

Abstract
A principal obstacle to completing maps and analyses of the human genome involves the genome’s “inaccessible” regions: sequences (often euchromatic and containing genes) that are isolated from the rest of the euchromatic genome by heterochromatin and other repeat-rich sequence. We describe a way to localize these sequences by using ancestry linkage disequilibrium in populations that derive ancestry from at least three continents, as is the case for Latinos. We used this approach to map the genomic locations of almost 20 megabases of sequence unlocalized or missing from the current human genome reference (NCBI Genome GRCh37)—a substantial fraction of the human genome’s remaining unmapped sequence. We show that the genomic locations of most sequences that originated from fosmids and larger clones can be admixture mapped in this way, by using publicly available whole-genome sequence data. Genome assembly efforts and future builds of the human genome reference will be strongly informed by this localization of genes and other euchromatic sequences that are embedded within highly repetitive pericentromeric regions.
Introduction
Studies of human genetic variation and genome biology, increasingly based on next-generation sequencing, utilize physical maps of the human genome’s sequence to interpret sequence data. The scope of such studies is therefore limited to those regions considered “accessible” in the maps of the human genome.
Approximately 200 Mbp of the human genome, mainly from the centromeres and the short arms of the acrocentric chromosomes, are missing from the human reference genome; a further 30 Mbp fall within ∼300 interstitial gaps mostly involving regions that could not be reliably cloned or assembled.1–3 Most of the interstitial 30 Mbp, and an unknown fraction of the other 200 Mbp, consists of complex euchromatic sequence. Sequence reads arising from these “missing pieces” are currently discarded or misaligned to paralogous sequences present in the human reference genome.4 We estimate that in whole-genome studies using short next-generation sequencing reads, ∼17 Mbp of the NCBI Genome GRCh37 human reference genome receives an excess of aligned reads that in fact arise from these missing pieces. To help address this problem in its own analyses, the 1000 Genomes Project Consortium now supplements the reference human genome, for the purpose of alignment, with a set of additional sequences, termed “decoy sequences,” consisting of ∼35.4 Mbp of partially assembled sequence that is missing from the human genome reference but is available from other sources (including GenBank,5 the HuRef alternate genome assembly,6 and the ALLPATH-LG assembly of NA128787).
There is ample reason to believe that unlocalized regions of the human genome contain biologically significant genes and other sequences. Genes missing from the human reference genome are often transcribed to mature mRNA,8 even when they reside within repeat-rich pericentromeric regions of the genome. A significant Mendelian neurological disease, thyrotoxic hypokalemic periodic paralysis (TTPP2 [MIM 613239]), was also shown to arise from mutations in KCNJ18 (MIM 613236),9 a pericentromeric gene missing from maps of the human genome.
We recently showed8 how long-range linkage disequilibrium information resulting from admixture in African Americans can be used to map the genomic location of assembled but unlocalized sequences that are missing from the human reference genome. We focused in that study on African Americans, the admixed population for which the most available genome-wide data were available, but genetic data from other admixed populations could also in principle be used for the same purpose. Sequence data from 242 Latino samples recently became publicly available from the 1000 Genomes Project Phase 1.10
Here we extend our admixture mapping approach to the three-way admixture present in Latino genomes. Surprisingly, we find that Latino genomes are particularly powerful for admixture mapping the human genome’s missing pieces. Notably, whereas African American genomes have an 80% ± 12% component of African descent,11,12 genomes from Latino samples have more evenly distributed amounts of ancestry,13–17 which could translate to increased power for mapping through admixture linkage disequilibrium. We show that low-coverage whole-genome sequence from even a limited number of Latino genomes (n = 242) already makes this strategy extremely effective, allowing the localization of most sequences originated from fosmids and larger clones. As the number of sequenced Latino genomes continues to increase, we expect admixture mapping to become increasingly valuable in helping to complete maps of the human genome.
Material and Methods
Mapping by Admixture Linkage Disequilibrium
We generalized our mapping method, first described in Genovese et al.,8 to populations that derive significant amounts of ancestry from three or more ancestral populations. We map an unlocalized scaffold by mapping a polymorphic marker known through sequence alignment to localize within the scaffold.
We model the observed genotype for the polymorphic marker as a function of the local ancestry deconvolution and the ancestral frequencies of the alternate allele that are estimated by maximizing this likelihood after marginalizing over the local ancestry deconvolution, which is modeled as a function of the global ancestry proportions of the sample.
We then compute the likelihood of the genotype for a biallelic marker of unknown localization marginalizing over the local ancestry deconvolution and we compare this to the genotype likelihood assuming that the marker localizes at a locus i around the genome for which the local ancestry is instead known.
Define the following variables:
-
•
p = (pE,pA,pN) as the alternate allele frequencies of the unlocalized marker in the ancestral European, West African, and Native American populations
-
•
j an admixed sample
-
•
gj∈{0,1,2} the genotype of the unlocalized marker for sample j
-
•
Ω = {(2,0,0), (1,1,0), (0,2,0), (1,0,1), (0,1,1), (0,0,2)} the set of all possible values for the local ancestry deconvolution, where each number corresponds to the number of haplotypes of European, West African, and Native American descent, respectively
-
•
i a locus around the genome
-
•
lij∈Ω the local ancestry deconvolution for sample j at locus i
-
•
P(lj = ω) a prior for the likelihood of the local ancestry deconvolution being equal to ω for sample j at a random or unknown locus. This is estimated separately for each sample by averaging the local ancestry calls across the genome.
The likelihood P(gj|lij,p) of observing a genotype gj given the local ancestry lij is given in Table 1. This is then compared to the marginal likelihood
and the odds ratio is computed to measure the evidence that the biallelic marker actually does indeed localize near locus i across the genome. Notice that P(lj = ω) is not just a function of the global ancestry proportions of sample j; because of population structure we do not assume that the parents had the same ancestry proportions, as could be the case for the offspring of an African American and a European American (for whom P(l = (0,2,0)) = 0), and therefore it is best estimated from the local ancestry deconvolution across the genome.
Table 1.
