

Abstract
The G protein-coupled receptor (GPCR) family is among the most druggable families in the human proteome. GPCRs are involved in most physiological processes, and our ability to modulate their activity is a hallmark of modern pharmacology. The means by which the activity of GPCRs can be modulated have been expanded by emerging data and concepts in pharmacology, which has created new strategies for their control. These new approaches will lead to the generation of more potent, selective, and efficient pharmaceutics, while reducing inappropriate actions and adverse effects. Herein, we review and comment on some recent advances in chemical and genetic approaches to the profiling of GPCR function, as well as the validation of orphan GPCRs as potential therapeutic targets using engineered receptors.
Keywords: G-protein coupled receptors (GPCRs), Drug screening, Drug discovery, DREADDs, RASSLs
Introduction
An important hallmark of all living organisms is their ability to react and respond to their environment. From unicellular to complex multicellular organisms, much of this sensory capability is achieved through cell membrane embedded receptors that can transduce extracellular signals into the intracellular milieu [1]. The nature of the extracellular signals is highly divergent, and includes photons (light), a wide range of small molecules, as well as larger molecules including peptides and proteins. Many of these stimuli exert their effects through a distinct family of transmembrane receptors called G protein-coupled receptors (GPCRs), which are characterized by having seven transmembrane helices linked by three extracellular and three intracellular loops. In vitro and in vivo pharmacological studies have consistently identified GPCRs as the most important class of druggable targets in the human genome; this is supported by the fact that nearly 50% of prescribed therapeutics act through modulation of GPCRs [2,3]. This high degree of druggability is derived both from the nature of GPCR ligands as well as the intrinsic capability of GPCRs to be modulated by small molecules at the canonical ligand-binding pocket (the orthosteric site) as well as additional allosteric sites [3]. Recent improvements in our capacity to generate high quality crystal structures of GPCRs portend important advances in drug discovery [3,4]. However, as in all drug development initiatives, drug discovery using these new insights is not guaranteed to be clinically successful. Most approved drugs have a broad range of off-target activity, which can account for the effectiveness of a medication through desirable effects, or can result in deleterious side effects [5]. Indeed, “polypharmacology” is now well documented [2,6]. In order to achieve the same clinical efficacy of known polypharmacological compounds with new drugs, it will be important to understand more completely their activities, especially with respect to identifying the specific target(s) essential for their effectiveness. This, combined with a more complete understanding of off-target activities, will allow for development of better therapeutics with lower side-effect profiles [7].
In order to achieve these goals, a wide range of pharmacological tools is required for designing and screening of novel drugs, as well as to more fully understand the pharmacology of already-prescribed drugs (e.g. see [5] and [6]). These tools will also lead to a more complete understanding of the receptors involved in drug actions, as well as the pairing of receptor activity with physiological and pathophysiological conditions. Herein, we will review some recent advances in chemical and genetic approaches to the study of GPCR function and activity.
From chemistry to pharmacology
Productive collaborations between chemists and biologists are now essential for successfully interrogating GPCRs using chemical-based approaches. Through the use of already-known drugs as templates, additional chemistry can help to identify specific essential determinants within the molecule for the optimization of desired activities. Although selectivity and efficacy have been the Holy Grail of pharmacologists for decades, it is now becoming apparent that an additional concept may also become important in future drug design. This concept, variously referred to as agonist trafficking, biased agonism, differential engagement, protean agonism or functional selectivity [8,9] involves different signaling outputs triggered by the same receptor, but using different modulators. Design of new drugs that preferentially activate a single specific signal transduction pathway has the potential to increase clinical effectiveness while decreasing undesirable activities or tolerance caused, for example, by receptor desensitization [9].
An example of the potential for advancing our understanding of the functional selectivity of drugs has resulted from the high resolution structure of a biased compound (ergotamine, Erg) bound to the 5-HT2B serotonin receptor [10]. Erg has full agonist activity at the arrestin recruitment pathway that is 100-fold more potent than its activity at Gq, while other compounds show considerably less bias (Fig. 1). The crystal structure of Erg bound to the 5-HT1B receptor, at which it acts as an unbiased full agonist, was also solved [11]. Comparison of these two crystal structures revealed specific molecular determinants responsible for the biased activity at the 5-HT2B receptor [10,11]. These studies represent the first description of the structural mechanism underlying the functional selectivity of a GPCR ligand. It can be anticipated that similar studies will lead to the development of more potent and selective GPCR ligands, with fewer side effects, even while taking into account the minimal chemical properties required for bioavailability and appropriate in vivo pharmacokinetics [4,6].
