIP [61]).
The insights provided by the presently characterized collection of small-molecule myosin inhibitors will be invaluable to future stages of inhibitor design. On a more general level, it is evident that most of the well-characterized myosin inhibitors at present function by binding to an allosteric pocket in the myosin motor domain and noncompetitively inhibiting the myosin ATPase (see ‘small-molecule inhibitors of myosins’ section). Since all myosins function via the same basic ATPase mechanism [24], future drug-design methods can apply this principle of noncompetitive inhibition to the development of inhibitors for previously untargeted myosins. On a more specific level, the presently characterized myosin inhibitors themselves can serve as templates for future drug development. Indeed, the chemical preparation of blebbistatin analogs via modification of its tricyclic core has already been evaluated as a method of developing myosin inhibitors with increased binding affinities and different selectivity [84,85]. This methodology was applied successfully in a recent study synthesizing an aryl azido derivative of blebbistatin that can be covalently cross-linked to myosin II, thereby reducing the concentration necessary for in vivo studies and reducing concerns due to the low in vivo binding affinity of blebbistatin [34]. Analysis of the precise binding mechanism of presently characterized myosin inhibitors may also assist with future drug-design efforts. An example of this is the evident continuity of the allosteric myosin binding pocket of the halogenated pseudilin inhibitors (PCIP, PBP) with the 50-kDa cleft in the myosin motor domain, which sets the stage for the future design of inhibitory molecules that extend further into this pocket and bind their myosin target over a larger area – a means of increasing both inhibitor affinity and specificity [58].
As an alternative strategy to motor domain/ATPase inhibitors, a potential avenue to explore in the future could also be the design/development of specific inhibitors targeting the cargo-binding regions of the different classes of ‘unconventional’ myosins. It is now known that these myosins, (e.g., in classes, I, V, VI, VII, IX and X) contain cargo-binding domains in their tail regions, which bind select adaptor proteins that target these myosins to specialized compartments/organelles within the cell where they perform specific functions. In the case of myosin V and VI there is structural data available on the cargo binding interfaces involved [86-88], which could serve as templates to design specific inhibitors. Thus, by targeting unique myosin-adaptor protein interactions, one could manipulate a specific adaptor protein-related motor function linked to distinct step(s) along a pathway within specialized cells.
As the field progresses towards the development of more potent and selective myosin inhibitors, it will be important to consider the complementary design of small-molecule myosin activators. Although small-molecule activators of proteins are well-established in the field as a tool for probing biological function and for therapeutic efforts [89-91], few small-molecule myosin activators have been developed at present. One notable exception is ‘omecamtiv mecarbil,’ a recently developed small-molecule that noncompetitively activates cardiac myosin by increasing the rate of inorganic phosphate release, thereby shortening the lifetime of the myosin–ADP–Pi intermediate and speeding the transition into a force-generating, actin-bound state [92]. This molecule has recently been cited as a therapeutic resource for the treatment of systolic heart failure [92] and sets the stage for the complement of small-molecule myosin inhibitor design with the new and promising field of small-molecule myosin activator development.
By providing an overview of the field of small-molecule myosin inhibitor design/discovery and detailing the characteristics and applications of currently characterized small molecule myosin inhibitors, this review will serve as a resource for future efforts in the design of small-molecule inhibitors and activators of myosins.
Executive summary.
Background
-
■
Design and discovery of small-molecule protein inhibitors has expanded with advances in high-throughput screening, structural analysis and computational methods.
-
■
The multifunctional family of myosin molecular motors represents an attractive target for inhibitor design efforts.
Small molecule inhibitors of myosins
-
■
Small-molecule inhibitors have been characterized for myosins I, II, V and VI.
-
■
The small-molecule myosin inhibitors characterized thus far are most commonly noncompetitive inhibitors that operate by hindering ADP/Pi release by the myosin ATPase.
