

Abstract
The recent introduction of highly pathogenic strains of West Nile virus (WNV) into naïve populations in Europe, Israel, and the USA has resulted in a marked increase in both the number of reported cases and the severity of disease compared to previous outbreaks. The impact of the increased virulence of recently emerged strains of WNV is exacerbated by the fact that antiviral therapies and vaccines are not currently available for use in humans. A greater understanding of the viral and host factors involved in WNV-mediated neuropathology is necessary to facilitate the development of novel therapeutic approaches. This review summarizes the current state of knowledge of the role of the cell-intrinsic innate immune responses as well as the cell-mediated innate and adaptive immune responses in promoting the detection and clearance of WNV from the CNS.
Keywords: West Nile virus, Immune response, Central nervous system
Re-emergence of WNV
West Nile virus (WNV) is a member of the Flaviviridae, which are enveloped, positive-stranded RNA viruses. This family includes several globally important emerging arthropod-borne viruses that cause neuroinvasive disease in humans such as Japanese encephalitis virus, tick-borne encephalitis, and WNV. Combined, these viruses are responsible for significant morbidity and mortality worldwide. Prior to the 1990s, WNV infections were typically asymptomatic or associated with a mild febrile illness known as West Nile fever. However, recent epidemics in Europe and North America have been associated with higher rates of severe disease, including meningitis, encephalitis, and acute flaccid paralysis. Since its introduction into the USA in 1999, yearly outbreaks of WNV in the USA have resulted in more than 15,000 reported cases with neurological complications and >1,500 deaths, making WNV the leading cause of mosquito-borne neuroinvasive disease in the USA (http://www.cdc.gov/ncidod/dvbid/westnile/index.htm).
WNV entry into the CNS
Neuroinvasive viruses enter the CNS via three main routes: (1) retrograde transport along axons, (2) spread from olfactory neurons and (3) hematogenous dissemination. Experimental evidence in animal models suggests that WNV can use all three methods to gain access to the CNS, depending on the route of infection (Diamond et al. 2003; Getts et al. 2008; Roe et al. 2012; Samuel et al. 2007; Wang et al. 2004). However, hematogenous spread is thought to be the primary route of WNV entry into the brain during a natural infection. Entry via this route requires WNV to cross the blood–brain barrier (BBB). The mechanism by which WNV circumvents the BBB remains controversial. Induction of peripheral expression of the proinflammatory cytokine TNFα was initially proposed to facilitate WNV entry into the CNS by triggering the breakdown of the BBB (Wang et al. 2004). However, in subsequent studies, low levels of TNFα detected in the serum of WNV-infected mice did not correlate with an increase in the permeability of the BBB or WNV entry into the CNS (Daffis et al. 2008; Dai et al. 2008; Shrestha et al. 2008). In addition, treatment of mice with neutralizing antibodies to TNFα enhanced susceptibility to WNV infection, suggesting that TNFα plays a role in controlling WNV replication rather that promoting viral dissemination (Shrestha et al. 2008). Moreover, WNV neuroinvasion in BALB/c mice can occur without an increase in the permeability of the BBB (Morrey et al. 2008). Taken together, these studies suggest that disruption of the BBB is not the primary mechanism by which WNV initially enters the CNS. Increasing evidence suggests that WNV entry into the CNS is a multistep process employing different mechanism as the infection progresses. Several groups have demonstrated that WNV replicates in brain microvascular endothelial cells and traverses an in vitro model of the BBB without compromising the integrity of the monolayer (Hussmann et al. 2013; Verma et al. 2009), suggesting that WNV may initially gain access to the CNS by directly infecting brain endothelial cells and releasing low levels of progeny virus from the abluminal surface. Alternatively, WNV may enter the CNS via a Trojan-horse mechanism through the diapedesis of an infected leukocyte (Bai et al. 2010; Dai et al. 2008). Circumstantial evidence suggests that the host response within the CNS and/or the process of leukocyte diapedesis itself contribute to the breakdown of the BBB late in infection (Roe et al. 2012). Thus, allowing the unrestricted entry of free virus and/or infected leukocytes into the CNS and promoting a second round of WNV dissemination.
WNV replication within the CNS
Within the CNS, intimate connections between neurons, astrocytes, and brain endothelial cells from the neurovascular unit (NVU; Fig. 1), which functions to regulate blood flow, the integrity of the BBB, and neuronal activity in response to environmental changes (Iadecola 2004). Because of its interconnectedness, perturbation of the function of any component within the NVU can have pathological effects on the other members. Studies in humans and mice have demonstrated that neurons are the primary cells targeted by WNV. However, WNV-positive brain microvascular endothelial cells and astrocytes have been detected in birds and humans, respectively, suggesting that these cells may serve as secondary targets in vivo (Lopes et al. 2007; van Marle et al. 2007; Wunschmann et al. 2004). In vitro studies conducted at both low and high MOIs demonstrated that pathogenic strains of WNV replicate within all three cell types, though replication in astrocytes was the most restricted (Cheeran et al. 2005; Diniz et al. 2006; Hussmann et al. 2013; van Marle et al. 2007; Verma et al. 2010).