Figure 1. Structure-activity comparison of compounds having biased signaling properties at the 5-HT2B receptor.

(A) Structure of different agonists having increasingly arrestin-biased activity (from left to right) used in the studies published by Wacker et al. and Wang et al. [10,11]. The green box highlights the common core shared by these agonists, including the endogenous ligand 5-HT (serotonin). The efficacy at recruiting arrestin and activating G protein signaling (Gq for 5HT2B) were measured and the bias factor was quantified. (B) Concentration-response curve of Ergotamine (Erg), which showed the most highly arrestin-biased activity at 5HT2B while being unbiased at 5HT1B. The black curve (squares) shows Gq potency measured by calcium release whereas the red curve illustrates arrestin recruitment. The arrow shows the leftward shift in potency (increased potency) of Erg at arrestin recruitment compared to Gq signaling. (C) Schematic representation of biased signaling induced by Erg at 5HT2B. The crystal structure revealed a structural change at helices 3 and 7 induced by Erg at the 5HT2B receptor generating an arrestin-polarized signaling.
The goal of chemistry-based pharmacology is to find, develop, and optimize compounds for particular desired outcomes, either as probes or drugs. With the maturation of concepts like functional selectivity, chemistry-based pharmacology will be essential for the discovery and refinement of drugs based on these newer concepts of drug action. In recent years, efforts have been made to develop a more mathematical approach to quantify functional selectivity, for example by comparing two or more assays of drug potency or pathways of drug action (review by Kenakin [9]). In its most usual implementation, we have compared canonical G protein signaling (cAMP or Ca2+) with β-arrestin recruitment to study the biased activity of a compound. However, since GPCR signaling, and the outcome of such signaling, may also be dependent on many other pathways, measurement of biased activity at these additional pathways will need to be considered for a full understanding of a drug's actions. Some caution in interpretation is warranted, however, since it is quite conceivable that compounds that exhibit bias in vitro might have considerably different behavior in vivo. It is also possible that biased compounds use a variety of mechanisms to achieve their signaling bias.
In vitro pharmacology generates much information regarding selectivity, potency, and efficacy—all of which are concepts essential to drug discovery. However, validation of the therapeutic potential of new compounds requires additional studies in vivo, including efficacy, toxicity and kinetic properties. In vivo studies can also provide novel means to decipher GPCR signaling and to validate the potential therapeutic properties of a targeted receptor. It is now feasible to do some of these studies even while lacking a known natural ligand (e.g., for study of orphan receptors), or without potent small molecule agonists. Advances such as these have initiated a “reverse pharmacology” approach, i.e., the study of the therapeutic effect of a target before compound screening.
When chemistry meets genetics
Signals triggered by the activation of GPCRs are typically not simple “input-single output” signals, but are rather an inter-regulated complex of signaling pathways that all lead to a subset of possible physiological responses. This network of pathways, and their complex effects, are dependent on both specific cellular micro-environments [12], and the distribution of particular cell types within tissues [13]. It is well established that the same receptor can elicit different responses in different environments, especially when considering the differences between in vivo and in vitro conditions [14,15]. The requirement for understanding of a receptor's function and regulation in the context of its normal environment has accelerated the development of tools that allow this type of study. Additionally, in response to the need to selectively activate only the targeted receptor, and not others, as well as to study receptors for which no small molecule mediators are known, a variety of engineered receptors have been developed [16,17].
The first generation of engineered receptors was dubbed `genetically engineered receptors' and could respond to otherwise inert compounds while lacking responsiveness to all known endogenous ligands [18]. The second generation was called RASSLs (Receptors Activated Solely by Synthetic Ligands) and was based on the κ-opioid receptor [17,19]. The first two generations of RASSLs had either low affinity towards their synthetic ligands [18], or had residual affinity for endogenous ligands [19], or the novel ligands had unknown pharmacokinetic properties limiting their use [18]. Although both technologies represented major advances, due to their limitations we designed a third generation of engineered receptors we called DREADDs (Designer Receptor Exclusively Activated by Designer Drugs).
The first DREADD used the M3-ACh muscarinic receptor as its template. Mutation of only two residues (Y149C/A239G) generated a receptor with essentially no affinity for the endogenous acetylcholine ligand (ACh), but responding to clozapine-N-oxide (CNO) [20]. CNO is an inert metabolite of clozapine, an atypical antipsychotic drug with known pharmacokinetic, pharmacodynamic and biodistribution properties [21]. From this initial effort, a full panel of DREADDs was developed covering all three of the major G protein signaling pathways – Gq (M3-DREADD), Gi (M4-DREADD) and Gs (Gs-DREADD), all based on muscarinic receptors [20,22].