Applications of small-molecule myosin inhibitors
-
■
The presently characterized small-molecule inhibitors have contributed to the scientific understanding of many myosin-based biological processes and aided in the optimization of tissue culture and imaging techniques.
-
■
Myosin inhibitors also serve as leads for the development of therapeutics for many human diseases, including renal disease and glaucoma.
Future perspective: small-molecule myosin inhibitors & activators
-
■
Future efforts in myosin inhibitor development can use the kinetic properties and physical structure of current inhibitors as templates for computer-aided design.
-
■
The development of small-molecule inhibitors for myosin-binding partners represents an attractive new technique for the depletion of myosin functionality on specific intracellular pathways.
-
■
The discovery and design of small-molecule myosin activators is an important and newly emerging field that will complement efforts in myosin inhibitor design.
Acknowledgments
This work was funded by the Wellcome Trust (F Buss) and supported by the Medical Research Council (J Kendrick-Jones). The CIMR is in receipt of a strategic award from the Wellcome Trust.
Key Terms
- ATPase
Enzyme that catalyzes the hydrolysis of ATP into ADP and inorganic phosphate (Pi). The energy gained from this hydrolysis pathway powers the directed stroke of myosin along actin.
- Rigor
Myosins free of ATP bind tightly to actin in a ‘rigor’ state. This rigor state is one of the basic steps in the actomyosin ATPase cycle. Upon ATP binding, myosins dissociate from actin and, thus, leave the rigor state.
- Fast twitch skeletal muscle
Characterized by their use of anaerobic metabolism to produce short bursts of strength/speed. They can be contrasted with slow twitch skeletal muscle fibers, which are characterized by their use of aerobic metabolism to fuel extended contraction over longer time periods.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Crews CM, Shotwell JB. Small-molecule inhibitors of the cell cycle: an overview. Prog. Cell Cycle Res. 2003;5:125–133. [PubMed] [Google Scholar]
- 2.Firestone AJ, Weinger JS, Maldonado M, et al. Small-molecule inhibitors of the AAA+ATPase motor cytoplasmic dynein. Nature. 2012;484(7392):125–129. doi: 10.1038/nature10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein–protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49(1):223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin. Cancer Res. 2012;18(10):2740–2753. doi: 10.1158/1078-0432.CCR-11-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persidis A. High-throughput screening. Advances in robotics and miniturization continue to accelerate drug lead identification. Nat. Biotechnol. 1998;16(5):488–489. doi: 10.1038/nbt0598-488. [DOI] [PubMed] [Google Scholar]
- 7.White RE. High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery. Annu. Rev. Pharmacol. Toxicol. 2000;40:133–157. doi: 10.1146/annurev.pharmtox.40.1.133. [DOI] [PubMed] [Google Scholar]
- 8.von Ahsen O, Bömer U. High-Throughput screening for kinase inhibitors. ChemBioChem. 2005;6(3):481–490. doi: 10.1002/cbic.200400211. [DOI] [PubMed] [Google Scholar]
- 9.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257(5073):1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 10.Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2012;27(6):759–772. doi: 10.3109/14756366.2012.672983. [DOI] [PubMed] [Google Scholar]
- 11.Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem. Biol. Interact. 2008;171(2):165–176. doi: 10.1016/j.cbi.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief. Bioinform. 2009;10(5):579–591. doi: 10.1093/bib/bbp023. [DOI] [PubMed] [Google Scholar]