Fig. 1.
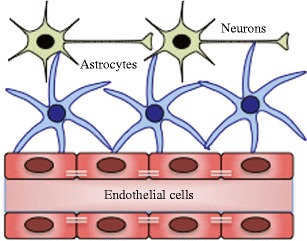
Neurovascular unit. Schematic of the composition of the neurovascular unit
Animal studies using intracranial inoculation indicate that most, if not all, strains of WNV can replicate within the CNS; nonetheless, the extent of neurovirulence is strain-dependent (Beasley et al. 2002; Shrestha et al. 2008). Recent studies have begun to define the host determinants for susceptibility to WNV in the various cell types comprising the NVU and the viral factors responsible for the strain-dependent differences in neuropathogenicity (Beasley et al. 2004; Beasley et al. 2005; Cho et al. 2013; Hussmann et al. 2013; Shirato et al. 2004b). Examination of WNV replication within the cells types comprising the NVU infected at low MOIs demonstrated that high and low neuropathogenic strains of WNV replicate with similar kinetics and to equivalent levels in brain microvascular endothelial cells and neurons (Hussmann et al. 2013). However, astrocytes exhibited a reduced susceptibility to the low neuropathogenic strain compared to the high neuropathogenic strain, suggesting a possible role for this cell type in limiting WNV replication within the CNS.
Neuroimmune response to WNV
Similar to peripheral tissues, the immune response within the CNS has both cell-intrinsic and cell-extrinsic components. However, the non-renewable nature of neurons necessitates the tight control of these responses to limit the possibility of irreversible immune-mediated neuronal destruction. Indeed, targeted disruption of various components of the immune system indicates that both branches of the immune response contribute to neuroprotection and neurodestruction during WNV infection depending on the context.
Cell-intrinsic response to WNV within the NVU
The cell-intrinsic innate antiviral immune response is a first line of defense against invading viral pathogens. It consists of multiple antiviral programs that work in concert to control viral replication and spread as well as promoting the cell-mediated innate and adaptive immune responses. The proper and controlled induction of this arm of the immune response is mediated by specialized cellular proteins termed pattern-recognition receptors (PRRs), which recognize specific pathogen-associated molecular patterns (PAMPs) produced during the course of infection (Fig. 2) (Janeway and Medzhitov 2002). Two classes of PRRs, Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), are involved in detecting WNV infection (Daffis et al. 2008; Suthar et al. 2013; Suthar et al. 2010; Town et al. 2009; Wang et al. 2004). TLRs are an evolutionarily conserved family of transmembrane molecules that are expressed on the cell surface or within endocytic vesicles and are therefore positioned to detect PAMPs in extracellular spaces and endosomes. TLR expression is typically restricted to immune cells, although expression is upregulated in many cell types in response to invading pathogens. In contrast, the RLR family members, RIG-I, MDA5, and LGP2, are cytoplasmic RNA helicases that are basally expressed at low levels in most cell types throughout the body. Upon detection of an invading viral pathogen, PRRs initiate signaling cascades that activate a number of latent transcription factors, such as IRF-3, IRF-7, and NFκB. Activation of these transcription factors results in the direct induction of antiviral effector proteins as well as proinflammatory cytokines and chemokines. The antiviral effector proteins function to control viral replication within an infected cell, while proinflammatory cytokines and chemokines amplify the cell-intrinsic antiviral programs and shape the nature of the cell-mediated immune responses. Recently, the NOD-like receptor (NLR) NLRP3 was shown to contribute to the cell-intrinsic response to WNV as well (Ramos et al. 2012); though, it remains to be determined whether NLRP3 plays a direct role in sensing WNV or an indirect role as a modulator of either the TLR or RLR pathways (Davis et al. 2011). NLRP3 functions as part of a large multicomponent protein complex termed the inflammasome, which serves as a molecular platform for the activation of the inflammatory caspase, Caspase 1 (Davis et al. 2011; Kanneganti 2010). Activated Caspase 1 cleaves pro-IL-1 and pro-IL-18 into biologically active forms that participate in the direct antiviral control of the invading pathogen and the regulation of the adaptive and inflammatory responses.
Fig. 2.