Genotype Likelihoods
| P(g|l,p = (pE,pA,pN)) | g = 0 | g = 1 | g = 2 |
|---|---|---|---|
| l = (2,0,0) | (1 − pE)2 | 2pE(1 − pE) | pE2 |
| l = (1,1,0) | (1 − pE)(1 − pA) | pE(1 − pA)+(1 − pE)pA | pEpA |
| l = (0,2,0) | (1 − pA)2 | 2pA(1 − pA) | pA2 |
| l = (1,0,1) | (1 − pE)(1 − pN) | pE(1 − pN)+(1 − pE)pN | pEpN |
| l = (0,1,1) | (1 − pA)(1 − pN) | pA(1 − pN)+(1 − pA)pN | pApN |
| l = (0,0,2) | (1 − pN)2 | 2pN(1 − pN) | pN2 |
Probabilities of observing genotype g for a biallelic marker with known ancestral allele frequencies p = (pE,pA,pN) and known local ancestry l at the marker.
To estimate the values for the ancestral allele frequencies p = (pE,pA,pN), we use a maximum-likelihood estimation approach of the marginal probabilities, by computing
For this computation, available nonadmixed samples that can act as proxies for the ancestral populations (e.g., CEU for Europeans and YRI for West Africans) can be used to improve the estimates of the ancestral allele frequencies. We empirically observed a limited but significant increase in power by doing so (despite this, CEU and YRI should not be considered optimal proxies for the European and African ancestral populations of Latinos). However, the estimates from the admixed genomes alone are still informative, and proxies for the ancestral populations are not required (in particular, Native American genomes are not needed).
For a single locus i, we combine the evidence across all samples for which the local ancestry has been estimated at the locus into a LOD score (logarithm base 10 of odds) as
For a given biallelic marker, we can then compute this LOD score for a dense and uniform set of loci i across the genome (say ∼10,000 in practice) and refine the best hits by searching near loci with the highest LOD scores. Notice that loci with missing local ancestry calls or with higher error rates in the calls will inevitably be less likely to achieve significant LOD scores.
In practice, because genotypes cannot be reliably inferred from low-pass sequencing data, we generalize this model to genotype likelihoods by marginalizing over the unknown genotypes, i.e., by replacing the probability of observing a given genotype with a weighted average of the probabilities for all three possible genotypes with the weights corresponding to the genotype likelihoods
The genotype likelihoods P(gj) are estimated from the sequence read data and are usually readily provided by genotyping software.
Very large LOD scores across the genome will be unlikely to be achieved by chance, though how large and unlikely is somewhat a function of the correlation structure in the local ancestry (which is mainly due to the number of generations since the admixing event of the ancestral populations), the level of polymorphism of the biallelic marker tested (which is related to demographics of the ancestral populations before the admixture), and the accuracy of the local ancestry calls. We consider LOD scores indicative of a correct mapping when those scores were larger than or equal to 6.
For each biallelic marker called by the 1000 Genomes Project, we computed the best LOD scores across all autosomes other than the autosome where the marker is localized by using the available local ancestry calls. We identified 666 SNPs with LOD scores greater than 6 across ∼2.6 Gbp of sequence from the autosomes, leading to an upper estimate of one SNP incorrectly mapped every ∼4 Mbp when using the available local ancestry deconvolution for the 242 Latino genomes from the 1000 Genomes Project. Assuming that SNPs across the genome behave, for the purpose of admixture mapping, in similar ways to SNPs from unplaced sequence, this suggests a low false-positive rate of less than ∼10 missmaped markers among the mappings for SNPs from the ∼41.5 Mbp of unplaced sequence analyzed here.
Coverage Analysis
To estimate the depth of coverage across GRCh37, we used low-coverage Illumina sequencing data from 820 samples from 1000 Genomes Project Phase 1. We first constructed a map of uniquely alignable positions on GRCh37 by aligning all k-mers of length 36 (the smallest read length in the sequencing data set) back to the reference to determine which positions have unique alignments. We then divided the reference into overlapping windows where each window contains 10,000 uniquely aligning positions and where adjacent windows overlap by 5,000 uniquely aligning positions.
For each window, we measured sequencing read depth and corrected for differential sequencing depth because of GC bias separately for each library. GC-bias correction factors were determined by measuring differences in read depth stratified by GC fraction in 400 bp sliding windows across 588 Mbp of the genome that have no annotated segmental duplications, repeats, or copy-number variants from the Database of Genomic Variants18 (DGV) in the UCSC genome browser.19 We then estimated each sample’s copy number from the GC-normalized sequencing read depth via a Gaussian mixture model20 extended to allow modeling of copy number greater than two.
Copy-number likelihoods were assigned to each sample for each possible copy-number genotype. These were then converted into diploid biallelic genotype likelihoods for the purpose of admixture mapping, by selecting the three modal copy-number likelihoods.
Decoy Sequences
Decoy sequences are composed of contiguous sequences from GRCh37.p4 patches, completely sequenced bacterial artificial chromosomes (BACs) and fosmids from GenBank, HuRef contigs, and NA12878 contigs with a length of at least 1,000 bp and which show less than 99% overall identity with paralogous sequence in the GRCh37 reference genome for stretches at least 20 kbp long or less than 95% for stretches at least 500 bp long. The whole resource consists of ∼35.4 Mbp of sequence (N50 = 22.9 kbp). Based on RepeatMasker-3.3.0 (RepBase 20110419) analysis, ∼50% of these sequences consist of satellite or simple repeats, and 23% consist of interspersed repeats. The human genome reference integrating GRCh37 and the decoy sequences is termed hs37d5.
Local Ancestry Deconvolution
Local ancestry deconvolution for African American (ASW, n = 61), Mexican (MXL, n = 66), Puerto Rican (PUR, n = 55), and Colombian (CLM, n = 60) samples from the 1000 Genomes Project Phase 1 (Figure 1) was computed from Illumina Omin2.5 genotype data and low-pass sequencing SNP calls via a consensus scheme from multiple algorithms: LAMP-LD,21 HAPMIX,22 RFMIX, and MULTIMIX.23 Ancestry calls are available as part of the 1000 Genomes Project Phase 1 release.
Figure 1.
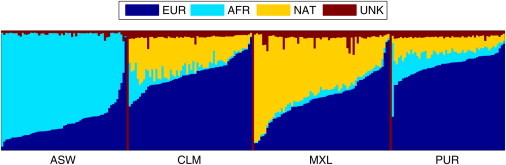
Ancestry Proportions for Admixed Samples from the 1000 Genomes Project Phase 1
Abbreviations for ancestral populations are as follows: Eur, European; AFR, West African; NAT, Native American; UNK, Unknown. Abbreviations from the 1000 Genomes Project: ASW, African American; CLM, Colombian; MXL, Mexican; PUR, Puerto Rican.