- 13.Odronitz F, Kollmar M. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biology. 2007;8(9):R196. doi: 10.1186/gb-2007-8-9-r196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proc. Natl Acad. Sci. USA. 2006;103(10):3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coluccio LM. Myosins: A Superfamily of Molecular Motors (Proteins and Cell Regulation) 1st edition Vol. 7. Springer; Dordrecht, The Netherlands: 2007. [Google Scholar]
- 16.Mohiddin SA, Ahmed ZM, Griffith AJ, et al. Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6) J. Med. Genet. 2004;41(4):309–314. doi: 10.1136/jmg.2003.011973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller M, Mazur AJ, Behrmann E, et al. Functional characterization of the human α-cardiac actin mutations Y166C and M305L involved in hypertrophic cardiomyopathy. Cell Mol. Life Sci. 2012;69(20):3457–3479. doi: 10.1007/s00018-012-1030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar M, Sackey K, Schmalstieg F, Trizna Z, Elghetany MT, Alter BP. Griscelli syndrome: rare neonatal syndrome of recurrent hemophagocytosis. J. Pediatr. Hematol. Oncol. 2001;23(7):464–468. doi: 10.1097/00043426-200110000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Gibson F, Walsh J, Mburu P, et al. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374(6517):62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 20.Topsakal V, Hilgert N, van Dinther J, et al. Genotype-phenotype correlation for DFNA22: characterization of non-syndromic, autosomal dominant, progressive sensorineural hearing loss due to MYO6 mutations. Audiol. Neurootol. 2010;15(4):211–220. doi: 10.1159/000255339. [DOI] [PubMed] [Google Scholar]
- 21.Dunn TA, Chen S, Faith DA, et al. A novel role of myosin VI in human prostate cancer. Am. J. Pathol. 2006;169(5):1843–1854. doi: 10.2353/ajpath.2006.060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vickaryous N, Polanco-Echeverry G, Morrow S. Smooth-muscle myosin mutations in hereditary non-polyposis colorectal cancer syndrome. Br. J. Cancer. 2008;99(10):1726–1728. doi: 10.1038/sj.bjc.6604737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berg JS, Powell BC, Cheney RE. A millennial myosin census. Mol. Biol. Cell. 2001;12(4):780–794. doi: 10.1091/mbc.12.4.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 25.Sweeney HL, Houdusse A. Structural and functional insights into the Myosin motor mechanism. Annu. Rev. Biophys. 2010;39:539–557. doi: 10.1146/annurev.biophys.050708.133751. [DOI] [PubMed] [Google Scholar]
- 26.Straight AF. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 2003;299(5613):1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- 27.Allingham JS, Smith R, Rayment I. The structural basis of blebbistatin inhibition and specificity for myosin II. Nat. Struct. Mol. Biol. 2005;12(4):378–379. doi: 10.1038/nsmb908. [DOI] [PubMed] [Google Scholar]
- 28.Coureux PD, Wells AL, Ménétrey J, et al. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425(6956):419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- 29.Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279(34):35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 30.Ramamurthy B, Yengo CM, Straight AF, Mitchison TJ, Sweeney HL. Kinetic mechanism of blebbistatin inhibition of nonmuscle myosin IIb. Biochemistry. 2004;43(46):14832–14839. doi: 10.1021/bi0490284. [DOI] [PubMed] [Google Scholar]
