

Abstract
Hypertension is a risk factor for other cardiovascular diseases and endothelial dysfunction was found in humans as well as in various commonly employed animal experimental models of arterial hypertension. Data from the literature indicate that, in general, endothelial dysfunction would not be the cause of experimental hypertension and may rather be secondary, that is, resulting from high blood pressure (BP). The initial mechanism of endothelial dysfunction itself may be associated with a lack of endothelium-derived relaxing factors (mainly nitric oxide) and/or accentuation of various endothelium-derived constricting factors. The involvement and role of endothelium-derived factors in the development of endothelial dysfunction in individual experimental models of hypertension may vary, depending on the triggering stimulus, strain, age, and vascular bed investigated. This brief review was focused on the participation of endothelial dysfunction, individual endothelium-derived factors, and their mechanisms of action in the development of high BP in the most frequently used rodent experimental models of arterial hypertension, including nitric oxide deficient models, spontaneous (pre)hypertension, stress-induced hypertension, and selected pharmacological and diet-induced models.
1. Introduction
Cardiovascular diseases account for about one-third of premature deaths in men and one-quarter of premature deaths in women, and arterial hypertension is one of the most significant risk factors for cardiovascular diseases. Despite current knowledge and extensive clinical and experimental research, the cause of hypertension remains unknown in about 95% of all cases. There are several factors that—alone or in combination—can increase the risk of developing primary hypertension in humans. In general, these include genetic and environmental factors.
Genetic association studies have identified polymorphisms in several candidate genes (e.g., angiotensinogen, angiotensin-converting enzyme, alpha-adducin, beta-adrenergic receptors, endothelial nitric oxide synthase (NOS), cytochrome P450 2C19, and nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase)) and several genomic sites that may include other genes contributing to primary hypertension [1–11]. However, none of these genetic abnormalities seems to be responsible for a significant portion of hypertension in the general population. Yet the influence of genetic factors may be accentuated and disease can be triggered by interaction of several gene polymorphisms or with environmental inputs such as sedentary life style, smoking, dietary factors (high salt, sugar, fat/cholesterol and alcohol intake, and low potassium and calcium intake) and chronic stress.
It is well known that pathophysiological characteristics of essential hypertension involve, besides other factors, increased total peripheral resistance. Thus, several experimental models of hypertension have been developed in rodents to study the mechanisms of blood pressure (BP) regulation in order to better understand the cause and consequences of human arterial hypertension [12]. These experimental models allow not only to modify all potential factors—diet, surrounding environment, and genetic information (by using specific gene knock-out or transgenic models)—but also to study the influence of interaction of specific risk factors in the etiology of hypertension. Moreover, these models are developed to allow investigation of endothelial function of various arteries, either in vivo or in vitro, including determination of mechanisms involved in loss of normal vascular function.
Endothelium, the inner layer of the blood vessels, was originally considered to be a passive barrier between blood and the vascular wall. This opinion was broken after the discovery of prostaglandin X by Bunting and coworkers in 1976 [13]. Their study showed that the arterial wall can synthetize and release a vasoactive substance that is able to relax arterial strips and to inhibit platelet aggregation. Prostaglandin (PG) X was soon identified as PGI2 (prostacyclin) synthetized and released by the endothelial cells [14]. However, only the discovery of the obligatory role of the endothelium in relaxation of the arterial wall by the chemically unknown “endothelium-derived relaxing factor (EDRF)” by Furchgott and Zawadski in 1980 [15] and identification of EDRF as nitric oxide (NO) started the real age of “endothelial research.” Soon it became clear that prostacyclin and NO are not the only vasoactive substances released by the endothelium.
Today it is known that the endothelium produces various substances collectively termed endothelium-derived relaxing factors (EDRFs) and endothelium-derived constricting factors (EDCFs), based on their function in modulation of the arterial wall. In addition to these factors there are vasorelaxing endothelium-derived hyperpolarizing factors (EDHFs), whose chemical moiety is still under discussion. This brief, though not exhausting, review is focused on main mechanisms involved in the development of endothelial dysfunction in selected models of experimental hypertension.
2. Endothelium-Derived Factors
2.1. Endothelium-Derived Relaxing Factors
NO, PGI2, and hydrogen sulfide (H2S) were described as EDRFs and NO is the best characterized EDRF. In addition, there is a group of EDRFs that produce relaxation due to hyperpolarization and they all go under the term EDHFs.
In mammals NO is produced by one of four nitric oxide synthase (NOS) isoenzymes (nNOS/NOS I, that is, neuronal NOS; iNOS/NOS II, that is, inducible NOS; eNOS/NOS III, that is, endothelial NOS; or mtNOS/NOS IV, that is, mitochondrial NOS). They are involved in the modulation of various pathophysiological or diseased states. Endothelial NOS is the main isoform expressed in the endothelium. It is localized in cellular plasma membranes in the caveolae and in the membrane of the Golgi body [16]. Briefly, the regulation of NO production in the endothelium depends on bioavailability of several cofactors, phosphorylation of eNOS in specific sites, co- and posttranslational lipid modifications, and it is a subject of negative feedback regulation by NO itself [17–20]. NO can diffuse from endothelial cells to vascular smooth muscle cells and guanylate cyclase (GC) has been identified as an intracellular receptor for NO (Figure 1). Its activation leads to the release of the second messenger cyclic guanosine monophosphate (cGMP) and activation of protein kinase (PK) G-dependent mechanisms resulting in reduction of intracellular calcium concentration ([Ca2+]i), followed by vasorelaxation. In addition, there are many other functions of NO participating in the regulation of gene transcription, mRNA translation, and protein modification of various enzymes involved in mitochondrial respiration, mitogenesis, and growth [19, 21, 22].
Figure 1.
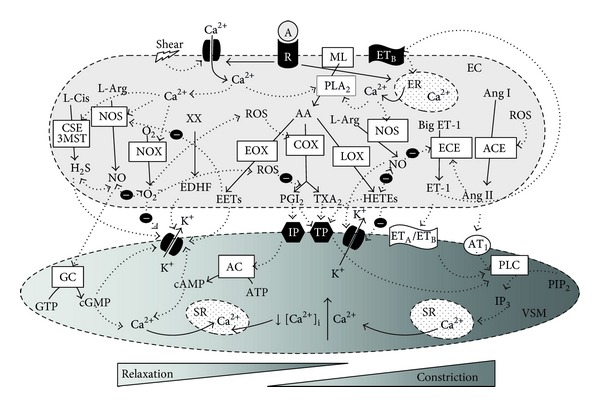
A brief scheme of interactions among the individual endothelium-derived factors and their mechanisms of action in the endothelial cells. Abbreviations are explained in the list of abbreviations. The activation of the appropriate receptor by its agonist as well as shear stress leads to alterations in the intracellular calcium concentration in the endothelial cells which affect the activity of all NOS, CSE, NOX, and PLA2 resulting in the release of NO, H2S, ROS, and AA-derived metabolites, respectively. ROS can further inhibit (marked by “−” sign) the production of PGI2 and to elevate TXA2 and Ang-II production. In addition, there are significant interactions among the NO, H2S, and superoxide as well as among NO, Ang-II, ET-1, and HETEs. Individual EDRFs then affect the vascular smooth muscle cells, via modulation of the appropriate receptors or channels, resulting in the respective vascular smooth muscle cell relaxation and constriction.
As mentioned above, PGI2 was the first endothelium-derived relaxing factor discovered. PGI2 is the main product of arachidonic acid (AA), cyclooxygenase- (COX-) mediated metabolism in vascular tissues. In normal conditions, PGI2 produced by prostacyclin synthase acts on the prostacyclin receptor (IP). Activation of the IP receptor leads to G protein-coupled increase in the second messenger cyclic adenosine monophosphate (cAMP) and PK A activation, resulting in decreased [Ca2+]i and vasorelaxation. In the vasculature, PGI2 inhibits cell adhesion, thrombosis, inflammation, apoptosis, and proliferation. It participates in vasorelaxation and BP regulation; however, its role in these functions is rather minor as compared to NO [23, 24]. In vitro studies have suggested that there is a cross-talk between NO and PGI2 production as NO was shown to activate the enzymes involved in PGI2 synthesis and vice versa [25, 26].
Hydrogen sulfide can be produced in the endothelial cells by cystathionine γ-lyase (CSE) and 3-mercaptopyruvate sulfur esterase (3MST) [27, 28]. Genetic deletion of CSE in mice markedly reduces H2S levels in the aorta and other tissues. Mutant mice lacking CSE display diminished endothelium-dependent vasorelaxation after muscarinic cholinergic stimulation of vascular endothelial cells and pronounced hypertension. CSE is physiologically activated by calcium-calmodulin, which is a mechanism for H2S formation in response to vascular activation [27].
2.2. Endothelium-Derived Hyperpolarizing Factors
Although many substances produced by the endothelium act as EDHFs, there is continuous discussion on the chemical moiety and molecular signaling of individual hyperpolarization-producing factors. Substances like NO, hydrogen peroxide (H2O2), carbon monoxide, adenosine [29], and K+ itself [30] possess the ability to induce hyperpolarization. Furthermore, recently published studies showed that H2S yielded significant hyperpolarization of vascular smooth muscle cells [31, 32].
In addition to these factors, several authors included epoxyeicosatrienoic acids (EETs) in EDHFs. EETs are synthesized in endothelial cells in the AA pathway, in which AA is converted by cytochrome P450 (CYP) epoxygenases to 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. The targets of EETs are large conductance calcium-activated K+ channels (BKCa) in vascular smooth muscle cells, as well as small (SKCa) and intermediate (IKCa) conductance KCa channels of endothelial cells, whose activation leads to hyperpolarization [33]. Besides regulation of vascular tone, EETs participate in vascular signaling processes involved in inflammation and angiogenesis.