SNP Calling
To identify SNPs over unlocalized hs37d5 contigs, we ran the Genome Analysis Toolkit24 (GATK), with default settings for the UnifiedGenotyper walker over aligned sequence data for European (CEU, n = 96), Yoruba (YRI, n = 88), African American (ASW, n = 61), Mexican (MXL, n = 66), Puerto Rican (PUR, n = 55), and Colombian (CLM, n = 60) samples.
To genotype SNPs over regions with excess coverage from 1000 Genomes Project Phase 1 alignments (that is, alignments by GRCh37 rather than hs37d5), presumed to have duplicated paralogous sequences, we run the UnifiedGenotyper walker over these regions with default settings other than the additional option “-ploidy 4,” which instructs the GATK to treat the reads at a single locus as if coming from four different haplotypes. We then used a custom python script to identify the three main modes for the tetraploid genotype likelihoods obtained with the UnifiedGenotyper walker, and subsequently recalibrate these to obtain standard diploid genotype likelihoods. This simple scheme significantly improves the genotype likelihoods within regions receiving excess coverage and better recapitulates the real genotype, which in turn leads to increased power for admixture mapping.
Validation of Ancestry Mappings by Alignments to Optical Restriction Maps
Genome-wide consensus restriction maps are high-resolution restriction maps obtained by combining restriction maps of many long, individual DNA molecules generated through optical mapping.25,26 We used available restriction maps for three cell lines (GM15510, GM10860, GM18994) with the SwaI restriction enzyme.27 To validate mapping through admixture, we attempted to match in silico generated restriction maps of unlocalized hs37d5 BAC clone sequences to these optical restriction maps. To perform this step in an automatized fashion, we used the Scaffolding using Optical Map Alignment (SOMA) software.28
Software for Admixture Mapping
To compute LOD scores from genotype likelihoods computed with GATK, we developed a software tool named LATOOLS that takes as input a file in variant call format29 (VCF) containing genotype likelihoods and a single file containing the local ancestry deconvolution for a group of admixed samples in the extended bedgraph format outputted by the unionbedg subcommand of the bedtools suite30 (Figure 2).
Figure 2.
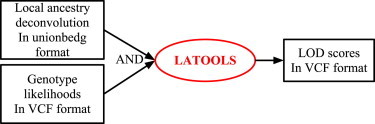
Admixture Mapping Flowchart for the LATOOLS Software Tool Described in This Study
Local ancestry deconvolution for multiple samples can be input in LATOOLS in unionbedg format, which can be easily generated with the bedtools suite starting from single sample deconvolutions in BedGraph format. Genotype likelihoods can be input from a VCF file, without further processing if directly generated with GATK.
The source code for the LATOOLS program is freely available and written in a combination of C and Python, using the PyVCF library, a flexible module to parse and output VCF files.
Results
Our approach utilizes the principle that Latino genomes are a mosaic of genomic segments derived from ancestors from three continents: Europeans, Native Americans, and West Africans. We first developed a statistical approach to estimate the likelihood that genotypes for a given marker are observed from a combination of local ancestry backgrounds permissible in three-way admixed genomes, such as Latino genomes. We then estimated this likelihood for different combinations of local ancestries previously estimated across the genome. By using data from 242 Latino genomes from the 1000 Genomes Project Phase 1, we identified loci whose estimated local ancestry explained the observed genotypes much better than chance, and we connected each marker to the genomic location at which it resides. We were thus able to infer the approximate genomic location of the previously unlocalized sequence from which the marker came.
Across all ∼41.5 Mbp of unlocalized human genome sequence included in the hs37d5 reference (the human genome reference integrating GRCh37 and the decoy sequences), we were able to localize 3,888 SNPs through admixture mapping with a LOD score greater than or equal to 6 (Table 2 and Table S1 available online); these SNPs arose from 569 distinct scaffolds (Table S2) spanning a total of ∼19.1 Mbp of sequence.
Table 2.
Statistics for Decoy Sequences Localized through Admixture Mapping
| Scaffold Type | SNPs LOD ≥ 6 | Mapped Scaffolds | Total Scaffolds | Mapped bp | Total bp |
|---|---|---|---|---|---|
| GRCh37 unlocalized | 255 | 22 | 59 | 2,798,503 | 6,110,758 |
| GRCh37.p4 patches | 67 | 3 | 6 | 186,895 | 222,135 |
| BAC and fosmids | 714 | 135 | 652 | 4,713,464 | 11,833,029 |
| HuRef (placed) | 982 | 94 | 310 | 5,263,708 | 7,417,706 |
| HuRef (unplaced) | 1,565 | 184 | 1,213 | 5,470,985 | 11,892,765 |
| NA12878 | 305 | 130 | 1,017 | 823,900 | 4,018,028 |
| TOTAL | 3,888 | 568 | 3,257 | 19,257,455 | 41,494,421 |
We sought to better understand this surprising finding that Latino genomes were so powerful for admixture mapping in this context. Remarkably, ∼25% of the admixture mapped SNPs in the current analysis were estimated to be polymorphic exclusively in the West African ancestral population, compared with 1%–2% polymorphic exclusively in the European ancestral population and 8%–10% polymorphic exclusively in the Native American ancestral population (Figures 3A–3C). We found that this was due to a mix of ancestry proportions of the samples analyzed, historical population demographics of the three ancestral populations, and informativeness of a SNP given its ancestral allele frequencies for the purpose of admixture mapping (Figures 3D–3F).
Figure 3.
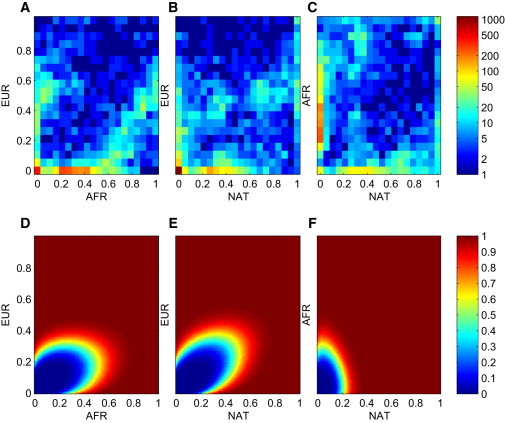
Ancestral Allele Frequencies Spectrum for Mapped SNPs and Power Estimates for the Mappability of a SNP Given Its Ancestral Allele Frequencies
(A–C) Ancestral frequencies estimates for SNPs from unlocalized scaffolds that mapped with a LOD score greater than or equal to 6.