To generate the Gs-DREADD, Guettier et al. [22] swapped the intracellular loops 2 and 3 (il2 and il3) of the original M3-DREADD receptor for those of the β1-adrenergic receptor, which changed its G protein coupling from Gq to Gs. This chimeric receptor responds to, and has affinity for, CNO similarly to its M3-DREADD parent, but signals through the Gs pathway. This approach serves as a “proof of concept” that opens the possibility that, by swapping the intracellular domains of a receptor of interest into a DREADD backbone, receptors could be designed that respond to CNO while retaining their native cellular signaling properties. This could be of particular interest when it is suspected that the target receptor signals by novel pathways as yet not well studied or characterized. Alternative domain-swapping strategies, for example by keeping the transmembrane portions of the receptor of interest, might be useful for studies of dimerization (such as for the CXCR4 receptor [23]), as well as of interactions with membrane cholesterol, such as for the β2-adrenergic receptor [24] and probably other GPCRs [25]. Since most GPCRs have increased intrinsic activity when overexpressed, the choice of the muscarinic receptor template, which has very low constitutive activity, was critical to detection of receptor activation by CNO. This was particularly important since it was found that, in some cases, overexpression of the previous generations of RASSLs induced a phenotype reflective of high constitutive activity [17,26]. Since constitutive activity can be conferred by the membrane-proximal amino acids at the end of il3 (near helix 6) [27], it should be possible by mutagenesis or domain swapping to reduce constitutive activity of the targeted engineered receptor. Thus, subsequent generations of RASSLs and DREADDs can be engineered to have all the properties required for the successful study of receptor signaling and regulation in natural cell and tissue environments.
Potential uses of DREADDs to profile GPCR function
By expressing DREADDs (or chimeric DREADDs) in the same environment as a targeted receptor of interest, the phenotype induced by CNO could then be used to measure adverse and/or beneficial effects of activation of signaling in a particular cell type or tissue in mouse models of disease, and thus could be used to validate the role and therapeutic potential of activation of receptors naturally expressed in such cells or tissues (see ref [22] for example). Additionally, by choosing different DREADDs, it would be possible to identify which signaling pathways need to be activated to overcome a phenotype, or to cause a phenotype [22].
DREADDs may also be of use in the study of functional selectivity. For example, by mutating the receptor in such a way that it will only stimulate a desired signaling pathway, it will be possible to create a biased system in which its activation, in a desired environment, will provide information about the potential of a biased compound to treat a disease, or to reduce the adverse effects of a compound (Fig. 2). For example, Nakajima and Wess have recently created a DREADD that only activates arrestin signaling pathways [28]. In a more general sense, identification of the minimal properties of a drug that are required for a beneficial outcome will better inform future drug development, as well as increase the druggability of many GPCRs. Further study of compounds already available, and indeed already prescribed, will identify the most beneficial signaling profiles, and will allow for the modification of these existing molecules to direct their properties more precisely towards their desired effect.
Figure 2. Schematic representation showing how DREADDs receptors could be used in a chemical-genetic approach to study GPCR function as well as in drug discovery.
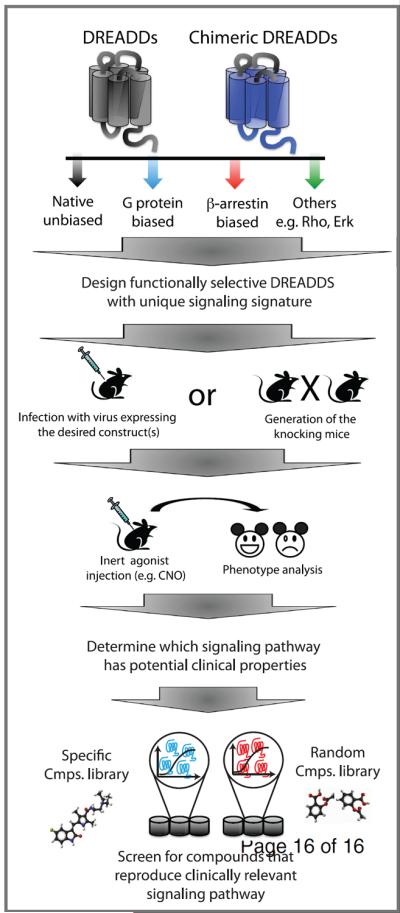
The engineered receptor or chimeric receptor can be mutated and so that there is a loss-of-function toward one or more specific pathways. This biased DREADDs is then incorporated into viral particles and injected into mice, or used to generate recombinant or knockin-mice; in both scenarios, expression of the receptor driven by a tissue- or cell-specific promoter, or the natural promoter for the targeted GPCR. The use of a natural promoter allows a more physiological expression level and spatiotemporal regulation. Once the receptor is expressed, the mice are injected with the inert agonist (e.g CNO) and specific outcomes and phenotypes are analyzed. Specific compound libraries (i.e. compounds known to interact with a targeted receptor) or random libraries are used to screen for drugs stimulating the signaling pathway previously identified by the DREADD that may have potentially clinically useful properties.