The importance of individual EDHFs may vary among various vascular beds and animal species. Regarding the issue of artery size, EDHF-mediated relaxation is a dominant component of acetylcholine- (ACh) induced relaxation in the small arteries, while its contribution to relaxation of the aorta is minor [23]. Differences in the mechanism of ACh-induced relaxation were observed also in mice with disruptions in the NOS genes, that is, eNOS−/−, n/eNOS−/−, and n/i/eNOS−/− mice [34]. Studies of Takaki et al. [34] revealed that EDHF-mediated relaxation and hyperpolarization in response to ACh in the small mesenteric arteries of mice were progressively reduced as the number of disrupted NOS genes increased, whereas vascular smooth muscle function was preserved, suggesting that loss of eNOS expression alone was compensated by other NOS genes. However, EDHF/H2O2-mediated responses were completely absent in n/i/eNOS−/− mice. Studies showed that NOS isoforms (nNOS, iNOS, and eNOS), especially eNOS, produce both NO and superoxide anions, and the latter is dismutated by superoxide dismutase (SOD) to EDHF/H2O2, which elicits hyperpolarization followed by vasodilation. In addition, the authors showed that NOS uncoupling was not involved in reactive oxygen species (ROS) production, as modulation of tetrahydrobiopterin (BH4) synthesis had no effect on EDHF-mediated relaxation, and the BH4/dihydrobiopterin ratio was comparable in the small mesenteric arteries and the aorta. Collectively, studies of Takaki et al. [34] provided a novel concept on the diverse roles of endothelial NOS system mainly contributing to the EDHF/H2O2 responses in microvessels while serving as NO-generating system in the large arteries.
The ratio of NO-dependent and NO-independent ACh-induced relaxation in various vascular beds is, in addition to artery size, affected by the cardiovascular genotype as well as by age. We found that NO-dependent and independent components of ACh-induced relaxation were approximately equal in the femoral artery of young (7 weeks' old) normotensive male Wistar-Kyoto (WKY) rats, while NO-dependent relaxation was dominant in age-matched spontaneously hypertensive rats (SHR). Furthermore, aging reduced NO production and NO-dependent relaxation in both normotensive and hypertensive rats [35–37].
2.3. Endothelium-Derived Constricting Factors
The endothelium can produce many constrictor factors including endothelin-1 (ET-1) and angiotensin II (Ang II), which appear to be the most powerful constrictors in the vasculature. Data in the literature suggest that ET-1 acts as mediator of Ang II-produced vasoconstriction [38–40].
However, experimental studies showed that significant effects of EDCFs in hypertensive rats were associated with COX products such as thromboxane A2 (TXA2), prostaglandins H2, E2, and F2α, isoprostanes, and monohydroxyeicosatetraenoic acids (HETEs), whose effect is associated mainly with thromboxane/prostaglandin endoperoxide receptor (TP) [41, 42]. EDCF-induced responses can be eliminated, at least partially, by COX-2 inhibitors and TP receptor blockers [43–45]. Interestingly, in conditions of lack or malfunction of the IP receptor, PGI2 can produce contraction via the TP receptor both in young [46] and aged [47] rats.
Furthermore, ROS such as H2O2, hydroxyl anion, and mainly superoxide were shown to constrict the arteries by modulation of the action of other endothelium-derived factors. ROS can be produced by mitochondria, uncoupled NOS, and various oxidases—xanthine oxidase, COX, lipoxygenases (LOX), and CYP monooxygenases. Yet NADPH oxidase was shown to serve as a primary source of so called “kindling radicals” [48]. The accentuated release of ROS is considered the main factor involved in vascular aging [37]. On the other hand, optimal ROS production is required for normal cell signaling as ROS serve as second messengers involved in activation of nuclear factor kappa B (NF-κB) and further in regulation of mitogen-activated protein kinase (MAPK) pathways including extracellular signal-regulated kinases 1 and 2 ERK1/2, p38 mitogen-activated protein kinase (p38MAPK), c-Jun N-terminal kinase (JNK), and extracellular-signal-regulated kinase 5 (ERK5), with their respective importance in cell growth, inflammation, apoptosis, and cell differentiation [49–52].
In the vasculature, ROS possess pleiotropic effects which might be variable depending on the oxidative status of the tissue. For example, H2O2-induced oxidative stress increased vascular TP sensitivity and predisposed segments of the small arteries to prostanoid-induced constriction. Conversely, H2O2-induced vasodilation was observed in the same segments in the presence of antioxidants targeting radicals downstream of H2O2 [53].
Thus, correct function of the endogenous antioxidant defense systems, in association with the exogenous antioxidants, is necessary for the maintenance of balance between EDRFs and EDCFs and for normal vascular function.
3. Endothelial Dysfunction and Its Classification
Besides the balance between ROS and NO, there is complex cross-talk among the individual endothelium-derived factors with the aim to maintain appropriate endothelial function (Figure 1). Dysregulation of this cross-talk can result in alteration of normal physiological processes carried out by the endothelium, including reduction of its anticoagulant and antithrombotic properties, acceleration of vascular growth and remodeling, and impairment of endothelium-dependent vasorelaxation, that is, in endothelial dysfunction (ED). Yet neither the factors and mechanisms that modulate the balance between relaxing, anticoagulant, antithrombotic, and anti-mitotic factors on the one side and constricting, proaggregatory, and promitogenic factors on the other, nor the processes in which endothelium loses its protective functions have been fully elucidated so far. Genetic predisposition and aging [35, 54], several environmental factors such as sedentary life style, smoking, improper diet, and stress might participate in the transformation of the endothelium from a protective to a “health-threatening” organ.
In the last two decades, enormous research on ED has been conducted; however, the issue whether ED is a cause (i.e., primary) or a consequence (i.e., secondary) of high BP remains still open. In fact, the increasing, but still not exhausting, knowledge on the etiology and pathophysiology of ED suggests the need of proper classification of this disorder that could facilitate the integration of current state of the art in this field. Such classification has been proposed by Evora [55] and it is briefly described in Table 1. It includes etiological, functional, and evolutionary or prognostic aspects of ED. It should however be noted that the proposed classification has not yet been widely discussed, opposed, or accepted among scientists. As mentioned later in this review, a correct etiological classification of ED might be very difficult in some experimental models of hypertension. Moreover, ED may occur in other diseased states independently of hypertension [56].
Table 1.
Classification of endothelial dysfunction proposed by Evora [55]. For more details and explanation see the original article.
| Endothelial dysfunction classification | |
| (I) Etiological classification | |
| (A) Primary or “genotypic”: demonstrated for example in normotensive patients with familial antecedents of essential arterial hypertension. | |
| (B) Secondary or “phenotypic”: present for example in cardiovascular diseases including arterial hypertension. | |
| (II) Functional classification | |
| (A) “Vasotonic”: implying a risk of vasospasm and thrombosis. | |
| (B) “Vasoplegic”: associated with a pathological release of endothelium-derived relaxing factors. | |
| (III) Evolutionary or prognostic classification | |
| (A) Reversible | |
| (B) Partially reversible | |
| (C) Irreversible |
4. Endothelial Dysfunction in Experimental Hypertension
4.1. Nitric Oxide-Deficient Models of Hypertension
One of the first diseases associated with reduced bioavailability of EDRF and altered vascular function was arterial hypertension. The importance of endogenous eNOS-derived NO production in regulation of vascular function underwent increasing investigation with implementation of NOS inhibitors. Several studies reported elevation of BP after intravenous administration of NOS inhibitors in rabbits [57], guinea pigs [58], dogs [59], monkeys [60], and rats [61, 62]. The increase in BP was confirmed also during long-term oral treatment with NOS inhibitors [63–67]. The experimental model of “NG-nitro-L-arginine methyl ester (L-NAME)-induced” or “NO-deficient” hypertension [68] was established to investigate not only the role of NO in vascular function and BP regulation but also in maintenance of homeostasis in the whole cardiovascular system.
Briefly, the complex mechanisms responsible for BP increase in this model of hypertension involve attenuated vascular relaxation and increased contraction in different parts of the vascular tree [69–72]. In addition, chronic NOS inhibition increases endothelium-dependent contractions of the rat aorta by inducing COX-2 expression and augmenting the production of EDCFs [73, 74] as well as by accentuation of ET-1 effect. The complex mechanism of BP increase in NO-deficient hypertension involves also accentuation of the sympathetic system tone, the renin-angiotensin system, and oxidative stress [75–78]. Furthermore, NO can modulate vascular remodeling independently of BP, which contributes to the maintenance of high BP [21, 79].
In the NO-deficient model of experimental hypertension, due to chronic nonspecific inhibition of NO production, the development of ED is associated with a gradual elevation of BP. Thus, regarding the abovementioned classification, ED in this model of hypertension might be classified as primary. Indeed, functional and morphological alterations observed in the model of L-NAME-induced hypertension [79] were similar to those observed in mice, in which three NOS isoforms were disrupted, that is, n/i/eNOS−/− [80]. Moreover, studies that used triple NOS disrupted n/i/eNOS−/− mice showed that the magnitude of hypertension in the triply NOS−/− mice was similar to that in mice with the eNOS gene disrupted singly (eNOS−/−) or doubly (n/eNOS−/− or i/eNOS−/−) [81]. Besides hypertension, ED due to lack of NO has been shown to be involved also in other pathological states. For example, mice with single disruption of the eNOS gene (eNOS−/−) were insulin resistant, displayed increased triglycerides and free fatty acid levels, defective mitochondrial β-oxidation, and renal dysfunction [82–85].
The abovementioned studies suggest that hypertension is a common characteristic of the lack of eNOS-produced NO. Moreover, studies pointed out the association between eNOS-derived NO, primary ED, hypertension, and metabolic disorders involved in the cluster of metabolic syndrome as well as the development of atherosclerosis. Indeed, elevated BP and altered vascular function were observed in various genetic and diet-induced models of metabolic syndrome [86].