(D–F) Probability of obtaining a LOD score greater than or equal to 6 for a SNP monomorphic for the reference allele for one ancestral population and with given alternate allele frequencies for the two other populations on the x and y axes.
Intuitively, genomic segments derived from West African ancestors are the segments most likely to contain ancestry-specific alleles, and the low levels of West African components in Latino genomes makes the observation of these alleles even more informative for the purpose of admixture mapping. Most of these West African-specific SNPs would have not had sufficient power to map in a similarly sized cohort of African Americans because of the 80% ± 12% West African component in the African American population. Conversely, European-specific markers are easier to map than West African-specific markers in African Americans (see Supplementary Note of Genovese et al.8).
Given our false-positive estimates, we expected less than ∼10 mismapped markers overall. Among scaffolds with multiple mapped SNPs (332 of the 569), we identified seven with discordant localizations. In one case (NW_001838929.1), this is due to a known misassembly between chromosomes 5 and 6 in HuRef over PRIM2 (MIM 176636). In another case (AL356585.7), the most parsimonious explanation is that one localization is a false positive. The other five cases relate to regions rich in satellite repeats where different SNPs localize to either chromosomes 1 and 14 (AEKP01218574.1, BX546479.5, and NW_001841051.1) or chromosomes 5 and 19 (NW_001841116.1 and NW_001840272.1). In these cases the most parsimonious explanation is alignment of reads originating from paralogous regions of different chromosomes for which at least one region was not modeled in hs37d5. Although these results are in line with the number of false-positive localizations estimated, they highlight the importance of relying on correct alignments, a problem that can be obviated by either using longer reads, including models for all paralogs in the reference used for alignment, or performing careful analysis of excess read coverage.
We sought to understand the statistical power of this approach, in part to evaluate its future contribution to completing physical maps of the human genome as more admixed genomes are sequenced. Empirically, with the available sequence from 242 admixed samples, approximately one SNP every 10 kbp achieved an admixture mapping LOD score greater than or equal to 6. Because larger contigs have a larger chance to contain a mapped SNP than smaller contigs, even if we were able to map only ∼17.5% of the contigs analyzed (569 scaffolds), we successfully mapped ∼46.4% of the sequence contained in all unlocalized contigs (∼19.1 Mbp of sequence). We therefore tried to estimate empirically the likelihood of mapping an unlocalized contig given its size. As expected, larger contigs were significantly more likely to map because they were more likely to contain a mapped SNP and contigs larger than ∼40 kbp were more likely to be mapped than not (Figure 4). Although these results are encouraging for the feasibility to map most unlocalized sequences originated from BAC and fosmid clones (20–180 kbp), we caution that the likelihood to identify a SNP that maps will also be a function of the sequence content and the LD structure of a given contig.
Figure 4.
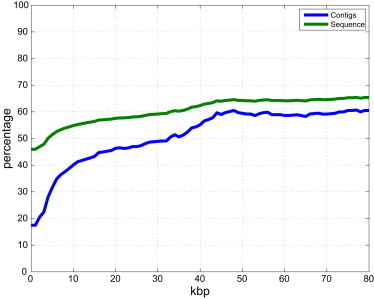
Percentage of hs37d5 Unlocalized Contigs that Were Localized in This Study as a Function of Contig Size
Percentage of localized contigs, in blue, and localized sequence, in green, admixture mapped among all unlocalized contigs from the hs37d5 reference larger than a given size using sequence data from the 242 admixed samples from the 1000 Genomes Project Phase 1.
Most of the mapped scaffolds localized to regions near gaps in the human reference genome (Figure 5). HuRef-unplaced scaffolds almost always mapped to pericentromeric regions, much more often than HuRef-placed scaffolds did. This is consistent with our earlier observation that these scaffolds often contain centromeric satellite sequence and that on many occasions they resemble euchromatic islands flanked by heterochromatic satellite sequence.8 Note that mappings within pericentromeric regions are unable to pinpoint the exact location of the sequence within pericentromeric gaps and often provide localizations to either side of the centromere.
Figure 5.

Regions of Excess Coverage and Mapping for Unlocalized Scaffolds from hs37d5
To critically evaluate these mappings by an independent molecular analysis, we utilized optical restriction maps for the human genome, previously obtained by visualizing the digestion patterns of a restriction enzyme on long, random, individual pieces of genomic DNA.25,26 We selected for analysis the 37 BAC clone sequences that we were able to localize and for which at least 7 SwaI restriction enzyme cuts were identified in silico. (The existence of at least 7 restriction sites was required for specificity; spurious matches with the restriction maps were observed for clones with fewer than 7 SwaI sites.) We compared the restriction maps for these clones to available consensus maps for the human genome to identify significant matches. We were able to connect 16 of these clones to a genomic location via the optical-map data, generally because a long, restriction-mapped segment of genomic DNA contained restriction fragments matching the clone and also restriction fragments matching an assembled part of the human genome. In each case (16/16), the optical map validated our admixture-based mapping of the same clone (Table 3).
Table 3.