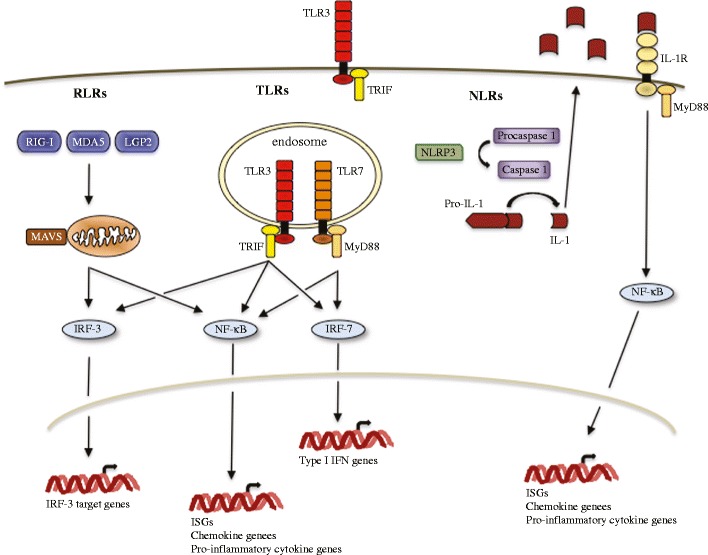
Cell-intrinsic innate antiviral response. Schematic of the key signaling pathways contributing to the innate antiviral response to WNV. Abbreviations: retinoic acid-inducible (RIG-I)-like receptors (RLRs); Toll-like receptor (TLRs); melanoma differentiation antigen 5 (MDA5); laboratory of genetics and physiology 2 (LGP2); nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs); NLR family PYD-containing 3 (NLPR3); TIR-domain-containing adapter-inducing interferon-β (TRIF); myeloid differentiation primary response 88 (MyD88); interferon stimulated genes (ISGs)
Transgenic mice with targeted deficiencies have been used to assess the contributions of the TLR, RLR, and NRL signaling pathways to controlling WNV replication within the CNS. Genetic depletion of TLR3, TLR7, or the receptor for IL-1β (IL-1R) resulted in increased viral loads within the CNS and enhanced susceptibility to lethal infection (Daffis et al. 2008; Durrant et al. 2013; Ramos et al. 2012; Town et al. 2009; Wang et al. 2004). Similar results were observed in mice deficient for MyD88, the adaptor molecule for TLR7 and IL-1R, or MAVS, a key downstream signaling molecule required for the RLR signal transduction pathway (Suthar et al. 2010; Szretter et al. 2010; Town et al. 2009). Thus, all three pathways contribute to the innate antiviral response to WNV within the CNS. However, the hierarchy of the involvement of these PRRs within the CNS remains to be determined and is likely to be cell-type specific.
Ex vivo studies have been used to investigate the cell-intrinsic innate antiviral response within neurons. Higher viral titers were detected in cortical neurons isolated from mice genetically deficient for TLR3 or MAVS, suggesting that both the TLR and RLR pathways play a role in restricting WNV replication within this cell type (Daffis et al. 2008; Suthar et al. 2010). Additionally, MAVS-dependent signaling was essential for the rapid and robust activation of innate antiviral programs and the induction of type I IFNs (Suthar et al. 2010). Disruption of IL-1β signaling also enhanced WNV replication within cortical neurons, suggesting that the NLR signaling pathway contributes to the antiviral response to WNV within neurons (Ramos et al. 2012). In addition, pretreatment of cortical neurons with IL-1β and IFN-β in combination substantially reduced WNV replication compared to either factor alone, suggesting that IL-1β functions synergistically with the type I IFNs. However, a separate study demonstrated that neutralization of IL-1β reduced WNV-induced apoptosis in a human neuroblastoma cell line (Kumar et al. 2010), suggesting that IL1-β can have toxic effects on neurons as well. Notably, a recent report comparing basal and WNV-induced innate immune signatures of cortical neurons isolated from the cerebral cortex and granule cell neurons isolated from the cerebellum demonstrated that neuronal subtypes have distinct innate immune responses (Cho et al. 2013). This analysis demonstrated that differences in basal levels of expression of key PRRs, transcription factors, and antiviral effector proteins resulted in the induction of unique innate immune profiles, which corresponded with the relative permissiveness of the two neuronal subtypes.