4.2. Models of Spontaneous Genetic Hypertension
Contrary to the NO-deficient model of hypertension, studies experimenting with the genetic model of spontaneously hypertensive rats produced inconsistent results regarding the role of NO and ED in the development of hypertension. The findings of ED in SHR depend on many factors, such as age, artery type, and methods used for determination of vascular function. A similar inconsistency was observed in NO production in SHR, in which reduced [87, 88], unchanged [89], and elevated [90–94] vascular NO synthesis and/or NOS expression were observed. Our previous reviewing of the literature showed that the enormous variability in the results concerning ED in SHR might result, on the one hand, from various methodologies used for determination of vascular function. On the other hand, regardless of methodology, aging seems to be an important factor in studying ED. ED was observed mainly in adult and aged (older than 25 weeks) and not in young (less than 6 weeks' old) SHR [95]. In our studies we observed elevated NOS activity in the aorta also in borderline hypertensive rats (BHR), which were the first filial generation of offspring of one normotensive and one hypertensive parent. In these rats, borderline hypertension (with systolic BP about 140 mmHg) was recorded in adulthood [92]. We also determined positive correlation between BP and NO-dependent component of ACh-induced relaxation in the femoral artery in adult rats [96]. We revealed that ED in the femoral artery in adult BHR and SHR males resulted rather from decreased NO-independent relaxation than from a lack of NO, a finding suggested also by other authors [47, 97]. In addition, we observed a decreased NO-independent component of ACh-induced relaxation already in young (7 weeks' old) male and female SHR [36, 98]. Interestingly, in young BHR and SHR females, ED was underlined also by a decrease of the NO-dependent component of relaxation and elevated superoxide production, which was not observed in males, suggesting sex-related differences in the mechanism of ED development.
To elucidate the causal relation between ED and high BP, we used young BHR rats whose BP is significantly elevated versus WKY as early as at the age of 7 weeks. However, despite higher BP and vascular ROS production, we did not observe ED in these rats as reduced NO-dependent relaxation was fully compensated be elevated NO-independent relaxation [98]. Thus, our findings in young BHR rats are not supportive of the idea that ED precedes hypertension in this particular model of hypertension. According to the suggested ED classification, ED in BHR (and supposedly also in SHR) seems to be rather secondary and other nonendothelial mechanisms are responsible for the induction of BP increase.
However, elevated BP itself can trigger damage of endothelial function and vascular remodeling [99]. Chronic presence of high BP per se was found to elicit increased arterial superoxide production by activating directly a PKC-dependent NADPH oxidase pathway, but also, in part, via activation of the local renin-angiotensin system [100].
Regarding the role of ROS in the development of hypertension, in addition to the reduction of bioavailable NO, ROS may also reduce hyperpolarization and PGI2 synthesis [35]. Elevated ROS production was observed in the aorta of young BHR and SHR [98] as well as in adult SHR [45, 101].
Recently, the role of excessive NO production by iNOS has been suggested in the development of ED. Elevated expression of iNOS was observed already in young SHR rats but not in WKY and inhibition of iNOS prevented BP increase [87, 102]. Yet whether iNOS activation participates in ED development has to be further investigated as normal iNOS expression was detected in the small mesenteric arteries from SHR [45]. However, in association with high NO production observed in SHR, elevated ROS may result in formation of peroxynitrite, thereby promoting nitrosative stress and ED. The interaction between NO and superoxide occurs at an extremely rapid rate of 6.7 × 109 mol/L−1·s−1 and is about 3-times faster than the reaction rate for superoxide with SOD [103].
ROS have been implicated also in the mechanism of AA-derived EDCFs-mediated development of ED in genetic hypertension due to activation of COX [104]. ACh-induced constrictions were observed in adult SHR and BHR as well as in aged WKY [105]. As ACh led to release of PGI2, PGH2, as well as PGE2 and PGF2α acting via TP receptors in the aorta [46, 105], all these substances may be involved in EDCF-induced ED in SHR. The contribution of EDCFs to ED was determined also in other arteries (mesenteric, renal, basilar, carotid, and others), yet the nature of EDCFs might be different from that observed in the aorta [105].
Regarding the mechanism of triggering elevated EDCFs release, accentuated accumulation of calcium in the endothelial cells was found to be an initial prerequisite. As intracellular calcium is required also for eNOS [106] and NADPH oxidase activation [107], it seems plausible that defective calcium handling or signaling in the endothelium [108], resulting in abnormal calcium accumulation, might account for the acceleration of EDCFs production resulting in ED. Then even mild ED, in association with increased calcium influx and sensitivity observed in the vascular smooth muscle cells of SHR [109], can markedly facilitate contractility and initiate BP increase.
4.3. Pharmacological and Diet-Induced Models of Hypertension
In addition to the abovementioned models of hypertension, there are various pharmacological and diet-induced models. These models allow investigating the involvement of specific factors (salt, corticoids, lead, sugar, etc.) and pathways which may affect endothelial function and thus trigger the development of hypertension. Although the exact disorders in signal transduction resulting in hypertension in the individual models of hypertension may not be known, involvement of oxidative stress has been suggested in Ang II-induced hypertension [110], ET-1-induced hypertension [111], Dahl salt-induced hypertension [112], deoxycorticosterone acetate- (DOCA-) salt induced hypertension [113, 114], fructose-induced [115, 116], and lead-induced hypertension [117].
In some models reduced NO bioavailability was determined as a consequence of elevated ROS [112, 117], yet in other models of hypertension the participation of NO deficiency in ED seems to be of rather minor importance.
Numerous mechanisms implicated in ED development were observed in the model of Dahl salt-sensitive rats. Regarding EDCFs (likely PGH2 and TXA2), their contribution to ED was observed in carotid rings [118]. Lukaszewicz and Lombard [119] demonstrated restoration of normal vascular function in Dahl salt-sensitive rats with inhibition of the CYP4A/20-hydroxyeicosatetraenoic acid pathway, suggesting a direct role for this pathway in vascular dysfunction. Involvement of the endothelin receptor type A (ETA)-mediated effect of ET-1 in the development of hypertension was observed in adult but not in young Dahl rats [120, 121]. Furthermore, high salt diet significantly reduced expression and activity of endothelial dimethylarginine dimethylaminohydrolase (DDAH-2), involved in asymmetric dimethylarginine (ADMA, endogenous NOS inhibitor) degradation and reduced eNOS expression, independently of BP [122]. Involvement of iNOS in modulation of endothelial function was not observed in salt-induced hypertension as W1400 (specific iNOS inhibitor) failed to modify aortic function [123]. Interestingly, in the same study NO produced by renal medullary iNOS was found to prevent excessive increases in arterial BP.
Endothelial dysfunction associated with increased aortic superoxide content, elevated NADPH oxidase activity, and decreased phosphorylated eNOS levels in aortic rings was observed also in DOCA-salt treated rats [114]. Similarly, elevated oxidative stress, yet due to activation of xanthine oxidase, was found in both the aorta and mesenteric arteries of DOCA-salt hypertensive rats in association with elevated expression of the ETA receptor [113] and ET-1 gene [124]. Participation of elevated COX-2-derived factors that enhanced rat aortic contractility was determined in DOCA-salt treated rats by Adeagbo et al. [125]. Moreover, involvement of COX-2 mediated free radicals that impaired EDHF-mediated relaxation in the mesenteric arteries of DOCA-salt induced hypertension was also observed, while NO-dependent relaxation remained unaltered [126].
In the model of fructose-induced hypertension, the development of hypertension was associated with insulin resistance, hyperinsulinemia, and NO-dependent ED with preserved endothelium-independent vasomotion [127]. On the other hand, Lee et al. [128] did not observe reduced NOx levels or protein expression of constitutive NOS and iNOS in the aorta of fructose-treated rats. However, their results indicate that an increased expression of vascular ET-1 may be causally related to the development of hypertension due to high fructose intake. This finding is in agreement with the observation of similar relaxing responses to ACh and sodium nitroprusside in the mesenteric arteries of fructose-treated rats in which accentuated Ang II-induced constriction was observed [129]. Finally, in contrast to salt-induced hypertension, in fructose-induced hypertension elevated ADMA levels seem to be secondary to the early development of NADPH oxidase-independent oxidative stress and elevated iNOS expression [116].
On balance, the findings in the abovementioned models of hypertension suggest that vascular NO deficiency would not be primarily involved in the hypertension development in Dahl salt-treated, DOCA-salt-treated, and fructose-treated rats while ET-1 seems to contribute significantly to ED in these models. Again, specific mechanisms can be involved in initial stages of ED in various arteries investigated.
4.4. Stress-Induced Hypertension
Stress is another important factor that has been suggested in the development of hypertension. Although the problem of stress-related hypertension has been addressed in several studies, there are still conflicting data regarding causal relation between stress and hypertension in humans [130, 131]. In animal studies, some models of psychosocial stress were able to induce hypertension in normotensive rats [132, 133], while other models did not produce changes in BP [134–136]. In addition, animal studies revealed the importance of genetic factors in the etiology of stress-induced hypertension [136–138], thus genetically consistent rats, either normotensive or SHR, may not be always suitable for investigation of cardiovascular effects of stress. The main mechanisms involved in BP elevation in stress are associated with sympathoadrenal activation and less information is available on the role of ED in stress-induced models of hypertension.
To investigate the influence of chronic stress on vascular function, we employed the model of chronic social stress produced by crowding resulting from reduced living space. This model seems to be more relevant to the human situation [139, 140] than other stress models.
In our studies chronic crowding stress failed to increase BP in normotensive rats, either by the use of 2-week crowding in young females or of 8- or 12-week crowding in adult male WKY. Interestingly, at all time points investigated, we always found elevated vascular NO production in the aorta of stress-exposed WKY [98, 141–143]. Despite the consistency in these findings, the effect on vascular function was variable. In adult males exposed to 8-week stress, endothelium-dependent relaxation was elevated versus control in both femoral and mesenteric arteries, which was associated with improvement of NO-dependent relaxation [143, 144]. Notably, maximal ACh-induced relaxation was reduced after 12 weeks, while NO-dependent relaxation was still elevated, suggesting the development of NO-independent ED in crowded WKY. Furthermore, 2-week stress reduced NO-dependent relaxation in the femoral artery in young WKY females, but this was fully compensated by increase of the NO-independent component [98].
In contrast to normotensive rats, the same stress model led to elevation of BP in SHR and SHR-mothered BHR but not in Wistar-mothered BHR after 8-week exposure. However, stress-reduced relaxation was seen only in SHR [92]. In young females, 2-week stress led to acceleration of time-related increase of BP in BHR yet relaxation in the femoral artery was not altered despite elevated NO production [98].