Optical Map Validation
| Accession | Clone/Scaffold Name | Length | Decoy | Mapping | Mapping Type | SwaI Cuts | OM Match |
|---|---|---|---|---|---|---|---|
| AC006359.3 | DJ1135M02 | 118,730 | 14,732 | 1p12 | pericentromeric | 12 | + |
| AC006453.3 | RP4-614C10 | 155,313 | 21,861 | 2q11.1 | pericentromeric | 9 | NA |
| AC010098.8 | RP11-400J9 | 176,043 | 35,075 | 1q21.1 | pericentromeric | 11 | NA |
| AC011850.12 | RP11-364J18 | 164,681 | 164,681 | 20q11.21 | pericentromeric | 13 | NA |
| AC018692.9 | RP11-555K2 | 189,789 | 189,789 | 21q11.2 | pericentromeric | 23 | NA |
| AC026273.7 | CTD-2314M3 | 144,645 | 118,722 | 2p11.2 | pericentromeric | 7 | + |
| AC040978.8 | RP11-570L14 | 180,983 | 2,300 | 8q24.3 | interstitial | 13 | + |
| AC092107.5 | RP11-755J8 | 137,617 | 135,288 | 20q11.21 | pericentromeric | 10 | NA |
| AC104301.2 | RP11-150N22 | 189,610 | 180,137 | 20q11.22 | pericentromeric | 11 | NA |
| AC109135.2 | RP11-240C17 | 175,099 | 117,528 | 1q21.1 | pericentromeric | 18 | + |
| AC114745.6 | RP11-116D16 | 142,666 | 1,718 | 2q31.1 | interstitial | 8 | NA |
| AC116618.4 | RP11-98L17 | 153,040 | 148,815 | 22q11.21 | pericentromeric | 10 | NA |
| AC127701.2 | RP11-79F18 | 161,405 | 77,395 | 7p12.3 | pericentromeric | 10 | NA |
| AC129531.8 | RP11-188B1 | 164,068 | 30,549 | 17q24.1 | interstitial | 8 | + |
| AC133920.2 | RP11-413O9 | 197,357 | 56,111 | 16p11.2 | pericentromeric | 14 | NA |
| AC137488.2 | CTD-2506I5 | 167,135 | 121,805 | 22q11.21 | pericentromeric | 11 | NA |
| AC138774.4 | RP11-1320P3 | 194,050 | 158,495 | 14q11.2 | pericentromeric | 12 | NA |
| AC233266.3 | CH17-257B11 | 200,859 | 52,993 | 2p11.2 | pericentromeric | 13 | + |
| AC233698.3 | CH17-16P3 | 205,905 | 6,425 | 17q12 | interstitial | 11 | + |
| AC233702.5 | CH17-53B9 | 237,913 | 32,188 | 17p11.2 | pericentromeric | 13 | + |
| AC234063.4 | RP11-281H18 | 171,953 | 97,656 | 17q24.1 | interstitial | 14 | + |
| AC239584.4 | CH17-186K1 | 188,924 | 7,642 | 4q13.3 | interstitial | 19 | NA |
| AC239860.3 | CH17-262O2 | 191,275 | 35,427 | 1q21.1 | pericentromeric | 10 | + |
| AC241586.3 | CH17-289G7 | 221,610 | 28,747 | 1p11.2 | pericentromeric | 23 | + |
| AC243974.2 | CH17-93H22 | 121,220 | 1,892 | 12p13.2 | interstitial | 15 | + |
| AL137861.5 | RP4-813B7 | 127,682 | 97,063 | 1q21.1 | pericentromeric | 9 | NA |
| AL163540.11 | RP11-348N17 | 166,566 | 166,566 | 9q21.12 | pericentromeric | 14 | NA |
| AL354926.17 | RP1-216J23 | 163,140 | 128,103 | 3p11.1 | pericentromeric | 17 | NA |
| AL356585.7 | RP11-341D18 | 186,858 | 186,858 | 13q12.11 | pericentromeric | 9 | NA |
| AL360154.30 | RP11-499D3 | 240,434 | 240,434 | 1q21.2 | pericentromeric | 17 | + |
| AL590523.5 | RP11-565G5 | 155,397 | 155,397 | 3q11.2 | pericentromeric | 11 | NA |
| AL592188.60 | RP11-337M7 | 161,802 | 161,802 | 1p36.11 | interstitial | 8 | NA |
| AL845331.2 | RP11-407P15 | 185,111 | 53,859 | 2p11.2 | pericentromeric | 21 | + |
| AL929347.8 | RP3-433O3 | 128,374 | 128,374 | 6p25.3 | telomeric | 11 | + |
| BX072566.10 | RP11-25L22 | 164,239 | 164,239 | 21q11.2 | pericentromeric | 10 | + |
| BX546479.5 | RP11-438N17 | 172,294 | 172,294 | 1q21.1 | pericentromeric | 9 | NA |
| BX640538.4 | RP6-238B6 | 165,731 | 162,788 | 9p13.1 | pericentromeric | 11 | NA |
List of admixture-mapped BAC clone sequences with at least seven SwaI restriction enzyme cuts. In the length column we report the length of the sequence associated with the clone, and in the decoy column we report the amount of sequence included in the decoy sequences. In the type column we indicate whether the clone was mapped to an interstitial or a pericentromeric region. Notice that interstitial clones usually report only a small fraction of their sequence as part of the decoy, because these usually are clones selected among the decoy sequences only for harboring a small insertion not present in GRCh37. In the last column we report whether we were able to match the in silico restriction map of the clone to the available consensus maps from optical mapping (+) or whether we were unable to find any match (NA).
We sought to more deeply understand the relationships among optical-mapping and ancestry-mapping results and the potential complementarity of these two new approaches to genome assembly. Several of the validated concordances involved the localization of the BAC clone sequences (e.g., AC026273.7) within a centromeric gap, confirming the potential of optical mapping to bridge euchromatic sequences separated by >100 kbp stretches of centromeric satellite repeats. Another case involved a BAC clone sequence (AL354926.17) that is part of a known ∼240 kbp euchromatic island resulting from a segmental duplication involving PRIM2 from chromosome 6 within the centromere of chromosome 3 (see Genovese et al.8); in this case, the optical restriction map was unable to localize the clone, despite the 17 restriction enzyme cuts predicted from the sequence. Consensus maps for chromosome 3 predict that this island must be separated by >400 kbp stretches of centromeric satellite repeats that are not bridged by current optical-mapping data sets. This example highlights the unique kind of long-range information that can be accessed through admixture mapping but not yet through currently available molecular technologies.
We hypothesized that our statistical approach could also be used to identify dispersed duplication polymorphisms, in which extra, polymorphic copies of a genomic locus reside far from the known copies. (Such dispersed duplications are in contrast to tandem duplications, the most common form of duplication polymorphism.) We therefore also attempted to remap the genomic locations of copy-number polymorphisms (CNPs) by the above approach and we identified four dispersed duplication events (Table S3). Two of these remapped polymorphisms relate to a large and common copy-number polymorphism in 16p11.2 that also affects missing sequence paralogous to sequence in 6p25.3, containing DUSP22, and 20q13.2 (Figure 6 and Table S4). Further analysis revealed the presence of an LD-related less common CNP also affecting sequence paralogous to 16p11.2 and 6p25.3 (Figure S1). Sequence coverage analysis shows that this missing piece is highly polymorphic in human populations, confirming previous observations about the DUSP22 paralog.8,31–33
Figure 6.