In contrast to neurons, the cell-intrinsic innate antiviral response in the other cell types comprising the NVU has not been extensively examined. Since the antiviral response contributes to the strain-specific control of replication of other neurotropic viruses (Daffis et al. 2011; Johnston et al. 2001; Ryman et al. 2000), our lab assessed whether the differential induction of the innate antiviral response contributed to the decreased susceptibility of astrocytes to a low neuropathogenic strain of WNV. Examination of the induction profiles of a panel of antiviral effector proteins (Fredericksen et al. 2004; Scherbik et al. 2006; Szretter et al. 2011) indicated that the kinetics and amplitude of the antiviral response to high and low neuropathogenic strains of WNV was similar in astrocytes (Hussmann and Fredericksen, unpublished observation). This suggests that the reduced level of replication of the low neuropathogenic strain of WNV within astrocytes was not due to the preferential induction of antiviral effector proteins. A detailed analysis of WNV replication within astrocytes indicated that the reduced susceptibility to low neuropathogenic strains was due to a delay in the synthesis of the viral genome and a reduction in the ability of this strain to spread from cell-to-cell (Hussmann et al. 2013). The restriction to viral spread was suggestive of the paracrine protection of type I IFNs and indeed, measureable levels of type I IFN were detected in supernatants recovered from cultures infected with either strain. However, neutralization of type I IFNs did not enhance the ability of the nonneuropathogenic strain to spread from cell-to-cell within the monolayer, suggesting that classical innate antiviral responses play only a modest role in restricting WNV replication within astrocytes.
One of the strategies that WNV uses to circumvent the cell-intrinsic antiviral host response is to evade detection by RIG-I at early times post-infection, thereby postponing induction of the response until after a productive infection has been established (Fredericksen and Gale 2006; Fredericksen et al. 2004). A similar delay in activation of the antiviral response was observed in WNV-infected astrocytes, brain endothelial cells, and neurons (Hussmann and Fredericksen, unpublished observation). The mechanism(s) by which WNV evades detect by RIG-I has yet to be determined. However, a systematic analysis of the WNV viral genome and antigenome identified multiple regions capable of rapidly activating RIG-I (Shipley et al. 2012). Yet, incorporation of these stimulatory regions into larger RNAs abrogated their capacity to rapidly activate RIG-I signaling, suggesting that WNV evades detection by RIG-I at early times post-infection by sequestering stimulatory RNA ligands within the intact viral genome and antigenome. Nonetheless, the eventual activation of the RIG-I pathway suggests that WNV PAMPs become exposed to the cell’s antiviral sensors over time. This unmasking of RIG-I-specific PAMPs buried within the WNV genome and antigenome may be mediated by the cell’s normal RNA processing pathways. Indeed, the OAS/RNAseL pathway has been shown liberate a potent RNA PAMP from the HCV genome (Malathi et al. 2007, 2010). Consistent with this model, larger nonstimulatory, nonreplicating fragments of the WNV genome induced a RIG-I-specific antiviral response over a longer time course (Shipely and Fredericksen, unpublished observation).
Cell-mediated immunity
The restrictive nature of the BBB ensures that under normal physiological conditions, the CNS is an immune-specialized compartment with limited leukocyte infiltration. However, WNV infection results in a multifocal, mixed leukocyte neuroinflammatory response consisting of monocytes, neutrophils, natural killer (NK) cells, dendritic cells (DC) as well as effector CD4+ and CD8+ T cells (Brehin et al. 2008; Glass et al. 2005; Lim et al. 2011; Omalu et al. 2003; Shrestha et al. 2006; Sitati et al. 2007). Leukocyte infiltration is critical for clearance of WNV from the CNS and is one of the major determinants for survival. CD8+ T cells in particular play a central role in the clearance of highly neuropathogenic strains of WNV from the CNS (Klein et al. 2005; Shrestha et al. 2006; Sitati et al. 2007). Mice deficient for CD8+ T cells have higher viral loads within the CNS and are more susceptible to a lethal infection with WNV compared to immunocompetent mice (Shrestha et al. 2006). Similar results were observed in mice deficient for CD4+ cells. However, further analysis indicated that CD4+ T cells are necessary to sustain the CD8+ T cell response within the CNS (Sitati and Diamond 2006), though CD4+ T cells may play a direct role in controlling the WNV replication within the periphery (Brien et al. 2008). Nevertheless, recruitment of effector T cells to the CNS is not sufficient to clear WNV infection. As discussed in later sections, T cells must be recruited to the proper location within the CNS and undergo additional stimulation in order to mediate viral clearance.
Monocytes, the most prevalent infiltrating cell type detected in the brains of WNV-infected mice (Getts et al. 2012; Glass et al. 2005), also play an important role in controlling WNV infection. Severely monocytopenic mice exhibited enhanced viral loads within the CNS and increased susceptibility to lethal infect with WNV (Lim et al. 2011). Once within the CNS, inflammatory monocytes differentiate into macrophages and microglia (Getts et al. 2008). Increasing evidence suggests that infiltrating monocyte-derived cells and resident microglia are functionally non-redundant (Shechter and Schwartz 2013). Therefore, these two cell types are likely to perform different roles in controlling WNV-mediated pathogenesis. A recent study suggests that resident microglia play a protective role during WNV infection, limiting neuronal death and/or promoting CNS repair, rather than contributing directly to the inhibition of viral replication or the stimulation of WNV-specific T cells (Wang et al. 2012b). The observation that mice deficient for MHC II expression are unable to clear WNV from the CNS underscores the importance of monocyte-derived antigen-presenting cells (APCs) and/or DCs to the neuroimmune response to WNV (Sitati and Diamond 2006). As with the peripheral immune response, APCs within the CNS have the potential for a spectrum of activities including the production and secretion of neurotropic factors, recruitment of T cells into the parenchyma of the brain and the stimulation of effector T cells (Savarin et al. 2010; Shechter and Schwartz 2013). While the precise role(s) of monocyte-derived cells in controlling WNV replication and spread within the CNS remains to be determined, a recent report suggests that DCs contribute to the optimal activation of effector T cells (Durrant et al. 2013).