Fuchs et al. [145] observed alterations in the mechanisms mediating endothelium-dependent relaxation to ACh in small mesenteric arteries isolated from adult male WKY and BHR rats after 10 days of repeated air-jet stress. In their study, decrease of NOS activity had a significantly larger inhibitory effect on ACh-induced relaxation in arteries from stressed compared with control BHR. COX-derived products contributed to ACh-induced relaxation of the small mesenteric arteries from stressed WKY rats, but not control WKY rats or BHR [145]. The same stress model led also to impairment of coronary artery relaxation in BHR [146]. In addition, the effect of air-jet on ACh-induced relaxation of the coronary artery was altered with aging in BHR. In young adult (3 months' old) BHR males exposure to stress produced an NOS-dependent increase in relaxation, while a decrease in relaxation was observed in aged (18 months' old) BHR. The impaired response to ACh observed in aged BHR was associated with superoxide anion, vasoconstrictor prostaglandins, and a loss of the component of relaxation that was NO-independent and K+ channel mediated [147].
In 129/SV mice exposed to 28 days of stress (consisting of exposure to rat, restraint stress, and tail suspension) impaired carbachol-induced endothelium-dependent vasorelaxation, increased superoxide production, and reduced aortic eNOS levels were observed. These changes were reversed by the glucocorticoid (GCC) receptor antagonist mifepristone [148].
The involvement of GCC in disruption of NO production and/or expression was reported in studies using cultured endothelial cells. Radomski et al. showed that GCC inhibited the expression of an iNOS but not of constitutive NOS in cultured porcine vascular endothelial cells [149]. The inhibitory effect of GCC on eNOS expression and NOx production was observed in cultured bovine coronary artery and aortic endothelial cells as well as in human umbilical vein endothelial cells [150, 151]. GCC was shown to downregulate NO production by limiting BH4 production also in cardiac microvascular endothelial cells [152]. Moreover, GCC response elements in the eNOS promoter region were demonstrated by Liu et al. [153]. In addition to modulation of NO, GCC was shown to downregulate COX-1 expression and PGI2 synthesis in the fetal pulmonary artery endothelial cells through activation of the glucocorticoid receptor (GR) and effects on COX-1 gene transcription [154].
Oxidative stress is another parameter that was shown to be increased by GCC. Decline in antioxidant defence by actions of corticosterone was evidenced by coordinate decreases in the activities of free-radical scavenging enzymes SOD, catalase, glutathione S-transferase, and glutathione reductase in the brain, liver, and heart of rats [155].
Surprisingly, in our studies in which elevated level of corticosterone resulted from chronic stress and not from pharmacological intervention, elevated corticosterone levels observed in adult WKY and young BHR and SHR failed to reduce NO-dependent relaxation compared to control rats [98, 143]. Our findings pointed out adapting mechanisms that play a role under chronic stress and may not be active in endothelial cell cultures and during acute stress exposure. We further considered the possible involvement of NOS isoforms different from eNOS.
NO has been previously suggested as a stress relieving molecule [156, 157] and activation of the vascular L-arginine/NO pathway may serve as an antistress system in vivo. Our results support the idea that elevated NO production in the vasculature can be considered one of the adaptation mechanisms protecting from sustained elevation of BP, at least in normotensive rats in allostasis. Long-term stress, however, may induce NO-independent ED that might be the initial step in the development of vascular remodelling followed by atherosclerosis and/or hypertension. Thus, in case of chronic stress-induced hypertension in genetically normotensive rats, NO-independent ED can precede hypertension and might be considered primary. Whether corticosterone is involved in the initiation of ED in stress-induced models of hypertension, or even in spontaneous hypertension, remains to be clarified.
5. Conclusion
The conclusion that can be drawn from reviewing the literature is that ED might be both a cause and consequence of high BP, depending on the model of hypertension used, strain, age, and vascular bed. The findings in the abovementioned models of hypertension suggest that ED, due to genetic or pharmacological disruption of eNOS-derived NO, observed before sustained elevation of BP, might be considered to be primary. Such ED is causally associated with reduced NO-dependent relaxation. On the other hand, ED observed in SHR and BHR seems to be EDCF associated and NO independent, and it can be considered to be secondary ED since numerous studies confirmed the presence of elevated BP before the development of ED. Furthermore, contribution of ET-1 to ED development was predominant in diet-induced models. Yet the most prevalent cause of ED seems to be oxidative stress that has been observed in all abovementioned experimental models of hypertension. As oxidative stress may result from a broad spectrum of genetic and environmental factors as well as from high PB per se, it is very difficult to distinguish between primary and secondary ED in experimental models of hypertension because the processes of endothelial function damage and elevation of BP are many times occurring simultaneously.
Acknowledgments
This study was supported by the Grants Nos. APVV-053-10 and VEGA 2/0084/14 and within the project of “ITMS 26240120020-Establishment of the Centre for the Research on Composite Materials for Structural, Engineering and Medical Applications-CEKOMAT II.”
Abbreviations
- A:
Agonist
- AA:
Arachidonic acid
- AC:
Adenylate Cyclase
- ACE:
Angiotensin-converting enzyme
- ADMA:
Asymmetric dimethylarginine
- ACh:
Acetylcholine
- Ang I:
Angiotensin I
- Ang II:
Angiotensin II
- AT1:
Angiotensin II receptor 1
- ATP:
Adenosine-5′-triphosphate
- BH4:
Tetrahydrobiopterin
- BHR:
Borderline hypertensive rats
- BKCa:
Large conductance calcium-activated K+ channel
- BP:
Blood pressure
- cAMP:
Cyclic adenosine monophosphate
- cGMP:
Cyclic guanosine monophosphate
- COX:
Cyclooxygenase
- CSE:
Cystathionine γ-lyase
- CYP:
Cytochrome P450
- DDAH-2:
Endothelial dimethylarginine dimethylaminohydrolase
- DOCA:
Deoxycorticosterone acetate
- EC:
Endothelial cell
- ECE:
Endothelin-1-converting enzyme
- ED:
Endothelial dysfunction
- EDCF(s):
Endothelium-derived constricting factor(s)
- EDHF(s):
Endothelium-derived hyperpolarizing factor(s)
- EDRF(s):
Endothelium-derived relaxing factor(s)
- EET(s):
Epoxyeicosatrienoic acid(s)
- eNOS/NOS III:
Endothelial nitric oxide synthase
- EOX:
Cytochrome P450 epoxygenases
- ER:
Endoplasmic reticulum
- ERK1/2:
Extracellular signal-regulated kinases 1 and 2
- ERK5:
Extracellular-signal-regulated kinase 5
- ET-1:
Endothelin-1
- ETA:
Endothelin receptor A
- ETB:
Endothelin receptor B
- GC:
Guanylate cyclase
- GCC:
Glucocorticoid
- GR:
Glucocorticoid receptor
- GTP:
Guanosine-5′-triphosphate
- HETE(s):
Monohydroxyeicosatetraenoic acid(s)
- H2O2:
Hydrogen peroxide
- H2S:
Hydrogen sulfide
- IKCa:
Intermediate conductance calcium-activated K+ channel
- iNOS/NOS II:
Inducible nitric oxide synthase
- IP:
Prostacyclin receptor
- IP3:
Inositol 1,4,5-trisphosphate
- JNK:
c-Jun N-terminal kinase
- KCa:
Calcium-activated K+ channel
- L-Arg:
L-arginine
- L-Cis:
L-cysteine
- L-NAME:
NG-nitro-L-arginine methyl ester
- LOX:
Lipoxygenase
- MAPK:
Mitogen-activated protein kinase
- ML:
Membrane lipids
- 3MST:
3-Mercaptopyruvate sulfuresterase
- mtNOS/NOS IV:
Mitochondrial nitric oxide synthase
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
Nuclear factor kappa B
- nNOS/NOS I:
Neuronal nitric oxide synthase
- NO:
Nitric oxide
- NOS:
Nitric oxide synthase
- NOX:
NADPH oxidase
- NOx:
Nitrate/nitrite
- PG:
Prostaglandin
- PGI2:
Prostacyclin
- PIP2:
Phosphatidylinositol 4,5-bisphosphate
- PK:
Protein kinase
- PLC:
Phospholipase C
- p38MAPK:
p38 mitogen-activated protein kinase
- R:
Receptor
- ROS:
Reactive oxygen species
- SHR:
Spontaneously hypertensive rats
- SKCa:
Small conductance calcium-activated K+ channel
- SOD:
Superoxide dismutase
- SR:
Sarcoplasmic reticulum
- TP:
Thromboxane/prostaglandin endoperoxide receptor
- TXA2:
Thromboxane A2
- VSM:
Vascular smooth muscle cell
- WKY:
Wistar-Kyoto rats
- XX:
Other, not specifically mentioned substances which may produce hyperpolarization.
Conflict of Interests
The author declares that there is no conflict of interests regarding the publication of this paper.