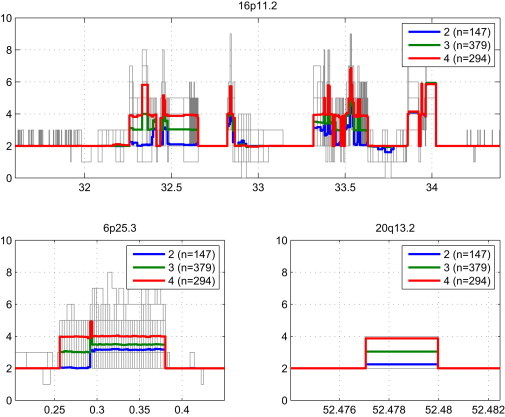
A Common Structural Polymorphism at 16p11.2
Sequence read coverage for 826 samples from the 1000 Genomes Project Phase 1 within regions 16p11.2, 6p25.3, and 20q11.2. For clarity, 42 samples that were not classified as copy number two over the two mostly unaffected windows chr16: 32,699,009–32,829,564 and chr16: 33,142,816–33,339,320 were excluded, because these may harbor larger and rarer CNVs. Median coverage for samples genotyped as CN = 2,3,4 over chr6: 257,000–295,000 is displayed. Notably, a strong correlation emerges between coverage over the genotyped region and sequence within window chr16: 32,258,540–32,659,102. Coordinates in Mbp on the horizontal axis are with respect to GRCh37.
We also mapped to a telomeric region of 20q13.33 the polymorphic, extra copy of sequence in 12p13.33, and we mapped to 21q11.2 a large CNP affecting missing sequence paralogous to sequence in 13q11. Notably, this last CNP also affects a region in 21q11.2, and it seems that samples with excess coverage in 13q11 have lower-than-expected sequence coverage in 21q11.2, possibly indicating the presence of a common polymorphism involving sequence exchange between chromosomes 13 and 21 (Figure S2).
Finally, we sought to identify cryptic missing paralogous sequences that are entirely missing from human reference genome sequences (i.e., that are not even described as unlocalized sequences in GRCh37) and exist as cryptic segmental duplications (or paralogs) of known genomic sequences. We found that ∼17 Mbp of sequence in the autosomes of the GRCh37 reference human genome receive an excessive number of reads (when aligning against GRCh37 rather than hs37d5), indicating the presence of missing paralogs (Tables 4 and S5). Notably, chromosome 1 contains an exceptional ∼2.6 Mbp of such sequence, mainly concentrated in the 1q21 region. Part of the reason is due to three large and recent segmental duplications of the region surrounding SRGAP2 from 1q32.1 to 1q21.1, 1p12, and proximal 1q21.1,34 which are not fully represented in GRCh37. Another autosome with a large amount of sequence paralogous to missing sequence is chromosome 16, containing ∼1.8 Mbp of such sequence, for which most of the excess coverage is due to missing paralogous sequence involved in the large CNP in 16p11.2 (Figures 6 and S1 and Table S4).
Table 4.
Amount of Autosomal Sequence in GRCh37 Estimated as Paralogous to Human Genome Sequence Missing from GRCh37
| Chromosome | Amount | Chromosome | Amount |
|---|---|---|---|
| 1 | 2,619,146 | 12 | 78,080 |
| 2 | 1,256,316 | 13 | 44,748 |
| 3 | 260,658 | 14 | 791,331 |
| 4 | 702,290 | 15 | 1,804,382 |
| 5 | 225,217 | 16 | 1,778,050 |
| 6 | 712,060 | 17 | 687,292 |
| 7 | 951,381 | 18 | 29,417 |
| 8 | 575,030 | 19 | 495,126 |
| 9 | 1,139,288 | 20 | 255,426 |
| 10 | 1,149,893 | 21 | 1,011,618 |
| 11 | 217,685 | 22 | 159,547 |
Having identified ∼17 Mbp of genomic sequence that harbors cryptic paralogs, we next sought to map the genomic locations of these cryptic paralogs by admixture mapping the genomic locations of variants in this sequence. After genotyping SNPs over the regions with observed excess coverage, we identified 175 SNPs that remapped to a chromosome different from the chromosome on which they were originally located (Table S6). Notably, 26 of these remapped SNPs were erroneously localized in the 1q21.1 region; 24 remapped SNPs related to the duplication from 6p25.3 to 16p11.2 containing DUSP22 (notice that because of the copy-number polymorphism at 16p11.2, these SNPs might actually be paralogous sequence variants); and 13 related to a known duplication from 6p11.2 to 3p11.1 containing a partial copy of PRIM2.8,35–38 Intriguingly, 21 of the remapped SNPs originated from the pericentromeric regions of chromosome 19 and remapped to pericentromeric regions of chromosome 5; the extent of apparent genetic exchange between these two genomic regions is probably due to the high degree of alpha satellite homology between them.39,40 Another 17 of the remapped SNPs originated from the short arm of chromosome 21 and mapped to chromosomes 13 and 14; this is consistent with the observation of recombination exchanges selective for sequences between the acrocentric chromosomes 13, 14, and 21 resulting from a common subfamily of alpha satellite DNA.40–42
Discussion
We successfully mapped much of the human genome’s remaining unlocalized sequence. By applying our three-way admixture mapping approach to whole-genome sequence data from 242 Latino genomes, we localized 569 scaffolds containing almost 20 Mbp of sequence currently unlocalized or missing from the current reference human genome. Only 38 of these scaffolds had been mapped in our previous work.8 Mappings in our studies based on African American and Latino admixture produced consistent results, with mappings agreeing at 37/38 scaffolds with only one exception (NW_001841149.1). Despite this effort, even more sequence remains unlocalized (much of it in smaller contigs) or missing from the current human reference genome.
Though our work was limited by the modest number of admixed samples for whom whole-genome sequence data are currently available (n = 242), Latino genomes turned out to be surprising powerful for admixture mapping the reference human genome’s missing sequence, as a result of the particular ancestral proportions that are present in Latino genomes (and in particular, because of the appreciable but still-modest contribution of African ancestry to the populations sampled). As more whole-genome sequence data sets from admixed samples become available (the 1000 Genomes Project alone is expected to include ∼500 admixed samples at the end of Phase 3), we predict that it will be possible to localize the great majority of fosmid-size or larger (>40 kbp) contigs by admixture mapping.