- 31.Limouze J, Straight AF, Mitchison T, Sellers JR. Specificity of blebbistatin, an inhibitor of myosin II. J. Muscle Res. Cell. Motil. 2004;25(4-5):337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- 32.Kolega J. Phototoxicity and photoinactivation of blebbistatin in UV and visible light. Biochem. Biophys. Res. Commun. 2004;320(3):1020–1025. doi: 10.1016/j.bbrc.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 33.Sakamoto T, Limouze J, Combs CA, Straight AF, Sellers JR. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry. 2005;44(2):584–588. doi: 10.1021/bi0483357. [DOI] [PubMed] [Google Scholar]
- 34.Képiró M, Várkuti BH, Bodor A, et al. Azidoblebbistatin, a photoreactive myosin inhibitor. Proc. Natl Acad. Sci. USA. 2012;109(24):9402–9407. doi: 10.1073/pnas.1202786109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheung A, Dantzig JA, Hollingworth S, et al. A small-molecule inhibitor of skeletal muscle myosin II. Nat. Cell Biol. 2002;4(1):83–88. doi: 10.1038/ncb734. [DOI] [PubMed] [Google Scholar]
- 36.Ramachandran I, Terry M, Ferrari MB. Skeletal muscle myosin cross-bridge cycling is necessary for myofibrillogenesis. Cell Motil. Cytoskeleton. 2003;55(1):61–72. doi: 10.1002/cm.10113. [DOI] [PubMed] [Google Scholar]
- 37.Shaw MA, Ostap EM, Goldman YE. Mechanism of inhibition of skeletal muscle actomyosin by N-benzyl-p-toluenesulfonamide. Biochemistry. 2003;42(20):6128–6135. doi: 10.1021/bi026964f. [DOI] [PubMed] [Google Scholar]
- 38.Wilson IB, Ginsburg B. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim. Biophys. Acta. 1955;18(1):168–170. doi: 10.1016/0006-3002(55)90040-8. [DOI] [PubMed] [Google Scholar]
- 39.Higuchi H, Takemori S. Butanedione monoxime suppresses contraction and ATPase activity of rabbit skeletal muscle. J. Biochem. 1989;105(4):638–643. doi: 10.1093/oxfordjournals.jbchem.a122717. [DOI] [PubMed] [Google Scholar]
- 40.Herrmann C, Wray J, Travers F, Barman T. Effect of 2,3-Butanedione monoxime on myosin and myofibrillar ATPases. An example of an uncompetitive inhibitor. Biochemistry. 1992;31(48):12227–12232. doi: 10.1021/bi00163a036. [DOI] [PubMed] [Google Scholar]
- 41.McKillop DF, Fortune NS, Ranatunga KW, Geeves MA. The influence of 2,3-Butanedione 2-monoxime (BDM) on the interaction between actin and myosin in solution and in skinned muscle fibres. J. Muscle Res. Cell. Motil. 1994;15(3):309–318. doi: 10.1007/BF00123483. [DOI] [PubMed] [Google Scholar]
- 42.Sellin LC, McArdle JJ. Multiple effects of 2,3-Butanedione monoxime. Pharmacol. Toxicol. 1994;74(6):305–313. doi: 10.1111/j.1600-0773.1994.tb01365.x. [DOI] [PubMed] [Google Scholar]
- 43.Cramer LP, Mitchison TJ. Myosin is involved in postmitotic cell spreading. J. Cell Biol. 1995;131(1):179–189. doi: 10.1083/jcb.131.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samaj J, Peters M, Volkmann D, Baluska F. Effects of myosin ATPase inhibitor 2,3-Butanedione 2-monoxime on distributions of myosins, F-actin, microtubules, and cortical endoplasmic reticulum in maize root apices. Plant Cell Physiol. 2000;41(5):571–582. doi: 10.1093/pcp/41.5.571. [DOI] [PubMed] [Google Scholar]
- 45.Borlak J, Zwadlo C. The myosin ATPase inhibitor 2,3-Butanedione monoxime dictates transcriptional activation of ion channels and Ca(2+)-handling proteins. Mol. Pharmacol. 2004;66(3):708–717. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 46.Pisarenko OI, Shul’zhenko VS, Studneva IM. The effect of myosin ATPase inhibition on metabolic and functional recovery of isolated rat heart after global ischemia. Biomed. Khim. 2009;55(4):451–461. [PubMed] [Google Scholar]
- 47.Hashimoto K, Yokota E, Shimmen T, Yoshida M. The myosin ATPase inhibitor, 2,3-Butanedione 2-monoxime, prevents protein secretion by the basidiomycete Coprinopsis cinerea. Biotechnol. Lett. 2011;33(4):769–775. doi: 10.1007/s10529-010-0497-0. [DOI] [PubMed] [Google Scholar]