References
- 1.Baker M, Rahman T, Hall D, et al. The C-532T polymorphism of the angiotensinogen gene is associated with pulse pressure: a possible explanation for heterogeneity in genetic association studies of AGT and hypertension. International Journal of Epidemiology. 2007;36(6):1356–1362. doi: 10.1093/ije/dym213. [DOI] [PubMed] [Google Scholar]
- 2.Niu W, Qi Y, Hou S, Zhou W, Qiu C. Correlation of angiotensin-converting enzyme 2 gene polymorphisms with stage 2 hypertension in Han Chinese. Translational Research. 2007;150(6):374–380. doi: 10.1016/j.trsl.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Manunta P, Citterio L, Lanzani C, Ferrandi M. Adducin polymorphisms and the treatment of hypertension. Pharmacogenomics. 2007;8(5):465–472. doi: 10.2217/14622416.8.5.465. [DOI] [PubMed] [Google Scholar]
- 4.Mo W, Zhang G, Yang T, et al. The genetic polymorphisms of β3-adrenergic receptor (AR) Trp64Arg and β2-AR Gln27Glu are associated with obesity in Chinese male hypertensive patients. Clinical Chemistry and Laboratory Medicine. 2007;45(4):493–498. doi: 10.1515/CCLM.2007.089. [DOI] [PubMed] [Google Scholar]
- 5.Nejatizadeh A, Kumar R, Stobdan T, et al. Endothelial nitric oxide synthase gene haplotypes and circulating nitric oxide levels significantly associate with risk of essential hypertension. Free Radical Biology and Medicine. 2008;44(11):1912–1918. doi: 10.1016/j.freeradbiomed.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Levy D, DeStefano AL, Larson MG, et al. Evidence for a gene influencing blood pressure on chromosome 17: genome scan linkage results for longitudinal blood pressure phenotypes in subjects from the Framingham Heart study. Hypertension. 2000;36(4):477–483. doi: 10.1161/01.hyp.36.4.477. [DOI] [PubMed] [Google Scholar]
- 7.Jurkovicova D, Sedlakova B, Riecansky I, et al. Cardiovascular diseases and molecular variants of the renin-angiotensin system components in Slovak population. General Physiology and Biophysics. 2007;26(1):27–32. [PubMed] [Google Scholar]
- 8.He Q, Fan C, Yu M, et al. Associations of ACE gene insertion/deletion polymorphism, ACE activity, and ACE mRNA expression with hypertension in a Chinese population. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0075870.e75870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma Y, Ni W, Zhu W, Xiong Y, Deng X. Association of genetic polymorphisms of CYP 2C19 with hypertension in a Chinese Han population. Blood Pressure. 2011;20(3):166–170. doi: 10.3109/08037051.2010.542640. [DOI] [PubMed] [Google Scholar]
- 10.Petrovic D. Association of the −262C/T polymorphism in the catalase gene promoter and the C242T polymorphism of the NADPH oxidase P22phox gene with essential arterial hypertension in patients with diabetes mellitus type 2. Clinical and Experimental Hypertension. 2014;36(1):36–39. doi: 10.3109/10641963.2013.783051. [DOI] [PubMed] [Google Scholar]
- 11.Zhang LN, Ji LD, Fei LJ, Yuan F, Zhang YM, Xu J. Association between polymorphisms of alpha-adducin gene and essential hypertension in Chinese population. BioMed Research International. 2013;2013:5 pages. doi: 10.1155/2013/451094.451094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Török J. Participation of nitric oxide in different models of experimental hypertension. Physiological Research. 2008;57(6):813–825. doi: 10.33549/physiolres.931581. [DOI] [PubMed] [Google Scholar]
- 13.Bunting S, Gryglewski R, Moncada S, Vane JR. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and coeliac arteries and inhibits platelet aggregation. Prostaglandins. 1976;12(6):897–913. doi: 10.1016/0090-6980(76)90125-8. [DOI] [PubMed] [Google Scholar]
- 14.Weksler BB, Marcus AJ, Jaffe EA. Synthesis of prostaglandin I2 (prostacyclin) by cultured human and bovine endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(9):3922–3926. doi: 10.1073/pnas.74.9.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(5):1015–1021. doi: 10.1161/01.ATV.0000216044.49494.c4. [DOI] [PubMed] [Google Scholar]
- 17.Rafikov R, Fonseca FV, Kumar S, et al. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. Journal of Endocrinology. 2011;210(3):271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopincova J, Puzserova A, Bernatova I. L-NAME in the cardiovascular system—nitric oxide synthase activator? Pharmacological Reports. 2012;64:511–520. doi: 10.1016/s1734-1140(12)70846-0. [DOI] [PubMed] [Google Scholar]
- 19.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. European Heart Journal. 2012;33(7):829d–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopincová J, Púzserová A, Bernátová I. Chronic low-dose L-NAME treatment effect on cardiovascular system of borderline hypertensive rats: feedback regulation? Neuroendocrinology Letters. 2008;29(5):784–789. [PubMed] [Google Scholar]
- 21.Bernátová I, Pecháňová O, Kristek F. Mechanism of structural remodelling of the rat aorta during long-term N(G)-nitro-L-arginine methyl ester treatment. Japanese Journal of Pharmacology. 1999;81(1):99–106. doi: 10.1254/jjp.81.99. [DOI] [PubMed] [Google Scholar]
- 22.Garg UC, Hassid A. Mechanisms of nitrosothio-induced antimitogenesis in aortic smooth muscle cells. European Journal of Pharmacology. 1993;237(2-3):243–249. doi: 10.1016/0014-2999(93)90275-m. [DOI] [PubMed] [Google Scholar]
- 23.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. Journal of Cardiovascular Pharmacology. 1996;28(5):703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- 24.Parkington HC, Coleman HA, Tare M. Prostacyclin and endothelium-dependent hyperpolarization. Pharmacological Research. 2004;49(6):509–514. doi: 10.1016/j.phrs.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 25.Davidge ST, Baker PN, McLaughlin MK, Roberts JM. Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circulation Research. 1995;77(2):274–283. doi: 10.1161/01.res.77.2.274. [DOI] [PubMed] [Google Scholar]
- 26.Niwano K, Arai M, Tomaru K, Uchiyama T, Ohyama Y, Kurabayashi M. Transcriptional stimulation of the eNOS gene by the stable prostacyclin analogue beraprost is mediated through cAMP-responsive element in vascular endothelial cells: close link between PGI2 signal and NO pathways. Circulation Research. 2003;93(6):523–530. doi: 10.1161/01.RES.0000091336.55487.F7. [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skovgaard N, Gouliaev A, Aalling M, Simonsen U. The role of endogenous H2S in cardiovascular physiology. Current Pharmaceutical Biotechnology. 2011;12(9):1385–1393. doi: 10.2174/138920111798280956. [DOI] [PubMed] [Google Scholar]
- 29.Félétou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Annals of Medicine. 2007;39(7):495–516. doi: 10.1080/07853890701491000. [DOI] [PubMed] [Google Scholar]
- 30.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396(6708):269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 31.Han J, Chen ZW, He GW. Acetylcholine- and sodium hydrosulfide-induced endothelium-dependent relaxation and hyperpolarization in cerebral vessels of global cerebral ischemia-reperfusion rat. Journal of Pharmacological Sciences. 2013;121:318–326. doi: 10.1254/jphs.12277fp. [DOI] [PubMed] [Google Scholar]
- 32.Tang G, Yang G, Jiang B, Ju Y, Wu L, Wang R. H2S is an endothelium-derived hyperpolarizing factor. Antioxidants and Redox Signaling. 2013;19:1634–1646. doi: 10.1089/ars.2012.4805. [DOI] [PubMed] [Google Scholar]
- 33.Fleming I, Busse R. Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hypertension. 2006;47(4):629–633. doi: 10.1161/01.HYP.0000208597.87957.89. [DOI] [PubMed] [Google Scholar]
- 34.Takaki A, Morikawa K, Tsutsui M, et al. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. Journal of Experimental Medicine. 2008;205(9):2053–2063. doi: 10.1084/jem.20080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bachschmid MM, Schildknecht S, Matsui R, et al. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Annals of Medicine. 2013;45:17–36. doi: 10.3109/07853890.2011.645498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slezak P, Puzserova A, Balis P, et al. Genotype-related effect of crowding stress on blood pressure and vascular function in young female rats. BioMed Research International. 2014;2014:11 pages. doi: 10.1155/2014/413629.413629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. Journal of Experimental Medicine. 2000;192(12):1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cediel E, Vázquez-Cruz B, Navarro-Cid J, et al. Role of endothelin-1 and thromboxane A2 in renal vasoconstriction induced by angiotensin II in diabetes and hypertension. Kidney International, Supplement. 2002;62(supplement 82):S2–S7. doi: 10.1046/j.1523-1755.62.s82.2.x. [DOI] [PubMed] [Google Scholar]
- 39.Konczalla J, Wanderer S, Mrosek J, et al. Crosstalk between the angiotensin and endothelin-system in the cerebrovasculature. Current Neurovascular Research. 2013;10:335–345. doi: 10.2174/15672026113109990030. [DOI] [PubMed] [Google Scholar]
- 40.D’Amours M, Lebel M, Grose JH, Larivière R. Renal and vascular effects of chronic nitric oxide synthase inhibition: involvement of endothelin 1 and angiotensin II. Canadian Journal of Physiology and Pharmacology. 1999;77(1):8–16. doi: 10.1139/cjpp-77-1-8. [DOI] [PubMed] [Google Scholar]
- 41.Luscher TF, Boulanger CM, Dohi Y, Yang Z. Endothelium-derived contracting factors. Hypertension. 1992;19(2):117–130. doi: 10.1161/01.hyp.19.2.117. [DOI] [PubMed] [Google Scholar]