The completion of physical maps of the human genome remains an important goal, in which the initial localization of novel sequences to their correct genomic locations is an important step. Most of the genetic findings from linkage, association, and CNVs have not yet been attached to specific functional variants or causal genes; knowing all of the eligible sequence and genes in a genomic region is key for informing follow-up strategies. The localization of pericentromeric sequences currently absent from the human reference genome will also help efforts to complete physical maps of the human genome in these regions, for example by informing the selection of clones for sequencing and the creation of tiling paths. Such resources will support the resolution of genetic signals, particularly in pericentromeric regions without a full and correct representation in the current human genome reference—for example, a risk factor for multiple sclerosis (MS [MIM 126200]) that is currently localized, but not yet identified, in the pericentromeric region of chromosome 1.43
The Genome Reference Consortium (GRC) is actively working to improve the current human genome assembly.44 At present, the whole 1q21 region has been resequenced by means of a haploid BAC library34 enabling analysis of many novel genes within it; this sequence will be incorporated into the reference assembly in GRCh38. Similar efforts are making progress in many other regions of the genome.
Admixture mapping the human genome’s missing pieces will complement current, clone-based efforts to finish the human genome assembly. We predict that interstitial gaps in the reference may be most completely closed through the use of more traditional tiling approaches by assembling sequences originated from fosmid or BAC clones. On the other hand, euchromatic islands isolated from the rest of the euchromatic genome in oceans of heterochromatic, repeat-rich sequence may be difficult or impossible to connect to the rest of the euchromatic genome by clone-based approaches. Admixture mapping may be critical for localizing such genomic sequences.
Acknowledgments
This work was supported by a grant from NHGRI (R01 HG006855-01 to S.A.M.) and by the Stanley Center for Psychiatric Research at the Broad Institute of MIT and Harvard.
Contributor Information
Giulio Genovese, Email: giulio.genovese@gmail.com.
Steven A. McCarroll, Email: mccarroll@genetics.med.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
BedGraph Format, http://genome.ucsc.edu/goldenPath/help/bedgraph.html
Decoy sequences, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/ or ftp://ftp.ncbi.nih.gov/1000genomes/ftp/technical/reference/phase2_reference_assembly_sequence/
GRC, Genome Reference Consortium, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/
LATOOLS software, http://www.broadinstitute.org/∼giulio/latools/
Local ancestry deconvolution, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/analysis_results/ancestry_deconvolution/ or ftp://ftp.ncbi.nlm.nih.gov/1000genomes/ftp/phase1/analysis_results/ancestry_deconvolution/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PyVCF, A Variant Call Format Parser for Python, http://pyvcf.readthedocs.org/en/latest/
RepeatMasker, http://www.repeatmasker.org
SOMA, Scaffolding using Optical Map Alignment, http://www.cbcb.umd.edu/soma/
UCSC Genome Browser, http://genome.ucsc.edu
unionbedg Format, http://bedtools.readthedocs.org/en/latest/content/tools/unionbedg.html
VCFtools, Variant Call Format, http://vcftools.sourceforge.net/specs.html
References
- 1.International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 2.Cole C.G., McCann O.T., Collins J.E., Oliver K., Willey D., Gribble S.M., Yang F., McLaren K., Rogers J., Ning Z. Finishing the finished human chromosome 22 sequence. Genome Biol. 2008;9:R78. doi: 10.1186/gb-2008-9-5-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander E.S. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 4.Pickrell J.K., Gaffney D.J., Gilad Y., Pritchard J.K. False positive peaks in ChIP-seq and other sequencing-based functional assays caused by unannotated high copy number regions. Bioinformatics. 2011;27:2144–2146. doi: 10.1093/bioinformatics/btr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2011;39(Database issue):D32–D37. doi: 10.1093/nar/gkq1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gnerre S., Maccallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genovese G., Handsaker R.E., Li H., Altemose N., Lindgren A.M., Chambert K., Pasaniuc B., Price A.L., Reich D., Morton C.C. Using population admixture to help complete maps of the human genome. Nat. Genet. 2013;45:406–414. doi: 10.1038/ng.2565. e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryan D.P., da Silva M.R., Soong T.W., Fontaine B., Donaldson M.R., Kung A.W., Jongjaroenprasert W., Liang M.C., Khoo D.H., Cheah J.S. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88–98. doi: 10.1016/j.cell.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parra E.J., Marcini A., Akey J., Martinson J., Batzer M.A., Cooper R., Forrester T., Allison D.B., Deka R., Ferrell R.E., Shriver M.D. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet. 1998;63:1839–1851. doi: 10.1086/302148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith M.W., Patterson N., Lautenberger J.A., Truelove A.L., McDonald G.J., Waliszewska A., Kessing B.D., Malasky M.J., Scafe C., Le E. A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhry S., Coyle N.E., Tang H., Salari K., Lind D., Clark S.L., Tsai H.-J., Naqvi M., Phong A., Ung N., Genetics of Asthma in Latino Americans GALA Study Population stratification confounds genetic association studies among Latinos. Hum. Genet. 2006;118:652–664. doi: 10.1007/s00439-005-0071-3. [DOI] [PubMed] [Google Scholar]