- 48.Ostap EM. 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J. Muscle Res. Cell. Motil. 2002;23(4):305–308. doi: 10.1023/a:1022047102064. [DOI] [PubMed] [Google Scholar]
- 49.Forer A, Fabian L. Does 2,3-Butanedione monoxime inhibit nonmuscle myosin? Protoplasma. 2005;225(1-2):1–4. doi: 10.1007/s00709-004-0077-z. [DOI] [PubMed] [Google Scholar]
- 50.Siegman MJ, Mooers SU, Warren TB, Warshaw DM, Ikebe M, Butler TM. Comparison of the effects of 2,3-Butanedione monoxime on force production, myosin light chain phosphorylation and chemical energy usage in intact and permeabilized smooth and skeletal muscles. J. Muscle Res. Cell. Motil. 1994;15(4):457–472. doi: 10.1007/BF00122119. [DOI] [PubMed] [Google Scholar]
- 51.Stapleton MT, Fuchsbauer CM, Allshire AP. BDM drives protein dephosphorylation and inhibits adenine nucleotide exchange in cardiomyocytes. Am. J. Physiol. 1998;275(4):H1260–H1266. doi: 10.1152/ajpheart.1998.275.4.H1260. Pt 2. [DOI] [PubMed] [Google Scholar]
- 52.Fryer MW, Gage PW, Neering IR, Dulhunty AF, Lamb GD. Paralysis of skeletal muscle by Butanedione monoxime, a chemical phosphatase. Pflugers Arch. 1988;411(1):76–79. doi: 10.1007/BF00581649. [DOI] [PubMed] [Google Scholar]
- 53.Lang RJ, Paul RJ. Effects of 2,3-Butanedione monoxime on whole-cell Ca2+ channel currents in single cells of the guinea-pig taenia caeci. J. Physiol. (Lond.) 1991;433:1–24. doi: 10.1113/jphysiol.1991.sp018411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chinthalapudi K, Taft MH, Martin R, et al. Mechanism and specificity of pentachloropseudilin-mediated inhibition of myosin motor activity. J. Biol. Chem. 2011;286(34):29700–29708. doi: 10.1074/jbc.M111.239210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Preller M, Chinthalapudi K, Martin R, Knolker H-J, Manstein DJ. Inhibition of Myosin ATPase activity by halogenated pseudilins: a structure-activity study. J. Med. Chem. 2011;54(11):3675–3685. doi: 10.1021/jm200259f. [DOI] [PubMed] [Google Scholar]
- 56.Burkholder PR, Pfister RM, Leitz FH. Production of a pyrrole antibiotic by a marine bacterium. Appl. Microbiol. 1966;14(4):649–653. doi: 10.1128/am.14.4.649-653.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin R, Jäger A, Böhl M, et al. Total synthesis of pentabromo- and pentachloropseudilin, and synthetic analogues–allosteric inhibitors of myosin ATPase. Angew. Chem. Int. Ed. Engl. 2009;48(43):8042–8046. doi: 10.1002/anie.200903743. [DOI] [PubMed] [Google Scholar]
- 58.Fedorov R, Böhl M, Tsiavaliaris G, et al. The mechanism of pentabromopseudilin inhibition of myosin motor activity. Nat. Struct. Mol. Biol. 2009;16(1):80–88. doi: 10.1038/nsmb.1542. [DOI] [PubMed] [Google Scholar]
- 59.Peters U, Cherian J, Kim JH, Kwok BH, Kapoor TM. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat. Chem. Biol. 2006;2(11):618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- 60.Islam K, Chin HF, Olivares AO, Saunders LP, De La Cruz EM, Kapoor TM. A myosin V inhibitor based on privileged chemical scaffolds. Angew. Chem. Int. Ed. Engl. 2010;49(45):8484–8488. doi: 10.1002/anie.201004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heissler SM, Selvadurai J, Bond LM, et al. Kinetic properties and small-molecule inhibition of human myosin VI. FEBS Lett. 2012;586(19):3208–3214. doi: 10.1016/j.febslet.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gavin CF, Rubio MD, Young E, Miller C, Rumbaugh G. Myosin II motor activity in the lateral amygdala is required for fear memory consolidation. Learn. Mem. 2012;19(1):9–14. doi: 10.1101/lm.024042.