- 42.Félétou M, Cohen RA, Vanhoutte PM, Verbeuren TJ. TP receptors and oxidative stress. Hand in hand from endothelial dysfunction to atherosclerosis. Advances in Pharmacology. 2010;60:90–106. doi: 10.1016/B978-0-12-385061-4.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, Bian KA, Bukoski RD. A non-cyclo-oxygenase, non-nitric oxide relaxing factor is present in resistance arteries of normotensive but not spontaneously hypertensive rats. American Journal of the Medical Sciences. 1994;307(1):7–14. doi: 10.1097/00000441-199401000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Vanhoutte PM, Feletou M, Taddei S. Endothelium-dependent contractions in hypertension. British Journal of Pharmacology. 2005;144(4):449–458. doi: 10.1038/sj.bjp.0706042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Virdis A, Colucci R, Versari D, et al. Atorvastatin prevents endothelial dysfunction in mesenteric arteries from spontaneously hypertensive rats role of cyclooxygenase 2-derived contracting prostanoids. Hypertension. 2009;53(6):1008–1016. doi: 10.1161/HYPERTENSIONAHA.109.132258. [DOI] [PubMed] [Google Scholar]
- 46.Félétou M, Verbeuren TJ, Vanhoutte PM. Endothelium-dependent contractions in SHR: a tale of prostanoid TP and IP receptors. British Journal of Pharmacology. 2009;156(4):563–574. doi: 10.1111/j.1476-5381.2008.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. British Journal of Pharmacology. 2005;146(6):834–845. doi: 10.1038/sj.bjp.0706390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majzunova M, Dovinova I, Barancik M, Chan JY. Redox signaling in pathophysiology of hypertension. Journal of Biomedical Science. 2013;20, article 69 doi: 10.1186/1423-0127-20-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Oberley LW. Redox regulation of transcriptional activators. Free Radical Biology and Medicine. 1996;21(3):335–348. doi: 10.1016/0891-5849(96)00109-8. [DOI] [PubMed] [Google Scholar]
- 50.Karin M, Takahashi T, Kapahi P, et al. Oxidative stress and gene expression: the AP-1 and NF-κB connections. BioFactors. 2001;15(2–4):87–89. doi: 10.1002/biof.5520150207. [DOI] [PubMed] [Google Scholar]
- 51.Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology: implications in hypertension. Histochemistry and Cell Biology. 2004;122(4):339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 52.Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. Journal of Biomedicine and Biotechnology. 2012;2012:14 pages. doi: 10.1155/2012/936486.936486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puri N, Zhang F, Monu SR, et al. Antioxidants condition pleiotropic vascular responses to exogenous H(2)O(2): role of modulation of vascular TP receptors and the heme oxygenase system. Antioxidants and Redox Signaling. 2013;18:471–480. doi: 10.1089/ars.2012.4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liskova S, Petrova M, Karen P, Kunes J, Zicha J. Effects of aging and hypertension on the participation of endothelium-derived constricting factor (EDCF) in norepinephrine-induced contraction of rat femoral artery. European Journal of Pharmacology. 2011;667(1–3):265–270. doi: 10.1016/j.ejphar.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 55.Evora PRB. An open discussion about endothelial dysfunction: is it timely to propose a classification? International Journal of Cardiology. 2000;73(3):289–292. doi: 10.1016/s0167-5273(00)00223-0. [DOI] [PubMed] [Google Scholar]
- 56.Rajendran P, Rengarajan T, Thangavel J, et al. The vascular endothelium and human diseases. International Journal of Biological Sciences. 2013;9:1057–1069. doi: 10.7150/ijbs.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rees DD, Palmer RMJ, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(9):3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aisaka K, Gross SS, Griffith OW, Levi R. N(G)-methylarginine, an inhibitor of endothelium-derived nitric oxide synthesis, is a potent pressor agent in the guinea pig: does nitric oxide regulate blood pressure in vivo? Biochemical and Biophysical Research Communications. 1989;160(2):881–886. doi: 10.1016/0006-291x(89)92517-5. [DOI] [PubMed] [Google Scholar]
- 59.Chu A, Chambers DE, Lin C-C, et al. Effects of inhibition of nitric oxide formation on basal vasomotion and endothelium-dependent responses of the coronary arteries in awake dogs. The Journal of Clinical Investigation. 1991;87(6):1964–1968. doi: 10.1172/JCI115223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weldon SM, Winquist RJ, Madwed JB. Differential effects of L-NAME on blood pressure and heart rate responses to acetylcholine and bradykinin in cynomolgus primates. Journal of Pharmacology and Experimental Therapeutics. 1995;272(1):126–133. [PubMed] [Google Scholar]
- 61.Hecker M, Mitchell JA, Harris HJ, Katsura M, Thiemermann C, Vane JR. Endothelial cells metabolize N(G)-monomethyl-L-arginine to L-citrulline and subsequently to L-arginine. Biochemical and Biophysical Research Communications. 1990;167(3):1037–1043. doi: 10.1016/0006-291x(90)90627-y. [DOI] [PubMed] [Google Scholar]
- 62.Vargas HM, Ignarro LJ, Chaudhuri G. Physiological release of nitric oxide is dependent on the level of vascular tone. European Journal of Pharmacology. 1990;190(3):393–397. doi: 10.1016/0014-2999(90)94204-b. [DOI] [PubMed] [Google Scholar]
- 63.Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis: a new model of arterial hypertension. Hypertension. 1992;20(3):298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- 64.Delacrétaz E, Hayoz D, Osterheld MC, Genton CY, Brunner HR, Waeber B. Long-term nitric oxide synthase inhibition and distensibility of carotid artery in intact rats. Hypertension. 1994;23(6):967–970. doi: 10.1161/01.hyp.23.6.967. [DOI] [PubMed] [Google Scholar]
- 65.Kristek F, Gerová M, Devát L, Varga I. Remodelling of septal branch of coronary artery and carotid artery in L-NAME treated rats. Physiological Research. 1996;45(4):329–333. [PubMed] [Google Scholar]
- 66.Bernátová I, Pecháňová O, Šimko F. Captopril prevents NO-deficient hypertension and left ventricular hypertrophy without affecting nitric oxide synthase activity in rats. Physiological Research. 1996;45(4):311–316. [PubMed] [Google Scholar]
- 67.Arnal J-F, Warin L, Michel J-B. Determinants of aortic cyclic guanosine monophosphate in hypertension induced by chronic inhibition of nitric oxide synthase. The Journal of Clinical Investigation. 1992;90(2):647–652. doi: 10.1172/JCI115906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dananberg J, Sider RS, Grekin RJ. Sustained hypertension induced by orally administered nitro-L-arginine. Hypertension. 1993;21(3):359–363. doi: 10.1161/01.hyp.21.3.359. [DOI] [PubMed] [Google Scholar]
- 69.Manning RD, Jr., Hu L, Mizelle HL, Montani J-P, Norton MW. Cardiovascular responses to long-term blockade of nitric oxide synthesis. Hypertension. 1993;22(1):40–48. doi: 10.1161/01.hyp.22.1.40. [DOI] [PubMed] [Google Scholar]
- 70.Török J, Gerová M. Vascular responses after long-term inhibition of nitric oxide synthesis in newborn dogs. Physiological Research. 1996;45(4):323–328. [PubMed] [Google Scholar]
- 71.Holécyová A, Török J, Bernátová I, Pecháňová O. Restriction of nitric oxide rather than elevated blood pressure is responsible for alterations of vascular responses in nitric oxide-deficient hypertension. Physiological Research. 1996;45(4):317–321. [PubMed] [Google Scholar]
- 72.Bernátová I, Pechánová O, Babál P, Kyselá S, Stvrtina S, Andriantsitohaina R. Wine polyphenols improve cardiovascular remodeling and vascular function in NO-deficient hypertension. American Journal of Physiology—Heart and Circulatory Physiology. 2002;282(3):H942–H948. doi: 10.1152/ajpheart.00724.2001. [DOI] [PubMed] [Google Scholar]
- 73.Qu C, Leung SWS, Vanhoutte PM, Man RYK. Chronic inhibition of nitric-oxide synthase potentiates endothelium-dependent contractions in the rat aorta by augmenting the expression of cyclooxygenase-2. Journal of Pharmacology and Experimental Therapeutics. 2010;334(2):373–380. doi: 10.1124/jpet.110.167098. [DOI] [PubMed] [Google Scholar]
- 74.Paulis L, Zicha J, Kunes J, et al. Regression of L-NAME-induced hypertension: the role of nitric oxide and endothelium-derived constricting factor. Hypertension Research. 2008;31(4):793–803. doi: 10.1291/hypres.31.793. [DOI] [PubMed] [Google Scholar]
- 75.Pecháňová O, Bernátová I, Pelouch V, Šimko F. Protein remodelling of the heart in NO-deficient hypertension: the effect of captopril. Journal of Molecular and Cellular Cardiology. 1997;29(12):3365–3374. doi: 10.1006/jmcc.1997.0566. [DOI] [PubMed] [Google Scholar]
- 76.Bernátová I, Pecháňová O, Šimko F. Effect of captopril in L-NAME-induced hypertension on the rat myocardium, aorta, brain and kidney. Experimental Physiology. 1999;84(6):1095–1105. [PubMed] [Google Scholar]
- 77.Dillenburg DR, Mostarda C, Moraes-Silva IC, et al. Resveratrol and grape juice differentially ameliorate cardiovascular autonomic modulation in L-NAME-treated rats. Autonomic Neuroscience. 2013;179(1-2):9–13. doi: 10.1016/j.autneu.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 78.Nakmareong S, Kukongviriyapan U, Pakdeechote P, et al. Tetrahydrocurcumin alleviates hypertension, aortic stiffening and oxidative stress in rats with nitric oxide deficiency. Hypertension Research. 2012;35(4):418–425. doi: 10.1038/hr.2011.180. [DOI] [PubMed] [Google Scholar]
- 79.Babál P. Chronic inhibition of NO synthesis produces myocardial fibrosis and arterial media hyperplasia. Histology and Histopathology. 1997;12(3):623–629. [PubMed] [Google Scholar]
- 80.Tsutsui M, Shimokawa H, Otsuji Y, Yanagihara N. Pathophysiological relevance of NO signaling in the cardiovascular system: novel insight from mice lacking all NO synthases. Pharmacology and Therapeutics. 2010;128(3):499–508. doi: 10.1016/j.pharmthera.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 81.Monshita T, Tsutsui M, Shimokawa H, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(30):10616–10621. doi: 10.1073/pnas.0502236102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang PL. Neuronal and endothelial nitric oxide synthase gene knockout mice. Brazilian Journal of Medical and Biological Research. 1999;32(11):1353–1359. doi: 10.1590/s0100-879x1999001100005. [DOI] [PubMed] [Google Scholar]