Conclusion
Our ability to design functionally selective compounds will allow us to fine-tune the outcomes of GPCR signaling to increase drug potency and efficacy while decreasing undesired, potentially harmful, side effects. These novel technologies have the potential to help close the current gap that exists between in vitro and in vivo findings. The generation of chimeric DREADDs using orphan GPCRs as partial template is likely to increase the number of GPCR druggable targets. The latter reverse pharmacology approaches, especially when combined with more and better GPCR structural information, can be predicted to accelerate discovery and development of new GPCR drugs for a variety of conditions.
Highlights
Chemistry-based pharmacology helps find, develop and optimize new compounds.
Functionally selective drugs reveal novel strategies for GPCR remote control.
Genetics pharmacology can validate the therapeutic potential of new compounds.
Engineered receptors provide a novel approach to decipher GPCR function in vivo.
Advances in chemical and genetic pharmacology will accelerate new drug development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- [1].King N, Hittinger CT, Carroll SB. Evolution of key cell signaling and adhesion protein families predates animal origins. Science. 2003;301:361–363. doi: 10.1126/science.1083853. [DOI] [PubMed] [Google Scholar]
- [2].Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- [3].Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- [4].Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011;51:117–144. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- [8].Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- [9].Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- ** [10].Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808..** Wacker D et al. have generated a high resolution crystal structure of the 5HT2B receptor bound to the biaised agonist Ergotamine. By comparing with the structure of the 5HT1B, they highlight molecular determinant regulating bias activity of the 5HT2B by Ergotamine.
- ** [11].Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, et al. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807..** Wang C et al. have generated a high resolution crystal structure of the 5HT1B receptor bound to Ergoatamine and dihydroergotamine. With the structure of the 5HT2B, they reveal important determinat regulating ligand specificity.
- [12].Ostrom RS, Insel PA. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol. 2004;143:235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hakak Y, Shrestha D, Goegel MC, Behan DP, Chalmers DT. Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett. 2003;550:11–17. doi: 10.1016/s0014-5793(03)00762-2. [DOI] [PubMed] [Google Scholar]
- [14].Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tobin AB, Butcher AJ, Kong KC. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nichols CD, Roth BL. Engineered G-protein Coupled Receptors are Powerful Tools to Investigate Biological Processes and Behaviors. Front Mol Neurosci. 2009;2:16. doi: 10.3389/neuro.02.016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Strader CD, Gaffney T, Sugg EE, Candelore MR, Keys R, Patchett AA, Dixon RA. Allele-specific activation of genetically engineered receptors. J Biol Chem. 1991;266:5–8. [PubMed] [Google Scholar]
- [19].Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Controlling signaling with a specifically designed Gi-coupled receptor. Proc Natl Acad Sci U S A. 1998;95:352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bender D, Holschbach M, Stocklin G. Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl Med Biol. 1994;21:921–925. doi: 10.1016/0969-8051(94)90080-9. [DOI] [PubMed] [Google Scholar]
- [22].Guettier JM, Gautam D, Scarselli M, Ruiz de Azua I, Li JH, Rosemond E, Ma X, Gonzalez FJ, Armbruster BN, Lu H, et al. A chemical-genetic approach to study G protein regulation of beta cell function in vivo. Proc Natl Acad Sci U S A. 2009;106:19197–19202. doi: 10.1073/pnas.0906593106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * [25].Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923..* Katritch V et al. review advance in GPCR structural biology gained from recent high resolution crystal structures.
- [26].Pei Y, Dong S, Roth BL. Generation of designer receptors exclusively activated by designer drugs (DREADDs) using directed molecular evolution. Curr Protoc Neurosci. 2010;Chapter 4(Unit 4):33. doi: 10.1002/0471142301.ns0433s50. [DOI] [PubMed] [Google Scholar]
- [27].Kjelsberg MA, Cotecchia S, Ostrowski J, Caron MG, Lefkowitz RJ. Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J Biol Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- * [28].Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol. 2012;82:575–582. doi: 10.1124/mol.112.080358..* Nakajima K et al. described the development of an arrestin-biased M3-DREADDs receptor. This biased receptor no longer activate G protein and reprensent an inovative tool to study arrestin signaling.