Neutrophils are hypothesized to play a biphasic role during WNV infection, enhancing viral spread to the CNS early in infection and attenuating viral pathogenesis at later times (Bai et al. 2010). Consistent with this model, depletion of neutrophils prior to infection resulted in decreased viral loads within the CNS and enhanced survival (Bai et al. 2010), while depletion after WNV infection shortened survival times. The ability of neutrophils to promote WNV entry into the CNS is further supported by the observation that disruption of neutrophil migration decreased viral loads within the CNS, lengthened survival times, and, in some circumstances, reduced susceptibility to a lethal infection (Bai et al. 2010; Wang et al. 2012a).
While neuroinflammation is essential for constraining WNV spread within the CNS and promoting viral clearance, several studies suggest that this process can have immunopathological consequences as well. Infection of CD8+ T cell-deficient mice with attenuated strains of WNV resulted in prolonged survival times and, depending on the viral dose that was administered, enhanced survival compared to WT mice (Szretter et al. 2012; Wang et al. 2003). Likewise, delaying monocyte migration to the CNS prolonged survival times and blocking migration decreased mortality in mice challenged intranasally with a low neuropathgenic strain of WNV (Getts et al. 2012, 2008). The effectiveness of the neuroinflammatory response in controlling an invading pathogen depends on a variety of factors such as the recruitment of immune cells to the appropriate location, at the right time and at the correct dose and elimination of the immune cell once the infection is cleared. The factors responsible for tipping the neuroinflammatory response to WNV from neuroprotective toward neuropathogenic are currently unknown.
Neuroinflammation
Because the inappropriate recruitment or retention of immune cells can have detrimental effects, the inflammatory response is, by necessity, a highly regulated, multistep process. This process is mediated in large part by the expression of chemoattractant cytokines, known as chemokines, and their cognate receptors on the targeted leukocyte. The first phase of leukocyte recruitment to the CNS is the initial attraction and movement of immune cells across the endothelial layer and into perivascular spaces. Since soluble gradients are rapidly dispersed by normal blood flow, the initial recruitment of leukocytes across the endothelial layer is mediated by solid phase gradients formed by chemokines binding to anchored glycosaminoglycans or scavenger receptors on endothelial cell surfaces (Middleton et al. 2002). Binding of immobilized chemokines to their cognate receptors on leukocytes triggers the activation of integrins on the surface of leukocytes, thereby promoting firm attachment to the endothelial cell layer. Leukocyte migration across an endothelial layer can occur via paracellular diapedesis, which requires disassembly of intercellular junctions, or transcellular diapedesis, which involves uptake and translocation through the endothelial cell itself (Carman 2009; Middleton et al. 2002). Due to the enhanced stability of the inter-endothelial tight junctions, transcellular diapedesis is the primary route of leukocyte infiltration across an intact BBB (Carman 2009; Greenwood et al. 1994; Wolburg et al. 2005). Therefore, the infiltration process at the BBB requires the establishment of a transcellular chemokine gradient within endothelial cells to facilitate the directional movement of immune cells into the perivascular space within the CNS (Carman 2009; Middleton et al. 2002). Once across the BBB, the chemokine profile within the perivascular space determines whether the infiltrating immune cells remain localized or migrate into the parenchyma of the CNS (McCandless et al. 2008; Wilson et al. 2010).