- 83.Duplain H, Burcelin Ŕ, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104(3):342–345. doi: 10.1161/01.cir.104.3.342. [DOI] [PubMed] [Google Scholar]
- 84.le Gouill E, Jimenez M, Binnert C, et al. Endothelial nitric oxide synthase (eNOS) knockout mice have defective mitochondrial β-oxidation. Diabetes. 2007;56(11):2690–2696. doi: 10.2337/db06-1228. [DOI] [PubMed] [Google Scholar]
- 85.Yamashita C, Tazawa N, Ohkita M, Matsumura Y. Exaggerated renal pathology of partial ablation-induced chronic renal failure in eNOS deficient mice. Biological and Pharmaceutical Bulletin. 2008;31(5):1029–1031. doi: 10.1248/bpb.31.1029. [DOI] [PubMed] [Google Scholar]
- 86.Lehnen AM, Rodrigues B, Irigoyen MC, de Angelis K, Schaan BD. Cardiovascular changes in animal models of metabolic syndrome. Journal of Diabetes Research. 2013;2013:11 pages. doi: 10.1155/2013/761314.761314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chou T, Yen M, Li C, Ding Y. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension. 1998;31(2):643–648. doi: 10.1161/01.hyp.31.2.643. [DOI] [PubMed] [Google Scholar]
- 88.Yang Q, Xue H, Wong W, et al. AVE3085, an enhancer of endothelial nitric oxide synthase, restores endothelial function and reduces blood pressure in spontaneously hypertensive rats. British Journal of Pharmacology. 2011;163(5):1078–1085. doi: 10.1111/j.1476-5381.2011.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kondrashov A, Vrankova S, Dovinova I, et al. The effects of new Alibernet red wine extract on nitric oxide and reactive oxygen species production in spontaneously hypertensive rats. Oxidative Medicine and Cellular Longevity. 2012;2012:8 pages. doi: 10.1155/2012/806285.806285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nava E, Noll G, Luscher TF. Increased activity of constitutive nitric oxide synthase in cardiac endothelium in spontaneous hypertension. Circulation. 1995;91(9):2310–2313. doi: 10.1161/01.cir.91.9.2310. [DOI] [PubMed] [Google Scholar]
- 91.Vaziri ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension. 2000;36(6):957–964. doi: 10.1161/01.hyp.36.6.957. [DOI] [PubMed] [Google Scholar]
- 92.Bernatova I, Csizmadiova Z, Kopincova J, Puzserova A. Vascular function and nitric oxide production in chronic social-stress-exposed rats with various family history of hypertension. Journal of Physiology and Pharmacology. 2007;58(3):487–501. [PubMed] [Google Scholar]
- 93.Púzserová A, Csizmadiová Z, Bernátová I. Effect of blood pressure on L-NAME-sensitive component of vasorelaxation in adult rats. Physiological Research. 2007;56(supplement 2):S77–S84. doi: 10.33549/physiolres.931401. [DOI] [PubMed] [Google Scholar]
- 94.Caniffi C, Elesgaray R, Gironacci M, Arranz C, Costa MÁ. C-type natriuretic peptide effects on cardiovascular nitric oxide system in spontaneously hypertensive rats. Peptides. 2010;31(7):1309–1318. doi: 10.1016/j.peptides.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 95.Bernatova I, Conde MV, Kopincova J, González MC, Puzserova A, Arribas SM. Endothelial dysfunction in spontaneously hypertensive rats: focus on methodological aspects. Journal of Hypertension. 2009;27:S27–S31. doi: 10.1097/01.hjh.0000358834.18311.fc. [DOI] [PubMed] [Google Scholar]
- 96.Puzserova A, Kopincova J, Slezak P, Balis P, Bernatova I. Endothelial dysfunction in femoral artery of the hypertensive rats is nitric oxide independent. Physiological Research. 2013;62(6):615–629. doi: 10.33549/physiolres.932517. [DOI] [PubMed] [Google Scholar]
- 97.Mori Y, Ohyanagi M, Koida S, Ueda A, Ishiko K, Iwasaki T. Effects of endothelium-derived hyperpolariziing factor and nitric oxide on endothelial function in femoral resistance arteries of spontaneously hypertensive rats. Hypertension Research. 2006;29(3):187–195. doi: 10.1291/hypres.29.187. [DOI] [PubMed] [Google Scholar]
- 98.Slezak P, Puzserova A, Balis P, et al. Genotype-related effect of crowding stress on blood pressure and vasular function in young female rats. doi: 10.1155/2014/413629. BioMed Research International. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van de Voorde J, Vanheel B, Leusen I. Depressed endothelium-dependent relaxation in hypertension: relation to increased blood pressure and reversibility. Pflugers Archiv European Journal of Physiology. 1988;411(5):500–504. doi: 10.1007/BF00582370. [DOI] [PubMed] [Google Scholar]
- 100.Ungvari Z, Csiszar A, Kaminski PM, Wolin MS, Koller A. Chronic high pressure-induced arterial oxidative stress: involvement of protein kinase C-dependent NAD(P)H oxidase and local renin-angiotensin system. American Journal of Pathology. 2004;165(1):219–226. doi: 10.1016/S0002-9440(10)63290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Condezo-Hoyos L, Arribas SM, Abderrahim F, et al. Liver growth factor treatment reverses vascular and plasmatic oxidative stress in spontaneously hypertensive rats. Journal of Hypertension. 2012;30:1185–1194. doi: 10.1097/HJH.0b013e328353824b. [DOI] [PubMed] [Google Scholar]
- 102.Huang CF, Hsu CN, Chien SJ, Lin YJ, Huang LT, Tain YL. Aminoguanidine attenuates hypertension, whereas 7-nitroindazole exacerbates kidney damage in spontaneously hypertensive rats: the role of nitric oxide. European Journal of Pharmacology. 2013;699:233–240. doi: 10.1016/j.ejphar.2012.11.034. [DOI] [PubMed] [Google Scholar]
- 103.Thomson L, Trujillo M, Telleri R, Radi R. Kinetics of cytochrome c2+ oxidation by peroxynitrite implications for superoxide measurements in nitric oxide-producing biological systems. Archives of Biochemistry and Biophysics. 1995;319(2):491–497. doi: 10.1006/abbi.1995.1321. [DOI] [PubMed] [Google Scholar]
- 104.Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. Journal of Physiology and Pharmacology. 2013;64:409–421. [PubMed] [Google Scholar]
- 105.Matz RL, de Sotomayor MA, Schott C, Stoclet J-C, Andriantsitohaina R. Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. British Journal of Pharmacology. 2000;131(2):303–311. doi: 10.1038/sj.bjp.0703568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Forstermann U, Pollock JS, Schmidt HHHW, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(5):1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pandey D, Gratton J, Rafikov R, Black SM, Fulton DJR. Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Molecular Pharmacology. 2011;80(3):407–415. doi: 10.1124/mol.110.070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336(6081):597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ungvari Z, Koller A. Endothelin and prostaglandin H2/thromboxane A2 enhance myogenic constriction in hypertension by increasing Ca2+ sensitivity of arteriolar smooth muscle. Hypertension. 2000;36(5):856–861. doi: 10.1161/01.hyp.36.5.856. [DOI] [PubMed] [Google Scholar]
- 110.Wakui H, Dejima T, Tamura K, et al. Activation of angiotensin II type 1 receptor-associated protein exerts an inhibitory effect on vascular hypertrophy and oxidative stress in angiotensin II-mediated hypertension. Cardiovascular Research. 2013;100:511–519. doi: 10.1093/cvr/cvt225. [DOI] [PubMed] [Google Scholar]
- 111.Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005;45(2):283–287. doi: 10.1161/01.HYP.0000153051.56460.6a. [DOI] [PubMed] [Google Scholar]
- 112.Zhou M, Schulman IH, Pagano PJ, Jaimes EA, Raij L. Reduced NAD(P)H oxidase in low renin hypertension: link among angiotensin II, atherogenesis, and blood pressure. Hypertension. 2006;47(1):81–86. doi: 10.1161/01.HYP.0000197182.65554.c7. [DOI] [PubMed] [Google Scholar]
- 113.Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. American Journal of Physiology—Heart and Circulatory Physiology. 2008;295(1):H281–H288. doi: 10.1152/ajpheart.00304.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gómez-Guzmán M, Jiménez R, Sánchez M, et al. Epicatechin lowers blood pressure, restores endothelial function, and decreases oxidative stress and endothelin-1 and NADPH oxidase activity in DOCA-salt hypertension. Free Radical Biology and Medicine. 2012;52(1):70–79. doi: 10.1016/j.freeradbiomed.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 115.Al-Awwadi NA, Araiz C, Bornet A, et al. Extracts enriched in different polyphenolic families normalize increased cardiac NADPH oxidase expression while having differential effects on insulin resistance, hypertension, and cardiac hypertrophy in high-fructose-fed rats. Journal of Agricultural and Food Chemistry. 2005;53(1):151–157. doi: 10.1021/jf048919f. [DOI] [PubMed] [Google Scholar]
- 116.Korandji C, Zeller M, Guilland JC, et al. Time course of asymmetric dimethylarginine (ADMA) and oxidative stress in fructose-hypertensive rats: a model related to metabolic syndrome. Atherosclerosis. 2011;214(2):310–315. doi: 10.1016/j.atherosclerosis.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 117.Vaziri ND, Sica DA. Lead-induced hypertension: role of oxidative stress. Current Hypertension Reports. 2004;6(4):314–320. doi: 10.1007/s11906-004-0027-3. [DOI] [PubMed] [Google Scholar]
- 118.Zhou M, Nishida Y, Chen Q, Kosaka H. Endothelium-derived contracting factor in carotid artery of hypertensive Dahl rats. Hypertension. 1999;34(1):39–43. doi: 10.1161/01.hyp.34.1.39. [DOI] [PubMed] [Google Scholar]
- 119.Lukaszewicz KM, Lombard JH. Role of the CYP4A/20-HETE pathway in vascular dysfunction of the Dahl salt-sensitive rat. Clinical Science. 2013;124:695–700. doi: 10.1042/CS20120483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barton M, D’Uscio LV, Shaw S, Meyer P, Moreau P, Lüscher TF. ET(A) receptor blockade prevents increased tissue endothelin-1, vascular hypertrophy, and endothelial dysfunction in salt-sensitive hypertension. Hypertension. 1998;31(1):499–504. doi: 10.1161/01.hyp.31.1.499. [DOI] [PubMed] [Google Scholar]