- 14.Price A.L., Patterson N., Yu F., Cox D.R., Waliszewska A., McDonald G.J., Tandon A., Schirmer C., Neubauer J., Bedoya G. A genomewide admixture map for Latino populations. Am. J. Hum. Genet. 2007;80:1024–1036. doi: 10.1086/518313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryc K., Velez C., Karafet T., Moreno-Estrada A., Reynolds A., Auton A., Hammer M., Bustamante C.D., Ostrer H. Colloquium paper: genome-wide patterns of population structure and admixture among Hispanic/Latino populations. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 2):8954–8961. doi: 10.1073/pnas.0914618107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winkler C.A., Nelson G.W., Smith M.W. Admixture mapping comes of age. Annu. Rev. Genomics Hum. Genet. 2010;11:65–89. doi: 10.1146/annurev-genom-082509-141523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Via M., Gignoux C.R., Roth L.A., Fejerman L., Galanter J., Choudhry S., Toro-Labrador G., Viera-Vera J., Oleksyk T.K., Beckman K. History shaped the geographic distribution of genomic admixture on the island of Puerto Rico. PLoS ONE. 2011;6:e16513. doi: 10.1371/journal.pone.0016513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Feuk L., Duggan G.E., Khaja R., Scherer S.W. Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet. Genome Res. 2006;115:205–214. doi: 10.1159/000095916. [DOI] [PubMed] [Google Scholar]
- 19.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Handsaker R.E., Korn J.M., Nemesh J., McCarroll S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baran Y., Pasaniuc B., Sankararaman S., Torgerson D.G., Gignoux C., Eng C., Rodriguez-Cintron W., Chapela R., Ford J.G., Avila P.C. Fast and accurate inference of local ancestry in Latino populations. Bioinformatics. 2012;28:1359–1367. doi: 10.1093/bioinformatics/bts144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price A.L., Tandon A., Patterson N., Barnes K.C., Rafaels N., Ruczinski I., Beaty T.H., Mathias R., Reich D., Myers S. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Churchhouse C., Marchini J. Multiway admixture deconvolution using phased or unphased ancestral panels. Genet. Epidemiol. 2013;37:1–12. doi: 10.1002/gepi.21692. [DOI] [PubMed] [Google Scholar]
- 24.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dimalanta E.T., Lim A., Runnheim R., Lamers C., Churas C., Forrest D.K., de Pablo J.J., Graham M.D., Coppersmith S.N., Goldstein S., Schwartz D.C. A microfluidic system for large DNA molecule arrays. Anal. Chem. 2004;76:5293–5301. doi: 10.1021/ac0496401. [DOI] [PubMed] [Google Scholar]
- 26.Valouev A., Schwartz D.C., Zhou S., Waterman M.S. An algorithm for assembly of ordered restriction maps from single DNA molecules. Proc. Natl. Acad. Sci. USA. 2006;103:15770–15775. doi: 10.1073/pnas.0604040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teague B., Waterman M.S., Goldstein S., Potamousis K., Zhou S., Reslewic S., Sarkar D., Valouev A., Churas C., Kidd J.M. High-resolution human genome structure by single-molecule analysis. Proc. Natl. Acad. Sci. USA. 2010;107:10848–10853. doi: 10.1073/pnas.0914638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagarajan N., Read T.D., Pop M. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics. 2008;24:1229–1235. doi: 10.1093/bioinformatics/btn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 32.Martin J., Han C., Gordon L.A., Terry A., Prabhakar S., She X., Xie G., Hellsten U., Chan Y.M., Altherr M. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004;432:988–994. doi: 10.1038/nature03187. [DOI] [PubMed] [Google Scholar]
- 33.Sudmant P.H., Kitzman J.O., Antonacci F., Alkan C., Malig M., Tsalenko A., Sampas N., Bruhn L., Shendure J., Eichler E.E., 1000 Genomes Project Diversity of human copy number variation and multicopy genes. Science. 2010;330:641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dennis M.Y., Nuttle X., Sudmant P.H., Antonacci F., Graves T.A., Nefedov M., Rosenfeld J.A., Sajjadian S., Malig M., Kotkiewicz H. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell. 2012;149:912–922. doi: 10.1016/j.cell.2012.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiratori A., Okumura K., Nogami M., Taguchi H., Onozaki T., Inoue T., Ando T., Shibata T., Izumi M., Miyazawa H. Assignment of the 49-kDa (PRIM1) and 58-kDa (PRIM2A and PRIM2B) subunit genes of the human DNA primase to chromosome bands 1q44 and 6p11.1-p12. Genomics. 1995;28:350–353. doi: 10.1006/geno.1995.1155. [DOI] [PubMed] [Google Scholar]
- 36.Alkan C., Sajjadian S., Eichler E.E. Limitations of next-generation genome sequence assembly. Nat. Methods. 2011;8:61–65. doi: 10.1038/nmeth.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ju Y.S., Kim J.-I., Kim S., Hong D., Park H., Shin J.-Y., Lee S., Lee W.-C., Kim S., Yu S.-B. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011;43:745–752. doi: 10.1038/ng.872. [DOI] [PubMed] [Google Scholar]
- 38.Chung J., Tsai S., James A.H., Thames B.H., Shytle S., Piedrahita J.A. Lack of genomic imprinting of DNA primase, polypeptide 2 (PRIM2) in human term placenta and white blood cells. Epigenetics. 2012;7:429–431. doi: 10.4161/epi.19777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finelli P., Antonacci R., Marzella R., Lonoce A., Archidiacono N., Rocchi M. Structural organization of multiple alphoid subsets coexisting on human chromosomes 1, 4, 5, 7, 9, 15, 18, and 19. Genomics. 1996;38:325–330. doi: 10.1006/geno.1996.0635. [DOI] [PubMed] [Google Scholar]
- 40.Hayden K.E. Human centromere genomics: now it’s personal. Chromosome Res. 2012;20:621–633. doi: 10.1007/s10577-012-9295-y. [DOI] [PubMed] [Google Scholar]
- 41.Choo K.H., Vissel B., Brown R., Filby R.G., Earle E. Homologous alpha satellite sequences on human acrocentric chromosomes with selectivity for chromosomes 13, 14 and 21: implications for recombination between nonhomologues and Robertsonian translocations. Nucleic Acids Res. 1988;16:1273–1284. doi: 10.1093/nar/16.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choo K.H., Vissel B., Earle E. Evolution of alpha-satellite DNA on human acrocentric chromosomes. Genomics. 1989;5:332–344. doi: 10.1016/0888-7543(89)90066-9. [DOI] [PubMed] [Google Scholar]
- 43.Reich D., Patterson N., De Jager P.L., McDonald G.J., Waliszewska A., Tandon A., Lincoln R.R., DeLoa C., Fruhan S.A., Cabre P. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 2005;37:1113–1118. doi: 10.1038/ng1646. [DOI] [PubMed] [Google Scholar]
- 44.Church D.M., Schneider V.A., Graves T., Auger K., Cunningham F., Bouk N., Chen H.-C., Agarwala R., McLaren W.M., Ritchie G.R.S. Modernizing reference genome assemblies. PLoS Biol. 2011;9:e1001091. doi: 10.1371/journal.pbio.1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.