Increased levels of several proinflammatory chemokines and their cognate receptors have been detected in the brains of WNV-infected mice (Fig. 3) (Glass et al. 2005; Klein et al. 2005; Lim et al. 2011; Shirato et al. 2004a). The role of several of these factors in WNV-induced neuroinflammation has been confirmed using transgenic mice with targeted deficiencies (Glass et al. 2005; Klein et al. 2005; Lim et al. 2011; Zhang et al. 2008). Genetic depletion of the chemokine receptor CCR5, expressed on subsets of T lymphocytes, NK cells, monocytes, and macrophages, impaired the trafficking of the corresponding leukocytes to the CNS in response to WNV infection (Glass et al. 2005; Lim et al. 2011). In addition, severe monocytopenia in CCR2−/− mice resulted in a monocyte-specific deficiency in the brains of WNV-infected animals (Lim et al. 2011). However, genetic depletion of either CXCL10 or its cognate receptor CXCR3 specifically abrogated CD8+ T cell migration to the cerebellum in WNV-infected mice without disrupting migration into other parts of the brain (Klein et al. 2005; Zhang et al. 2008). In all three cases, perturbation of the neuroinflammatory response resulted in higher viral loads and a marked increase in mortality, demonstrating the importance of regulated leukocyte infiltration in controlling WNV infection in mice. Whether the neuroinflammatory response plays a similar role in controlling disease in humans remains to be determined; however, genetic evidence suggests that individuals lacking functional CCR5 are at an increased risk for symptomatic infection with WNV (Glass et al. 2006; Lim et al. 2008; Lim et al. 2010).
Fig. 3.
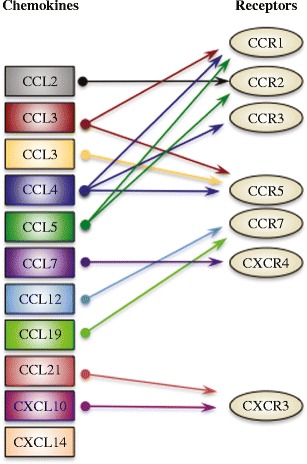
Chemokines and chemokine receptors involved in the neuroinflammatory response to WNV. Chemokines and the chemokine receptors upregulated in the brains of WNV-infected mice
Recent studies suggest that chemokine expression by the different cell types within the CNS can have counterbalancing effects on leukocyte localization within brains of WNV-infected mice. Neuronal production of CXCL10 promotes T cell migration into the parenchyma of the CNS (Klein et al. 2005; Zhang et al. 2008); however, the polarized expression of CXCL12 at the basal lateral surface of the endothelial cells can impede migration by retaining T cells in perivascular spaces (McCandless et al. 2008). Normally, the restriction of leukocytes to perivascular spaces within the CNS is a protective measure that limits the neuropathological consequences of immune cell infiltration. However, in the case of WNV, this restriction is counterproductive to viral clearance and survival. Indeed, McCandless et al. demonstrated that antagonism of CXCR4, the receptor for CXCL12, enhanced intraparenchymal migration of CD8+ T cells and resulted in decreased viral loads within the CNS, reduced immunopathology and enhanced survival (McCandless et al. 2008).
While chemokines control the recruitment and distribution of immune cells within the CNS, cytokines play a central role in regulating the activation of these cells. A recent study suggests that the cytokine IL-1 plays an important, though indirect role, in controlling the activation of effector T cells within the CNS of WNV-infected mice (Durrant et al. 2013). Mice deficient for IL-1 receptor (IL-1R1) exhibited enhanced susceptibility to WNV infection compared to WT mice despite the fact that CD8+ and CD4+ T cells were efficiently recruited to the parenchyma of the CNS. Ex vivo analysis of T cells recovered from IL-1R1 deficient mice indicated that CD8+ T cells were capable of responding to WNV, but the effector function of CD4+ T cells was impaired. However, the lack of IL1-R1 expression on CD8+ or CD4+ T cells in WT mice suggested that IL-1 did not directly act on either of these cell types. Further characterization of immune cells within the CNS of WNV-infected IL-1R1−/− mice demonstrated that IL-1 was required for the full activation of DC-like APCs within the CNS. Moreover, adoptive transfer of WT DC-like APCs restored T cell activity in the CNS of WNV-infected IL-1R1−/− mice, confirming the requirement of IL-1 responsive DCs for optimal activation of the effector T cell response during WNV neuroinvasion and demonstrating the importance of professional APCs to the neuroimmune response to WNV.
Conclusions
The dramatic increase in the number of WNV infections during the most recent transmission season within the USA and the outbreaks in Eastern Europe serve as a reminder that WNV is likely to remain a public health concern in the coming years. It is now clear that the neuroimmune response to WNV is a dynamic, multifactorial process involving the cell-intrinsic immune response as well as the cell-mediated innate and adaptive responses. A more complete understanding of these immune processes will be essential for the development of novel therapeutic agents for treating the severe neurological symptoms associated with WNV infection. Experiments in mice suggest that therapies modulating the neuroinflammatory can improve the outcome of WNV infection (Getts et al. 2012; McCandless et al. 2008). However, the dual nature of neuroinflammatory responses will necessitate the development of highly directed therapies capable of maximizing the neuroprotective effects of infiltrating immune cells while minimizing the potential for immunopathology.