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moore CC, Lakner AM, Yengo CM, Schrum LW. Nonmuscle myosin II regulates migration but not contraction in rat hepatic stellate cells. World J. Hepatol. 2011;3(7):184–197. doi: 10.4254/wjh.v3.i7.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim JH, Wang A, Conti MA, Adelstein RS. Nonmuscle myosin II is required for internalization of the epidermal growth factor receptor and modulation of downstream signalling. J. .Biol. Chem. 2012;287(33):27345–58. doi: 10.1074/jbc.M111.304824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamada N, Suetsugu N, Wada M, Kadota A. Phototropin-dependent biased relocalization of cp-actin filaments can be induced even when chloroplast movement is inhibited. Plant Signal Behav. 2011;6(11):1651–1653. doi: 10.4161/psb.6.11.17767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Z, Friedman H, Meir S, Belausov E, Philosoph-Hadas S. Actomyosin mediates gravisensing and early transduction events in reoriented cut snapdragon spikes. J. Plant Physiol. 2011;168(11):1176–1183. doi: 10.1016/j.jplph.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 67.Perry CG, Kane DA, Lin CT, et al. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 2011;437(2):215–222. doi: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu X, Sun Z, Foskett A, Trzeciakowski JP, Meininger GA, Muthuchamy M. Cardiomyocyte contractile status is associated with differences in fibronectin and integrin interactions. Am. J. Physiol. Heart Circ. Physiol. 2010;298(6):H2071–2081. doi: 10.1152/ajpheart.01156.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dou Y, Andersson-Lendahl M, Arner A. Structure and function of skeletal muscle in zebrafish early larvae. J. Gen. Physiol. 2008;131(5):445–453. doi: 10.1085/jgp.200809982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thum T, Borlak J. Butanedione monoxime increases the viability and yield of adult cardiomyocytes in primary cultures. Cardiovasc. Toxicol. 2001;1(1):61–72. doi: 10.1385/ct:1:1:61. [DOI] [PubMed] [Google Scholar]
- 71.Kabaeva Z, Zhao M, Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am. J. Physiol. Heart Circ. Physiol. 2008;294(4):H1667–H1674. doi: 10.1152/ajpheart.01144.2007. [DOI] [PubMed] [Google Scholar]
- 72.Matsui Y, Nakayama Y, Okamoto M, Fukumoto Y, Yamaguchi N. Enrichment of cell populations in metaphase, anaphase, and telophase by synchronization using nocodazole and blebbistatin: a novel method suitable for examining dynamic changes in proteins during mitotic progression. Eur. J. Cell Biol. 2012;91(5):413–419. doi: 10.1016/j.ejcb.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 73.Fedorov VV, Lozinsky IT, Sosunov EA, et al. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4(5):619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 74.Li D, Nattel S. Pharmacological elimination of motion artifacts during optical imaging of cardiac tissues: is blebbistatin the answer? Heart Rhythm. 2007;4(5):627–628. doi: 10.1016/j.hrthm.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 75.Si J, Ge Y, Zhuang S, Gong R. Inhibiting nonmuscle myosin II impedes inflammatory infiltration and ameliorates progressive renal disease. Lab. Invest. 2010;90(3):448–458. doi: 10.1038/labinvest.2009.142. [DOI] [PubMed] [Google Scholar]
- 76.Mikulich A, Kavaliauskiene S, Juzenas P. Blebbistatin, a myosin inhibitor, is phototoxic to human cancer cells under exposure to blue light. Biochim. Biophys. Acta. 2012;1820(7):870–877. doi: 10.1016/j.bbagen.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 77.Derycke L, Stove C, Vercoutter-Edouart AS, et al. The role of non-muscle myosin IIA in aggregation and invasion of human MCF-7 breast cancer cells. Int. J. Dev. Biol. 2011;55(7-9):835–840. doi: 10.1387/ijdb.113336ld. [DOI] [PubMed] [Google Scholar]