- 121.Zicha J, Dobešová Z, Kuneš J, Vaněčková I. Chronic endothelin A receptor blockade attenuates contribution of sympathetic nervous system to salt hypertension development in adult but not in young Dahl rats. Acta Physiologica. 2012;205(1):124–132. doi: 10.1111/j.1748-1716.2012.02395.x. [DOI] [PubMed] [Google Scholar]
- 122.Cao Y, Mu JJ, Fang Y, Yuan ZY, Liu FQ. Impact of high salt independent of blood pressure on PRMT/ADMA/DDAH pathway in the aorta of Dahl salt-sensitive rats. International Journal of Molecular Sciences. 2013;14:8062–8072. doi: 10.3390/ijms14048062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tian N, Gannon AW, Khalil RA, Manning RD., Jr. Mechanisms of salt-sensitive hypertension: role of renal medullary inducible nitric oxide synthase. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2003;284(2):R372–R379. doi: 10.1152/ajpregu.00509.2002. [DOI] [PubMed] [Google Scholar]
- 124.Lariviere R, Deng LY, Day R, Sventek P, Thibault G, Schiffrin EL. Increased endothelin-1 gene expression in the endothelium of coronary arteries and endocardium in the DOCA-salt hypertensive rat. Journal of Molecular and Cellular Cardiology. 1995;27(10):2123–2131. doi: 10.1016/s0022-2828(95)91263-0. [DOI] [PubMed] [Google Scholar]
- 125.Adeagbo ASO, Zhang X, Patel D, et al. Cyclo-oxygenase-2, endothelium and aortic reactivity during deoxycorticosterone acetate salt-induced hypertension. Journal of Hypertension. 2005;23(5):1025–1036. doi: 10.1097/01.hjh.0000166844.42227.5c. [DOI] [PubMed] [Google Scholar]
- 126.Adeagbo ASO, Joshua IG, Falkner C, Matheson PJ. Tempol, an antioxidant, restores endothelium-derived hyperpolarizing factor-mediated vasodilation during hypertension. European Journal of Pharmacology. 2003;481(1):91–100. doi: 10.1016/j.ejphar.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 127.Dumont AS, Hyndman ME, Dumont RJ, et al. Improvement of endothelial function in insulin-resistant carotid arteries treated with pravastatin. Journal of Neurosurgery. 2001;95(3):466–471. doi: 10.3171/jns.2001.95.3.0466. [DOI] [PubMed] [Google Scholar]
- 128.Lee DH, Lee J, Kang DG, Paek YW, Chung DJ, Chung MY. Increased vascular endothelin-1 gene expression with unaltered nitric oxide synthase levels in fructose-induced hypertensive rats. Metabolism. 2001;50(1):74–78. doi: 10.1053/meta.2001.19527. [DOI] [PubMed] [Google Scholar]
- 129.Navarro-Cid J, Maeso R, Perez-Vizcaino F, et al. Effects of losartan on blood pressure, metabolic alterations, and vascular reactivity in the fructose-induced hypertensive rat. Hypertension. 1995;26(6):1074–1078. doi: 10.1161/01.hyp.26.6.1074. [DOI] [PubMed] [Google Scholar]
- 130.Esler M, Eikelis N, Schlaich M, et al. Chronic mental stress is a cause of essential hypertension: presence of biological markers of stress. Clinical and Experimental Pharmacology and Physiology. 2008;35(4):498–502. doi: 10.1111/j.1440-1681.2008.04904.x. [DOI] [PubMed] [Google Scholar]
- 131.Sparrenberger F, Fuchs SC, Moreira LB, Fuchs FD. Stressful life events and current psychological distress are associated with self-reported hypertension but not with true hypertension: results from a cross-sectional population-based study. BMC Public Health. 2008;8, article 357 doi: 10.1186/1471-2458-8-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fokkema DS, Koolhaas JM, van der Meulen J, Schoemaker R. Social stress induced pressure breathing and consequent blood pressure oscillation. Life Sciences. 1986;38(6):569–575. doi: 10.1016/0024-3205(86)90036-6. [DOI] [PubMed] [Google Scholar]
- 133.Andrews E, Jenkins C, Seachrist D, Dunphy G, Ely D. Social stress increases blood pressure and cardiovascular pathology in a normotensive rat model. Clinical and Experimental Hypertension. 2003;25(2):85–101. doi: 10.1081/ceh-120017930. [DOI] [PubMed] [Google Scholar]
- 134.Harrap SB, Louis WJ, Doyle AE. Failure of psychosocial stress to induce chronic hypertension in the rat. Journal of Hypertension. 1984;2(6):653–662. doi: 10.1097/00004872-198412000-00011. [DOI] [PubMed] [Google Scholar]
- 135.Lemaire V, Mormède P. Telemetered recording of blood pressure and heart rate in different strains of rats during chronic social stress. Physiology and Behavior. 1995;58(6):1181–1188. doi: 10.1016/0031-9384(95)02064-0. [DOI] [PubMed] [Google Scholar]
- 136.Mansi JA, Drolet G. Chronic stress induces sensitization in sympathoadrenal responses to stress in borderline hypertensive rats. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1997;272(3):R813–R820. doi: 10.1152/ajpregu.1997.272.3.R813. [DOI] [PubMed] [Google Scholar]
- 137.Sanders BJ, Lawler JE. The borderline hypertensive rat (BHR) as a model for environmentally- induced hypertension: a review and update. Neuroscience and Biobehavioral Reviews. 1992;16(2):207–217. doi: 10.1016/s0149-7634(05)80181-2. [DOI] [PubMed] [Google Scholar]
- 138.Bernatova I, Csizmadiova Z. Effect of chronic social stress on nitric oxide synthesis and vascular function in rats with family history of hypertension. Life Sciences. 2006;78(15):1726–1732. doi: 10.1016/j.lfs.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 139.Fleming I, Baum A, Davidson LM, Rectanus E, McArdle S. Chronic stress as a factor in physiologic reactivity to challenge. Health Psychology. 1987;6(3):221–237. doi: 10.1037//0278-6133.6.3.221. [DOI] [PubMed] [Google Scholar]
- 140.D’Atri DA, Fitzgerald EF, Kasl SV, Ostfeld AM. Crowding in prison: the relationship between changes in housing mode and blood pressure. Psychosomatic Medicine. 1981;43(2):95–105. doi: 10.1097/00006842-198104000-00001. [DOI] [PubMed] [Google Scholar]
- 141.Púzserová A, Csizmadiová Z, Andriantsitohaina R, Bernátová I. Vascular effects of red wine polyphenols in chronic stress-exposed Wistar-Kyoto rats. Physiological Research. 2006;55(supplement 1):S39–S47. doi: 10.33549/physiolres.930000.55.S1.39. [DOI] [PubMed] [Google Scholar]
- 142.Bernatova I, Puzserova A, Dubovicky M. Sex differences in social stress-induced pressor and behavioral responses in normotensive and prehypertensive rats. General Physiology and Biophysics. 2010;29(4):346–354. doi: 10.4149/gpb_2010_04_346. [DOI] [PubMed] [Google Scholar]
- 143.Puzserova A, Slezak P, Balis P, Bernatova I. Long-term social stress induces nitric oxide-independent endothelial dysfunction in normotensive rats. Stress. 2013;16:331–339. doi: 10.3109/10253890.2012.725116. [DOI] [PubMed] [Google Scholar]
- 144.Bernátová I, Púzserová A, Navarová J, Csizmadiová Z, Zeman M. Crowding-induced alterations in vascular system of Wistar-Kyoto rats: role of nitric oxide. Physiological Research. 2007;56(5):667–669. doi: 10.33549/physiolres.931386. [DOI] [PubMed] [Google Scholar]
- 145.Fuchs LC, Hoque AM, Clarke NL. Vascular and hemodynamic effects of behavioral stress in borderline hypertensive and Wistar-Kyoto rats. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1998;274(2):R375–R382. doi: 10.1152/ajpregu.1998.274.2.R375. [DOI] [PubMed] [Google Scholar]
- 146.Fuchs LC, Landas SK, Johnson AK. Behavioral stress alters coronary vascular reactivity in borderline hypertensive rats. Journal of Hypertension. 1997;15(3):301–307. doi: 10.1097/00004872-199715030-00012. [DOI] [PubMed] [Google Scholar]
- 147.Giulumian AD, Clark SG, Fuchs LC. Effect of behavioral stress on coronary artery relaxation altered with aging in BHR. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 1999;276(2):R435–R440. doi: 10.1152/ajpregu.1999.276.2.R435. [DOI] [PubMed] [Google Scholar]
- 148.Balkaya M, Prinz V, Custodis F, et al. Stress worsens endothelial function and ischemic stroke via glucocorticoids. Stroke. 2011;42(11):3258–3264. doi: 10.1161/STROKEAHA.110.607705. [DOI] [PubMed] [Google Scholar]
- 149.Radomski MW, Palmer RMJ, Moncada S. Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(24):10043–10047. doi: 10.1073/pnas.87.24.10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rogers KM, Bonar CA, Estrella JL, Yang S. Inhibitory effect of glucocorticoid on coronary artery endothelial function. American Journal of Physiology—Heart and Circulatory Physiology. 2002;283(5):H1922–H1928. doi: 10.1152/ajpheart.00364.2002. [DOI] [PubMed] [Google Scholar]
- 151.Wallerath T, Witte K, Schäfer SC, et al. Down-regulation of the expression of endothelial NO synthase is likely to contribute to qlucocorticoid-mediated hypertension. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13357–13362. doi: 10.1073/pnas.96.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Simmons WW, Ungureanu-Longrois D, Smith GK, Smith TW, Kelly RA. Glucocorticoids regulate inducible nitric oxide synthase by inhibiting tetrahydrobiopterin synthesis and L-arginine transport. The Journal of Biological Chemistry. 1996;271(39):23928–23937. doi: 10.1074/jbc.271.39.23928. [DOI] [PubMed] [Google Scholar]
- 153.Liu Y, Mladinov D, Pietrusz JL, Usa K, Liang M. Glucocorticoid response elements and 11β-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovascular Research. 2009;81(1):140–147. doi: 10.1093/cvr/cvn231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jun SS, Chen Z, Pace MC, Shaul PW. Glucocorticoids downregulate cyclooxygenase-1 gene expression and prostacyclin synthesis in fetal pulmonary artery endothelium. Circulation Research. 1999;84(2):193–200. doi: 10.1161/01.res.84.2.193. [DOI] [PubMed] [Google Scholar]
- 155.Zafir A, Banu N. Induction of oxidative stress by restraint stress and corticosterone treatments in rats. Indian Journal of Biochemistry and Biophysics. 2009;46(1):53–58. [PubMed] [Google Scholar]
- 156.Malyshev IY, Manukhina EB. Stress, adaptation, and nitric oxide. Biochemistry. 1998;63(7):840–853. [PubMed] [Google Scholar]
- 157.Stefano GB, Fricchione GL, Esch T. Relaxation: molecular and physiological significance. Medical Science Monitor. 2006;12(9):HY21–HY31. [PubMed] [Google Scholar]