References
- Bai F, Kong KF, Dai J, Qian F, Zhang L, Brown CR, Fikrig E, Montgomery RR. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J Infect Dis. 2010;202:1804–1812. doi: 10.1086/657416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley DW, Li L, Suderman MT, Barrett AD. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology. 2002;296:17–23. doi: 10.1006/viro.2002.1372. [DOI] [PubMed] [Google Scholar]
- Beasley DW, Davis CT, Whiteman M, Granwehr B, Kinney RM, Barrett AD (2004) Molecular determinants of virulence of West Nile virus in North America. Archives of virology. Supplementum (18):35–41 [DOI] [PubMed]
- Beasley DW, Whiteman MC, Zhang S, Huang CY, Schneider BS, Smith DR, Gromowski GD, Higgs S, Kinney RM, Barrett AD. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J Virol. 2005;79:8339–8347. doi: 10.1128/JVI.79.13.8339-8347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehin AC, Mouries J, Frenkiel MP, Dadaglio G, Despres P, Lafon M, Couderc T. Dynamics of immune cell recruitment during West Nile encephalitis and identification of a new CD19+ B220-BST-2+ leukocyte population. J Immunol. 2008;180:6760–6767. doi: 10.4049/jimmunol.180.10.6760. [DOI] [PubMed] [Google Scholar]
- Brien JD, Uhrlaub JL, Nikolich-Zugich J. West Nile virus-specific CD4 T cells exhibit direct antiviral cytokine secretion and cytotoxicity and are sufficient for antiviral protection. J Immunol. 2008;181:8568–8575. doi: 10.4049/jimmunol.181.12.8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci. 2009;122:3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- Cheeran MC, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. Differential responses of human brain cells to West Nile virus infection. J Neurovirol. 2005;11:512–524. doi: 10.1080/13550280500384982. [DOI] [PubMed] [Google Scholar]
- Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nature Medicine. 2013;19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Gale M, Jr, Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349–10358. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, Khromykh AA, Diamond MS. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol. 2011;85:5664–5668. doi: 10.1128/JVI.00232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Wang P, Bai F, Town T, Fikrig E. Icam-1 participates in the entry of West Nile virus into the central nervous system. J Virol. 2008;82:4164–4168. doi: 10.1128/JVI.02621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz JA, Da Rosa AP, Guzman H, Xu F, Xiao SY, Popov VL, Vasconcelos PF, Tesh RB. West Nile virus infection of primary mouse neuronal and neuroglial cells: the role of astrocytes in chronic infection. AmJTrop Med Hyg. 2006;75:691–696. [PubMed] [Google Scholar]
- Durrant DM, Robinette ML, Klein RS. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med. 2013;210:503–516. doi: 10.1084/jem.20121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Gale M., Jr West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J Virol. 2006;80:2913–2923. doi: 10.1128/JVI.80.6.2913-2923.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M., Jr The host response to West Nile Virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Terry RL, Getts MT, Muller M, Rana S, Deffrasnes C, Ashhurst TM, Radford J, Hofer M, Thomas S, Campbell IL, King NJ. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflammation. 2012;9:246–254. doi: 10.1186/1742-2094-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J, Howes R, Lightman S. The blood-retinal barrier in experimental autoimmune uveoretinitis. Leukocyte interactions and functional damage. Laboratory investigation. J Tech methods Pathol. 1994;70:39–52. [PubMed] [Google Scholar]
- Hussmann KL, Samuel MA, Kim KS, Diamond MS, Fredericksen BL. Differential replication of pathogenic and nonpathogenic strains of West Nile virus within astrocytes. J Virol. 2013;87:2814–2822. doi: 10.1128/JVI.02577-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nature reviews. Neuroscience. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Johnston C, Jiang W, Chu T, Levine B. Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol. 2001;75:10431–10445. doi: 10.1128/JVI.75.21.10431-10445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Verma S, Nerurkar VR. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J Neuroinflammation. 2010;7:73–87. doi: 10.1186/1742-2094-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, McDermott DH, Murphy PM. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis. 2008;197:262–265. doi: 10.1086/524691. [DOI] [PubMed] [Google Scholar]
- Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, Stramer SL, Murphy PM. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis. 2010;201:178–185. doi: 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol. 2011;186:471–478. doi: 10.4049/jimmunol.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes H, Redig P, Glaser A, Armien A, Wunschmann A. Clinical findings, lesions, and viral antigen distribution in great gray owls (Strix nebulosa) and barred owls (Strix varia) with spontaneous West Nile virus infection. Avian Dis. 2007;51:140–145. doi: 10.1637/0005-2086(2007)051[0140:CFLAVA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Saito T, Crochet N, Barton DJ, Gale M, Jr, Silverman RH. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA. 2010;16:2108–2119. doi: 10.1261/rna.2244210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci U S A. 2008;105:11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- Morrey JD, Olsen AL, Siddharthan V, Motter NE, Wang H, Taro BS, Chen D, Ruffner D, Hall JO. Increased blood–brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J Gen Virol. 2008;89:467–473. doi: 10.1099/vir.0.83345-0. [DOI] [PubMed] [Google Scholar]