- 78.Duxbury MS, Ashley SW, Whang EE. Inhibition of pancreatic adenocarcinoma cellular invasiveness by blebbistatin: a novel myosin II inhibitor. Biochem. Biophys. Res. Commun. 2004;313(4):992–997. doi: 10.1016/j.bbrc.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 79.Epstein DL, Rowlette LL, Roberts BC. Acto-myosin drug effects and aqueous outflow function. Invest. Ophthalmol. Vis. Sci. 1999;40(1):74–81. [PubMed] [Google Scholar]
- 80.Zhang M, Rao PV. Blebbistatin, a novel inhibitor of myosin II ATPase activity, increases aqueous humor outflow facility in perfused enucleated porcine eyes. Invest. Ophthalmol. Vis. Sci. 2005;46(11):4130–4138. doi: 10.1167/iovs.05-0164. [DOI] [PubMed] [Google Scholar]
- 81.Wetzel DM, Schmidt J, Kuhlenschmidt MS, Dubey JP, Sibley LD. Gliding motility leads to active cellular invasion by Cryptosporidium parvum sporozoites. Infect. Immun. 2005;73(9):5379–5387. doi: 10.1128/IAI.73.9.5379-5387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dobrowolski JM, Carruthers VB, Sibley LD. Participation of myosin in gliding motility and host cell invasion by Toxoplasma gondii. Mol. Microbiol. 1997;26(1):163–173. doi: 10.1046/j.1365-2958.1997.5671913.x. [DOI] [PubMed] [Google Scholar]
- 83.Thum T, Borlak J. Reprogramming of gene expression in cultured cardiomyocytes and in explanted hearts by the myosin ATPase inhibitor Butanedione monoxime. Transplantation. 2001;71(4):543–552. doi: 10.1097/00007890-200102270-00010. [DOI] [PubMed] [Google Scholar]
- 84.Lucas-Lopez C, Allingham JS, Lebl T, et al. The small molecule tool (S)-(-)-blebbistatin: novel insights of relevance to myosin inhibitor design. Org. Biomol. Chem. 2008;6(12):2076–2084. doi: 10.1039/b801223g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lawson CPAT, Slawin AMZ, Westwood JN. Application of the copper catalysed N-arylation of amidines in the synthesis of analogues of the chemical tool, blebbistatin. Chem. Commun. (Camb.) 2011;47(3):1057–1059. doi: 10.1039/c0cc03624b. [DOI] [PubMed] [Google Scholar]
- 86.Pashkova N, Jin Y, Ramaswamy S, Weisman LS. Structural basis for myosin V discrimination between distinct cargoes. EMBO J. 2006;25(4):693–700. doi: 10.1038/sj.emboj.7600965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu C, Feng W, Wei Z. Myosin VI undergoes cargo-mediated dimerization. Cell. 2009;138(3):537–548. doi: 10.1016/j.cell.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 88.Eves PT, Jin Y, Brunner M, Weisman LS. Overlap of cargo binding sites on myosin V coordinates the inheritance of diverse cargoes. J. Cell Biol. 2012;198(1):69–85. doi: 10.1083/jcb.201201024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zorn JA, Wells JA. Turning enzymes ON with small molecules. Nat. Chem. Biol. 2010;6(3):179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- 90.Galloway WR, Hodgkinson JT, Bowden S, Welch M, Spring DR. Applications of small molecule activators and inhibitors of quorum sensing in Gram-negative bacteria. Trends Microbiol. 2012;20(9):449–458. doi: 10.1016/j.tim.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 91.West JD, Wang Y, Morano KA. Small molecule activators of the heat shock response: chemical properties, molecular targets, and therapeutic promise. Chem. Res. Toxicol. 2012;25(10):2036–2053. doi: 10.1021/tx300264x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Malik FI, Hartman JJ, Elias KA. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331(6023):1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]