- Omalu BI, Shakir AA, Wang G, Lipkin WI, Wiley CA. Fatal fulminant pan-meningo-polioencephalitis due to West Nile virus. Brain Pathol. 2003;13:465–472. doi: 10.1111/j.1750-3639.2003.tb00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M Jr (2012) IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS pathogens 8, e1003039 [DOI] [PMC free article] [PubMed]
- Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S. West Nile virus-induced disruption of the blood–brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol. 2012;93:1193–1203. doi: 10.1099/vir.0.040899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/JVI.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci U S A. 2007;104:17140–17145. doi: 10.1073/pnas.0705837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarin C, Stohlman SA, Atkinson R, Ransohoff RM, Bergmann CC. Monocytes regulate T cell migration through the glia limitans during acute viral encephalitis. J Virol. 2010;84:4878–4888. doi: 10.1128/JVI.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. RNase L plays a role in the antiviral response to West Nile virus. J Virol. 2006;80:2987–2999. doi: 10.1128/JVI.80.6.2987-2999.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Schwartz M. Harnessing monocyte-derived macrophages to control central nervous system pathologies: no longer 'if' but 'how'. J Pathol. 2013;229:332–346. doi: 10.1002/path.4106. [DOI] [PubMed] [Google Scholar]
- Shipley JG, Vandergaast R, Deng L, Mariuzza RA, Fredericksen BL. Identification of multiple RIG-I-specific pathogen associated molecular patterns within the West Nile virus genome and antigenome. Virology. 2012;432:232–238. doi: 10.1016/j.virol.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Kimura T, Mizutani T, Kariwa H, Takashima I. Different chemokine expression in lethal and non-lethal murine West Nile virus infection. J Med Virol. 2004;74:507–513. doi: 10.1002/jmv.20205. [DOI] [PubMed] [Google Scholar]
- Shirato K, Miyoshi H, Goto A, Ako Y, Ueki T, Kariwa H, Takashima I. Viral envelope protein glycosylation is a molecular determinant of the neuroinvasiveness of the New York strain of West Nile virus. J Gen Virol. 2004;85:3637–3645. doi: 10.1099/vir.0.80247-0. [DOI] [PubMed] [Google Scholar]
- Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J Virol. 2008;82:8956–8964. doi: 10.1128/JVI.01118-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati E, McCandless EE, Klein RS, Diamond MS. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol. 2007;81:9801–9811. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja MK, Diamond MS, Gale M Jr (2010) IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS pathogens 6, e1000757 [DOI] [PMC free article] [PubMed]
- Suthar MS, Diamond MS, Gale M., Jr West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11:115–128. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M, Jr, Diamond MS. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J Virol. 2010;84:12125–12138. doi: 10.1128/JVI.01026-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Brien JD, Thackray LB, Virgin HW, Cresswell P, Diamond MS. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J Virol. 2011;85:11557–11566. doi: 10.1128/JVI.05519-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M Jr, Virgin HW, Klein RS, Sen GC, Diamond MS (2012) 2'-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS pathogens 8, e1002698 [DOI] [PMC free article] [PubMed]
- Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity. 2009;30:242–253. doi: 10.1016/j.immuni.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol. 2007;81:10933–10949. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood–brain barrier. Virology. 2009;385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. Reversal of West Nile virus-induced blood–brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology. 2010;397:130–138. doi: 10.1016/j.virol.2009.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lobigs M, Lee E, Mullbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003;77:13323–13334. doi: 10.1128/JVI.77.24.13323-13334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Wang P, Bai F, Zenewicz LA, Dai J, Gate D, Cheng G, Yang L, Qian F, Yuan X, Montgomery RR, Flavell RA, Town T, Fikrig E (2012a) IL-22 signaling contributes to West Nile encephalitis pathogenesis. PloS one 7, e44153 [DOI] [PMC free article] [PubMed]
- Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13:753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Investig. 2010;120:1368–1379. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005;109:181–190. doi: 10.1007/s00401-004-0928-x. [DOI] [PubMed] [Google Scholar]
- Wunschmann A, Shivers J, Bender J, Carroll L, Fuller S, Saggese M, van Wettere A, Redig P. Pathologic findings in red-tailed hawks (Buteo jamaicensis) and Cooper's hawks (Accipiter cooper) naturally infected with West Nile virus. Avian Dis. 2004;48:570–580. doi: 10.1637/7170-022004R. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol. 2008;180:2641–2649. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]